
Influence of Asphaltene Modification on Structure of P3HT/Asphaltene Blends: Molecular Dynamics Simulations


Abstract
:1. Introduction
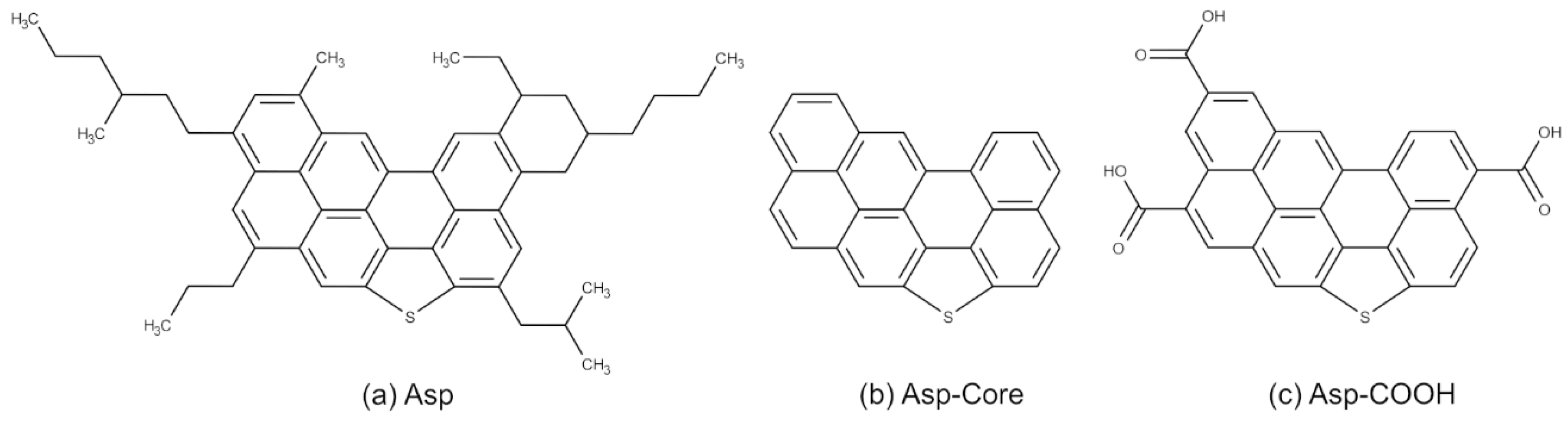
2. Models and Methods
3. Results and Discussion
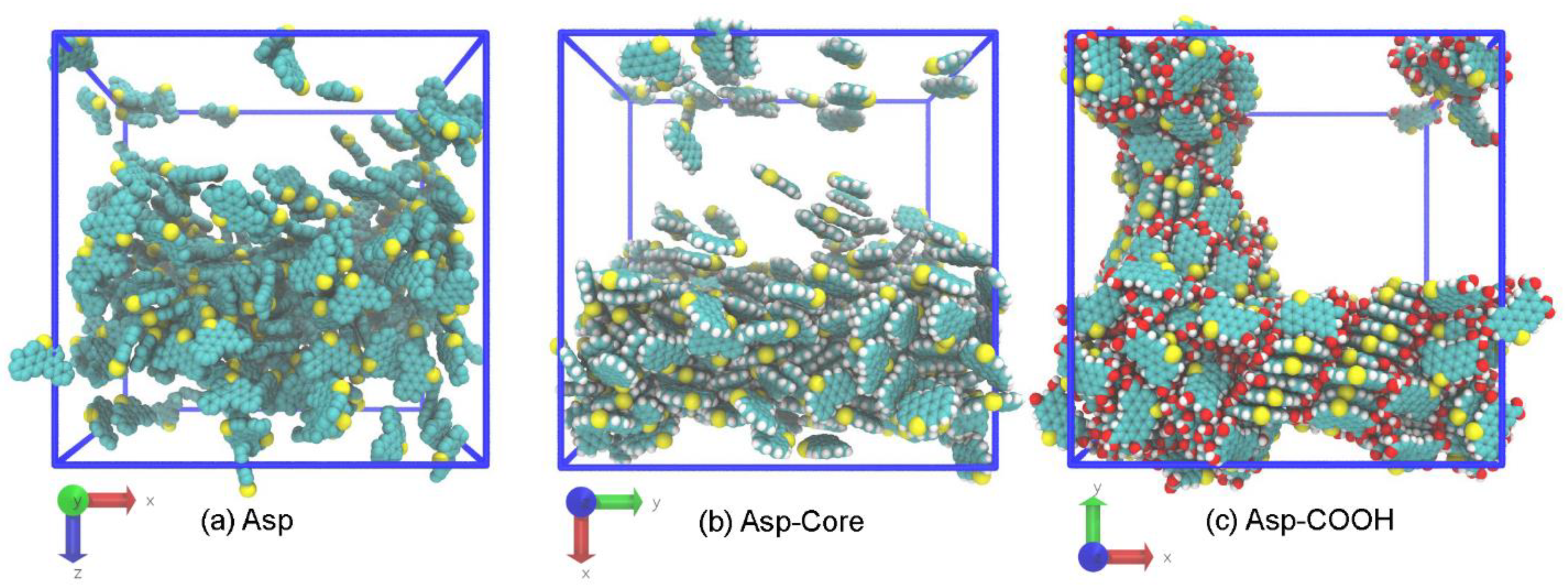
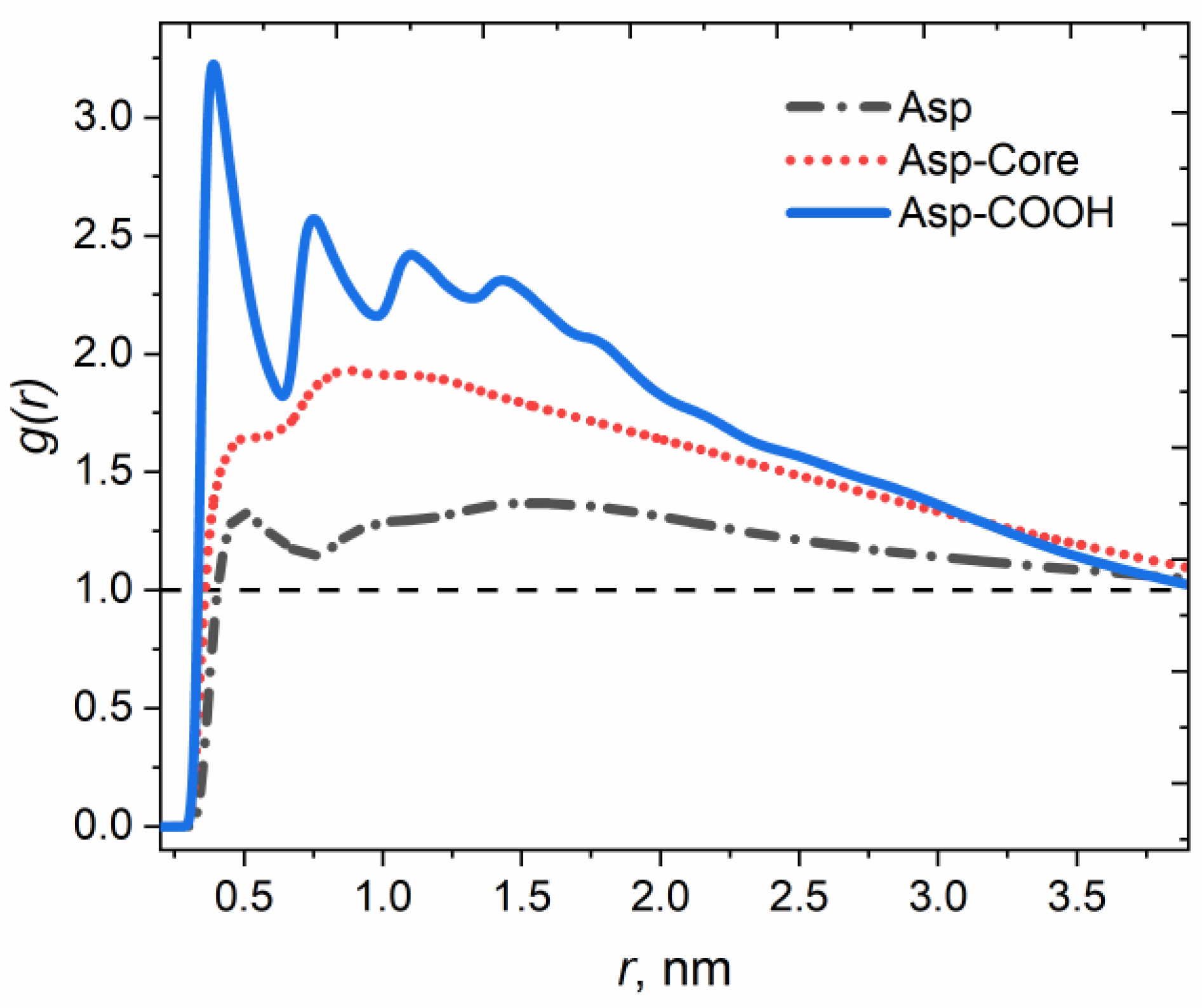
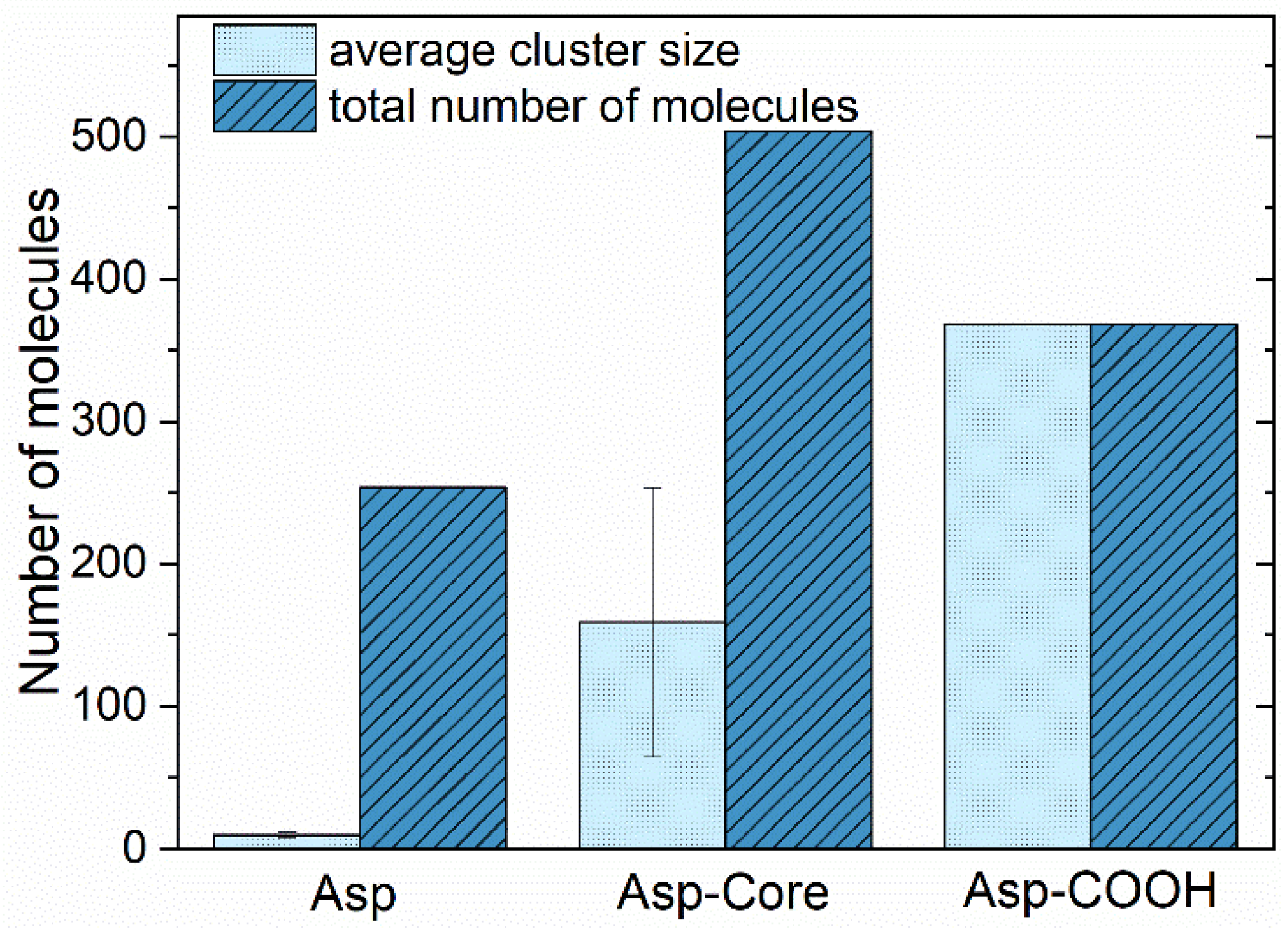
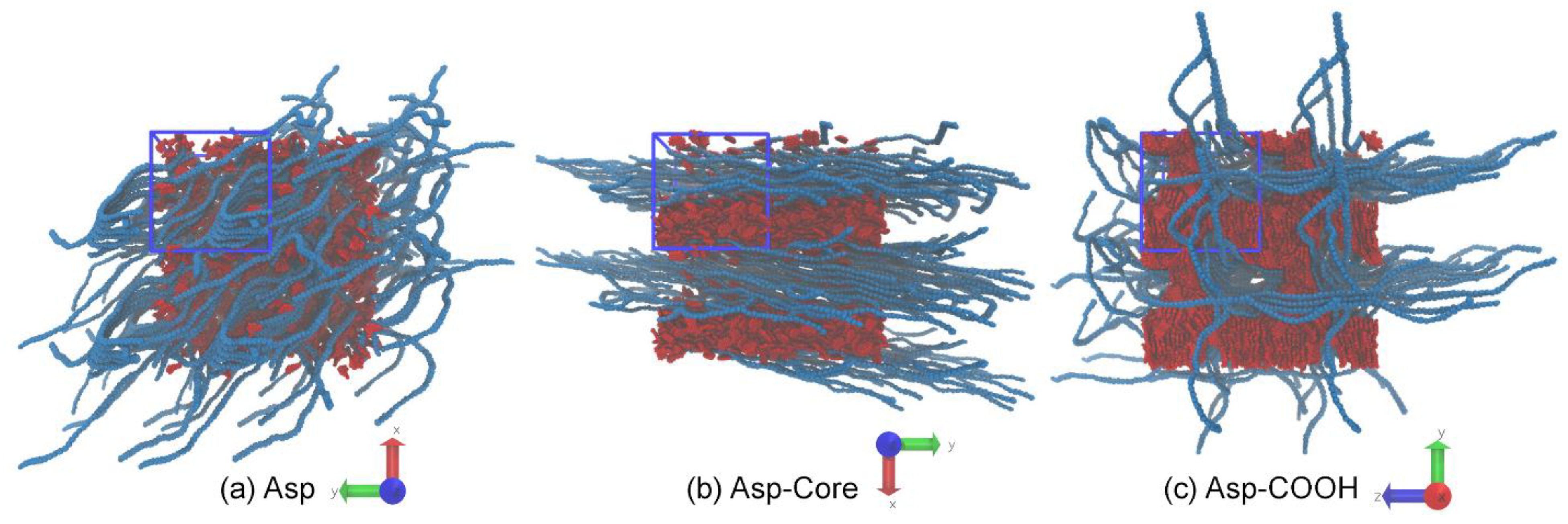
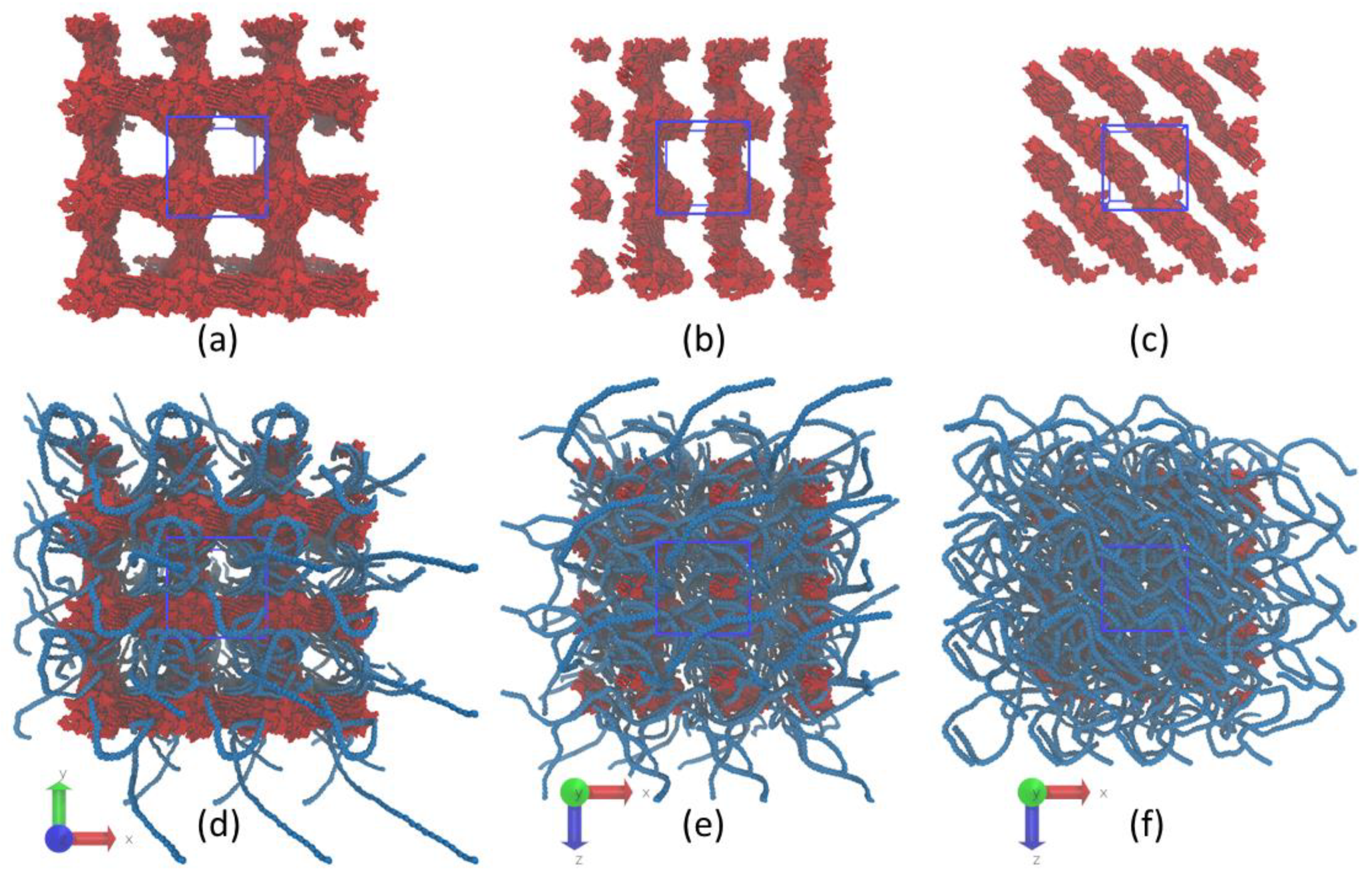
3.1. Structure of Asphaltene Phase
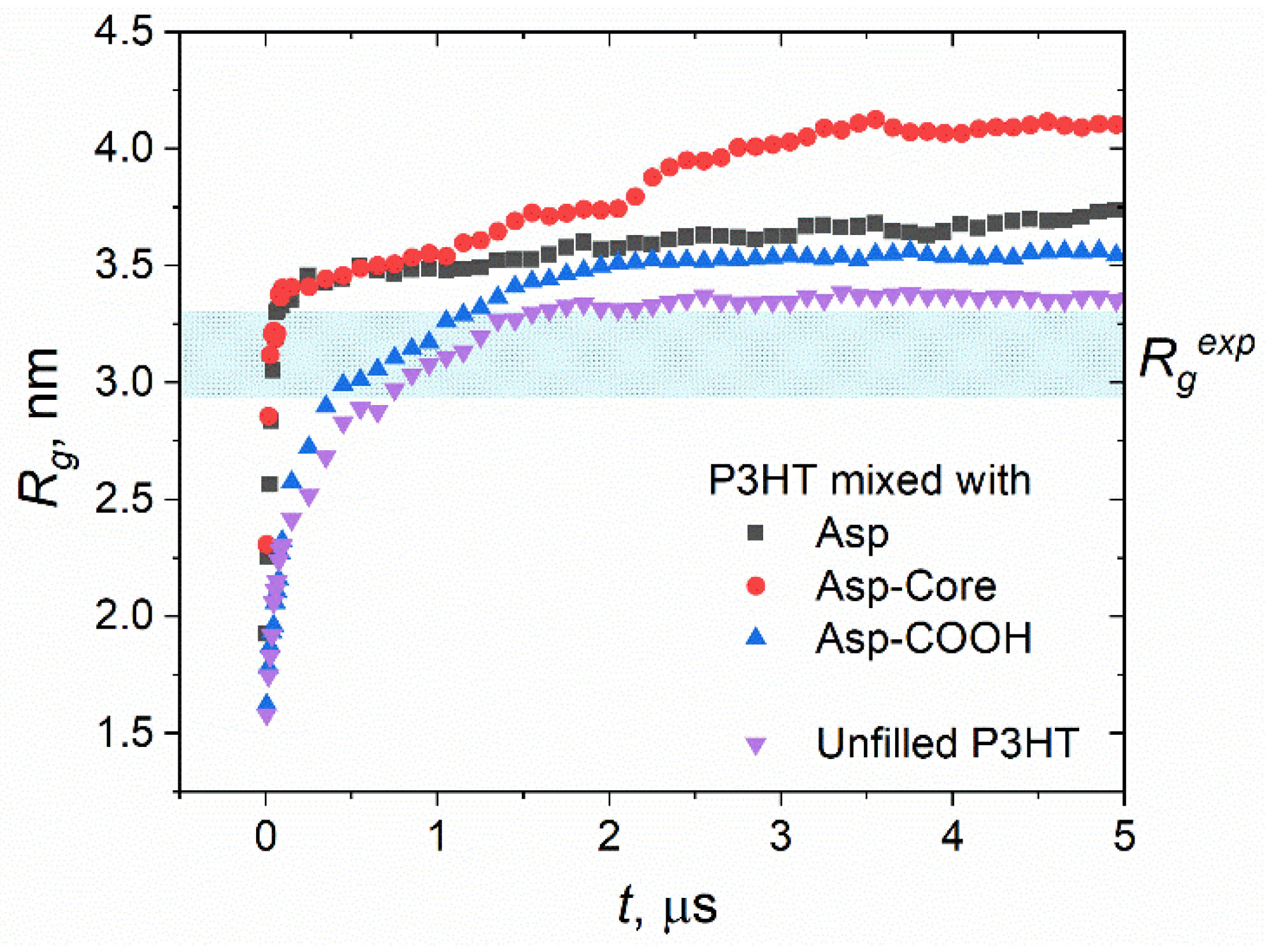
3.2. Structure of Polymer Phase
3.3. Concentration Effects
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rafique, S.; Abdullah, S.M.; Sulaiman, K.; Iwamoto, M. Fundamentals of Bulk Heterojunction Organic Solar Cells: An Overview of Stability/Degradation Issues and Strategies for Improvement. Renew. Sustain. Energy Rev. 2018, 84, 43–53. [Google Scholar] [CrossRef]
- Fukuda, K.; Yu, K.; Someya, T. The Future of Flexible Organic Solar Cells. Adv. Energy Mater. 2020, 10, 2000765. [Google Scholar] [CrossRef]
- Yin, Z.; Zhu, J.; He, Q.; Cao, X.; Tan, C.; Chen, H.; Yan, Q.; Zhang, H. Graphene-Based Materials for Solar Cell Applications. Adv. Energy Mater. 2014, 4, 1300574. [Google Scholar] [CrossRef]
- Adams, J.J. Asphaltene Adsorption, a Literature Review. Energy Fuels 2014, 28, 2831–2856. [Google Scholar] [CrossRef]
- Wu, H.; Thakur, V.K.; Kessler, M.R. Novel Low-Cost Hybrid Composites from Asphaltene/SBS Tri-Block Copolymer with Improved Thermal and Mechanical Properties. J. Mater. Sci. 2016, 51, 2394–2403. [Google Scholar] [CrossRef]
- Guzmán, R.; Ancheyta, J.; Trejo, F.; Rodríguez, S. Methods for Determining Asphaltene Stability in Crude Oils. Fuel 2017, 188, 530–543. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Glova, A.D.; Falkovich, S.G.; Ivanov, V.A.; Nazarychev, V.M.; Lyulin, A.V.; Larin, S.V.; Antonov, S.V.; Ganan, P.; Kenny, J.M. Computer Simulation of Asphaltenes. Pet. Chem. 2018, 58, 983–1004. [Google Scholar] [CrossRef] [Green Version]
- Fotland, P.; Anfindsen, H. Conductivity of Asphaltenes. In Structures and Dynamics of Asphaltenes; Springer: Boston, MA, USA, 1998; pp. 247–266. [Google Scholar]
- Evdokimov, I.N.; Losev, A.P. Electrical Conductivity and Dielectric Properties of Solid Asphaltenes. Energy Fuels 2010, 24, 3959–3969. [Google Scholar] [CrossRef]
- Dilabio, G.; Mackie, I.; Dettman, H. Asphaltene Components as Organic Electronic Materials 2015. U.S. Patent No. 9,065,059, 2015. [Google Scholar]
- Irwin, M.D.; Chianelli, R.R.; Maher, R.D., III. Methods and Apparatus Using Asphaltenes in Solid-State Organic Solar Cells 2013. U.S. Patent Application No. 13/588,737, 2013. [Google Scholar]
- Abujnah, R.E.; Sharif, H.; Torres, B.; Castillo, K.; Gupta, V.; Chaielli, R.R. Asphaltene as Light Harvesting Material in Dye-Sensitized Solar Cell: Resurrection of Ancient Leaves. J. Environ. Anal. Toxicol. 2016, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Deemer, E.M.; Chianelli, R.R. Novel Applications with Asphaltene Electronic Structure. In Modified Asphalt; Intech Open: London, UK, 2018. [Google Scholar]
- Bhui, U.K.; Ray, A.; Joshi, M.P. Feasibility Study of Crude Oil Asphaltenes as Light-Harvesting Materials for Organic Photovoltaics: Light Absorption Characteristics of the Thin Film with P3HT. In Macromolecular Characterization of Hydrocarbons for Sustainable Future; Springer: Singapore, 2021; pp. 129–139. [Google Scholar]
- He, Y.; Li, Y. Fullerene Derivative Acceptors for High Performance Polymer Solar Cells. Phys. Chem. Chem. Phys. 2011, 13, 1970–1983. [Google Scholar] [CrossRef]
- Cui, C.; Li, Y.; Li, Y. Fullerene Derivatives for the Applications as Acceptor and Cathode Buffer Layer Materials for Organic and Perovskite Solar Cells. Adv. Energy Mater. 2017, 7, 1601251. [Google Scholar] [CrossRef]
- Cui, C.; Li, Y. High-Performance Conjugated Polymer Donor Materials for Polymer Solar Cells with Narrow-Bandgap Nonfullerene Acceptors. Energy Environ. Sci. 2019, 12, 3225–3246. [Google Scholar] [CrossRef]
- Clarke, A.J.; Luke, J.; Meitzner, R.; Wu, J.; Wang, Y.; Lee, H.K.H.; Speller, E.M.; Bristow, H.; Cha, H.; Newman, M.J.; et al. Non-Fullerene Acceptor Photostability and Its Impact on Organic Solar Cell Lifetime. Cell Rep. Phys. Sci. 2021, 2, 100498. [Google Scholar] [CrossRef]
- Holliday, S.; Ashraf, R.S.; Wadsworth, A.; Baran, D.; Yousaf, S.A.; Nielsen, C.B.; Tan, C.-H.; Dimitrov, S.D.; Shang, Z.; Gasparini, N.; et al. High-Efficiency and Air-Stable P3HT-Based Polymer Solar Cells with a New Non-Fullerene Acceptor. Nat. Commun. 2016, 7, 11585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Wang, J.; Zhang, Z.-G.; Bai, H.; Li, Y.; Zhu, D.; Zhan, X. An Electron Acceptor Challenging Fullerenes for Efficient Polymer Solar Cells. Adv. Mater. 2015, 27, 1170–1174. [Google Scholar] [CrossRef]
- Kim, T.H.; Lee, H.J.; Saeed, M.A.; Son, J.H.; Woo, H.Y.; Kim, T.G.; Shim, J.W. Elastomeric Indoor Organic Photovoltaics with Superb Photothermal Endurance. Adv. Funct. Mater. 2022, 32, 2201921. [Google Scholar] [CrossRef]
- Ahsan Saeed, M.; Hyeon Kim, S.; Baek, K.; Hyun, J.K.; Youn Lee, S.; Won Shim, J. PEDOT:PSS: CuNW-Based Transparent Composite Electrodes for High-Performance and Flexible Organic Photovoltaics under Indoor Lighting. Appl. Surf. Sci. 2021, 567, 150852. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, C.H.; Saeed, M.A.; Ko, D.-H.; Lee, J.-H.; Shim, J.W. β-Cyclodextrin–Polyacryloyl Hydrazide-Based Surface Modification for Efficient Electron-Collecting Electrodes of Indoor Organic Photovoltaics. J. Mater. Res. Technol. 2022, 16, 1659–1666. [Google Scholar] [CrossRef]
- Borzdun, N.I.; Ramazanov, R.R.; Glova, A.D.; Larin, S.V.; Lyulin, S.V. Model Carboxyl-Containing Asphaltenes as Potential Acceptor Materials for Bulk Heterojunction Solar Cells. Energy Fuels 2021, 35, 8423–8429. [Google Scholar] [CrossRef]
- Tsourtou, F.D.; Peristeras, L.D.; Apostolov, R.; Mavrantzas, V.G. Molecular Dynamics Simulation of Amorphous Poly(3-Hexylthiophene). Macromolecules 2020, 53, 7810–7824. [Google Scholar] [CrossRef]
- Tummala, N.R.; Risko, C.; Bruner, C.; Dauskardt, R.H.; Brédas, J.-L. Entanglements in P3HT and Their Influence on Thin-Film Mechanical Properties: Insights from Molecular Dynamics Simulations. J. Polym. Sci. Part B Polym. Phys. 2015, 53, 934–942. [Google Scholar] [CrossRef] [Green Version]
- Poelking, C.; Andrienko, D. Effect of Polymorphism, Regioregularity and Paracrystallinity on Charge Transport in Poly(3-Hexylthiophene) [P3HT] Nanofibers. Macromolecules 2013, 46, 8941–8956. [Google Scholar] [CrossRef]
- Mehmood, U.; Al-Ahmed, A.; Hussein, I.A. Review on Recent Advances in Polythiophene Based Photovoltaic Devices. Renew. Sustain. Energy Rev. 2016, 57, 550–561. [Google Scholar] [CrossRef]
- R. Murad, A.; Iraqi, A.; Aziz, S.B.; N. Abdullah, S.; Brza, M.A. Conducting Polymers for Optoelectronic Devices and Organic Solar Cells: A Review. Polymers 2020, 12, 2627. [Google Scholar] [CrossRef] [PubMed]
- Borzdun, N.I.; Nazarychev, V.M.; Larin, S.V.; Reiter, G.; Lyulin, S.V. Self-Assembly of Oligo(Phenylene-Thiophene)s on Monolayer Graphene: Molecular Dynamics Simulations. J. Phys. Chem. C 2019, 123, 859–867. [Google Scholar] [CrossRef]
- Borzdun, N.I.; Larin, S.V.; Falkovich, S.G.; Nazarychev, V.M.; Volgin, I.V.; Yakimansky, A.V.; Lyulin, A.V.; Negi, V.; Bobbert, P.A.; Lyulin, S.V. Molecular Dynamics Simulation of Poly(3-Hexylthiophene) Helical Structure In Vacuo and in Amorphous Polymer Surrounding. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 2448–2456. [Google Scholar] [CrossRef] [Green Version]
- Greenstein, P.D.; Casabianca, L.B. Interplay Between π-Stacking and Hydrogen Bonding in the Self-Association of Different Isomers of Naphthalenedicarboxylic Acid. J. Phys. Chem. B 2017, 121, 5086–5093. [Google Scholar] [CrossRef]
- Mullins, O.C. The Asphaltenes. Annu. Rev. Anal. Chem. 2011, 4, 393–418. [Google Scholar] [CrossRef]
- Li, D.D.; Greenfield, M.L. Chemical Compositions of Improved Model Asphalt Systems for Molecular Simulations. Fuel 2014, 115, 347–356. [Google Scholar] [CrossRef]
- Glova, A.D.; Nazarychev, V.M.; Larin, S.V.; Lyulin, A.V.; Lyulin, S.V.; Gurtovenko, A.A. Asphaltenes as Novel Thermal Conductivity Enhancers for Liquid Paraffin: Insight from in Silico Modeling. J. Mol. Liq. 2022, 346, 117112. [Google Scholar] [CrossRef]
- Rahimi, K.; Botiz, I.; Stingelin, N.; Kayunkid, N.; Sommer, M.; Koch, F.P.V.; Nguyen, H.; Coulembier, O.; Dubois, P.; Brinkmann, M.; et al. Controllable Processes for Generating Large Single Crystals of Poly(3-Hexylthiophene). Angew. Chem. Int. Ed. 2012, 51, 11131–11135. [Google Scholar] [CrossRef] [PubMed]
- Agbolaghi, S.; Zenoozi, S. A Comprehensive Review on Poly(3-Alkylthiophene)-Based Crystalline Structures, Protocols and Electronic Applications. Org. Electron. 2017, 51, 362–403. [Google Scholar] [CrossRef]
- Koch, F.P.V.; Heeney, M.; Smith, P. Thermal and Structural Characteristics of Oligo(3-Hexylthiophene)s (3HT) n, n = 4–36. J. Am. Chem. Soc. 2013, 135, 13699–13709. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M. Structure and Morphology Control in Thin Films of Regioregular Poly(3-Hexylthiophene). J. Polym. Sci. Part B Polym. Phys. 2011, 49, 1218–1233. [Google Scholar] [CrossRef]
- Wright, M.; Lin, R.; Tayebjee, M.J.Y.; Conibeer, G. Effect of Blend Composition on Bulk Heterojunction Organic Solar Cells: A Review. Sol. RRL 2017, 1, 1700035. [Google Scholar] [CrossRef]
- Ji, D.; Liu, G.; Zhang, X.; Zhang, C.; Yuan, S. Molecular Dynamics Study on the Adsorption of Heavy Oil Drops on a Silica Surface with Different Hydrophobicity. Energy Fuels 2020, 34, 7019–7028. [Google Scholar] [CrossRef]
- Ahmadi, M.; Chen, Z. Comprehensive Molecular Scale Modeling of Anionic Surfactant-Asphaltene Interactions. Fuel 2021, 288, 119729. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE-AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Glova, A.D.; Larin, S.V.; Nazarychev, V.M.; Kenny, J.M.; Lyulin, A.V.; Lyulin, S.V. Toward Predictive Molecular Dynamics Simulations of Asphaltenes in Toluene and Heptane. ACS Omega 2019, 4, 20005–20014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, J.S.; Lemarchand, C.A.; Nielsen, E.; Dyre, J.C.; Schrøder, T. Four-Component United-Atom Model of Bitumen. J. Chem. Phys. 2013, 138, 094508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Taylor, C.; Hu, H.; Humphries, K.L.; Jaini, A.; Kitimet, M.; Scott, T.; Stewart, Z.; Ulep, K.J.; Houck, S.; et al. Nanoaggregates of Diverse Asphaltenes by Mass Spectrometry and Molecular Dynamics. Energy Fuels 2017, 31, 9140–9151. [Google Scholar] [CrossRef]
- Venkataraman, P.; Zygourakis, K.; Chapman, W.G.; Wellington, S.L.; Shammai, M. Molecular Insights into Glass Transition in Condensed Core Asphaltenes. Energy Fuels 2017, 31, 1182–1192. [Google Scholar] [CrossRef]
- Obata, S.; Shimoi, Y. Control of Molecular Orientations of Poly(3-Hexylthiophene) on Self-Assembled Monolayers: Molecular Dynamics Simulations. Phys. Chem. Chem. Phys. 2013, 15, 9265. [Google Scholar] [CrossRef]
- Trapalis, C.; Lidorikis, E.; Papageorgiou, D.G. Structural and Energetic Properties of P3HT and PCBM Layers on the Ag(1 1 1) Surface. Comput. Theor. Chem. 2020, 1190, 112997. [Google Scholar] [CrossRef]
- Pan, Q.-Q.; Zhao, Z.-W.; Wu, Y.; Geng, Y.; Zhang, M.; Su, Z.-M. A Theoretical Exploration on Why the Replacement of Hexyl Group by Alkoxycarbonyl in P3HT Could Greatly Improve the Performance of Non-Fullerene Organic Solar Cell. J. Taiwan Inst. Chem. Eng. 2019, 100, 160–167. [Google Scholar] [CrossRef]
- Pani, R.C.; Bond, B.D.; Krishnan, G.; Yingling, Y.G. Correlating Fullerene Diffusion with the Polythiophene Morphology: Molecular Dynamics Simulations. Soft Matter 2013, 9, 10048. [Google Scholar] [CrossRef]
- Mulunda, M.M.; Zhang, Z.; Nies, E.; van Goethem, C.; Vankelecom, I.F.J.; Koeckelberghs, G. Influence of Branching of Polythiophenes on the Microporosity. Macromol. Chem. Phys. 2018, 219, 1800024. [Google Scholar] [CrossRef]
- Wildman, J.; Repiščák, P.; Paterson, M.J.; Galbraith, I. General Force-Field Parametrization Scheme for Molecular Dynamics Simulations of Conjugated Materials in Solution. J. Chem. Theory Comput. 2016, 12, 3813–3824. [Google Scholar] [CrossRef]
- Bhatta, R.S.; Yimer, Y.Y.; Perry, D.S.; Tsige, M. Improved Force Field for Molecular Modeling of Poly(3-Hexylthiophene). J. Phys. Chem. B 2013, 117, 10035–10045. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Casalegno, M.; Raos, G.; Meille, S.V.; Po, R. Molecular Modeling of Crystalline Alkylthiophene Oligomers and Polymers. J. Phys. Chem. B 2010, 114, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Cheung, D.L.; Troisi, A. Structural Variability and Dynamics of the P3HT/PCBM Interface and Its Effects on the Electronic Structure and the Charge-Transfer Rates in Solar Cells. Phys. Chem. Chem. Phys. 2011, 13, 21461–21470. [Google Scholar] [CrossRef]
- Bernardi, M.; Giulianini, M.; Grossman, J.C. Self-Assembly and Its Impact on Interfacial Charge Transfer in Carbon Nanotube/P3HT Solar Cells. ACS Nano 2010, 4, 6599–6606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yimer, Y.Y.; Tsige, M. Static and Dynamic Properties of Poly(3-Hexylthiophene) Films at Liquid/Vacuum Interfaces. J. Chem. Phys. 2012, 137, 204701. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Gartner, T.E.; Jayaraman, A. Modeling and Simulations of Polymers: A Roadmap. Macromolecules 2019, 52, 755–786. [Google Scholar] [CrossRef] [Green Version]
- Lyulin, S.V.; Gurtovenko, A.A.; Larin, S.V.; Nazarychev, V.M.; Lyulin, A.V. Microsecond Atomic-Scale Molecular Dynamics Simulations of Polyimides. Macromolecules 2013, 46, 6357–6363. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Hoppe, H.; Erb, T.; Günes, S.; Gobsch, G.; Sariciftci, N.S. Effects of Annealing on the Nanomorphology and Performance of Poly(Alkylthiophene):Fullerene Bulk-Heterojunction Solar Cells. Adv. Funct. Mater. 2007, 17, 1071–1078. [Google Scholar] [CrossRef]
- Park, M.; Kim, F. Synergistic Effects of Processing Additives and Thermal Annealing on Nanomorphology and Hole Mobility of Poly(3-Hexylthiophene) Thin Films. Polymers 2019, 11, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Yuan, G.; Roche, P.; Leclerc, M. A Calorimetric Study of the Phase Transitions in Poly(3-Hexylthiophene). Polymer 1995, 36, 2211–2214. [Google Scholar] [CrossRef]
- Müller, C.; Ferenczi, T.A.M.; Campoy-Quiles, M.; Frost, J.M.; Bradley, D.D.C.; Smith, P.; Stingelin-Stutzmann, N.; Nelson, J. Binary Organic Photovoltaic Blends: A Simple Rationale for Optimum Compositions. Adv. Mater. 2008, 20, 3510–3515. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Wu, B.; Pang, S.; Yuan, X.; Duan, C.; Huang, F.; Cao, Y. High-Efficiency P3HT-Based All-Polymer Solar Cells with a Thermodynamically Miscible Polymer Acceptor. Sol. RRL 2022, 6, 2200073. [Google Scholar] [CrossRef]
- Singh, C.R.; Gupta, G.; Lohwasser, R.; Engmann, S.; Balko, J.; Thelakkat, M.; Thurn-Albrecht, T.; Hoppe, H. Correlation of Charge Transport with Structural Order in Highly Ordered Melt-Crystallized Poly(3-Hexylthiophene) Thin Films. J. Polym. Sci. Part B Polym. Phys. 2013, 51, 943–951. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Larin, S.V.; Gurtovenko, A.A.; Nazarychev, V.M.; Falkovich, S.G.; Yudin, V.E.; Svetlichnyi, V.M.; Gofman, I.V.; Lyulin, A.V. Thermal Properties of Bulk Polyimides: Insights from Computer Modeling versus Experiment. Soft Matter 2014, 10, 224–1232. [Google Scholar] [CrossRef]
- Wadsworth, A.; Hamid, Z.; Kosco, J.; Gasparini, N.; McCulloch, I. The Bulk Heterojunction in Organic Photovoltaic, Photodetector, and Photocatalytic Applications. Adv. Mater. 2020, 32, 2001763. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, R.; Cheng, J.; Chang, Y.; Huo, M.; Yu, J.; Li, L.; Zhang, J.-P. Appropriate Donor-Acceptor Phase Separation Structure for the Enhancement of Charge Generation and Transport in Polymer Solar Cells. Polymers 2018, 10, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Grohens, Y.; Jyotishkumar, P. Characterization of Polymer Blends; Thomas, S., Grohens, Y., Jyotishkumar, P., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; Volume 9783527331, ISBN 9783527645602. [Google Scholar]
- Belmares, M.; Blanco, M.; Goddard, W.A.; Ross, R.B.; Caldwell, G.; Chou, S.H.; Pham, J.; Olofson, P.M.; Thomas, C. Hildebrand and Hansen Solubility Parameters from Molecular Dynamics with Applications to Electronic Nose Polymer Sensors. J. Comput. Chem. 2004, 25, 1814–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glova, A.D.; Falkovich, S.G.; Dmitrienko, D.I.; Lyulin, A.V.; Larin, S.V.; Nazarychev, V.M.; Karttunen, M.; Lyulin, S.V. Scale-Dependent Miscibility of Polylactide and Polyhydroxybutyrate: Molecular Dynamics Simulations. Macromolecules 2018, 51, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.; Hess, B.; van der Spoel, D.; Lindahl, E. Gromacs User Manual Version 5.1.4.; Royal Institute of Technology and Uppsala University: Uppsala, Sweden, 2016. [Google Scholar]
- Nazarychev, V.M.; Larin, S.V.; Yakimansky, A.V.; Lukasheva, N.V.; Gurtovenko, A.A.; Gofman, I.V.; Yudin, V.E.; Svetlichnyi, V.M.; Kenny, J.M.; Lyulin, S.V. Parameterization of Electrostatic Interactions for Molecular Dynamics Simulations of Heterocyclic Polymers. J. Polym. Sci. Part B Polym. Phys. 2015, 53, 912–923. [Google Scholar] [CrossRef]
- Hourani, W.; Rahimi, K.; Botiz, I.; Vinzenz Koch, F.P.; Reiter, G.; Lienerth, P.; Heiser, T.; Bubendorff, J.-L.; Simon, L. Anisotropic Charge Transport in Large Single Crystals of π-Conjugated Organic Molecules. Nanoscale 2014, 6, 4774. [Google Scholar] [CrossRef]
- Yakimansky, A.V.; Bushin, S.V.; Bezrukova, M.A.; Lezov, A.A.; Gubarev, A.S.; Lebedeva, E.V.; Akhmadeeva, L.I.; Podseval’nikova, A.N.; Tsvetkov, N.V.; Koeckelberghs, G.; et al. Hydrodynamic Properties and Conformation of Poly(3-Hexylthiophene) in Dilute Solutions. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 875–883. [Google Scholar] [CrossRef]
- McCulloch, B.; Ho, V.; Hoarfrost, M.; Stanley, C.; Do, C.; Heller, W.T.; Segalman, R.A. Polymer Chain Shape of Poly(3-Alkylthiophenes) in Solution Using Small-Angle Neutron Scattering. Macromolecules 2013, 46, 1899–1907. [Google Scholar] [CrossRef]
- Anwar, M.; Turci, F.; Schilling, T. Crystallization Mechanism in Melts of Short N-Alkane Chains. J. Chem. Phys. 2013, 139, 214904. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.; Berryman, J.T.; Schilling, T. Crystal Nucleation Mechanism in Melts of Short Polymer Chains under Quiescent Conditions and under Shear Flow. J. Chem. Phys. 2014, 141, 9. [Google Scholar] [CrossRef] [Green Version]
- Larin, S.V.; Falkovich, S.G.; Nazarychev, V.M.; Gurtovenko, A.A.; Lyulin, A.V.; Lyulin, S.V. Molecular-Dynamics Simulation of Polyimide Matrix Pre-Crystallization near the Surface of a Single-Walled Carbon Nanotube. RSC Adv. 2014, 4, 830–844. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | δ, (J/cm3)0.5 |
|---|---|
| P3HT | 12.6 |
| Asp | 12.3 |
| Asp-Core | 15.8 |
| Asp-COOH | 26.7 |
| System | Rg, nm | b | c | SN | |
|---|---|---|---|---|---|
| Asp-COOH/P3HT | 1:1 | 3.53 ± 0.03 | 4.0 ± 0.4 | 0.6 ± 0.3 | 0.22 ± 0.03 |
| 2:3 | 3.3 ± 0.1 | 1.9 ± 0.3 | 2.1 ± 0.2 | 0.17 ± 0.04 | |
| 1:3 | 3.04 ± 0.04 | 2.4 ± 0.2 | 3.2 ± 0.2 | 0.26 ± 0.05 | |
| Asp/P3HT | 3.6 ± 0.1 | 8.2 ± 1.1 | 2.6 ± 0.6 | 0.66 ± 0.08 | |
| Asp-Core/P3HT | 4.0 ± 0.1 | 11.6 ± 3.5 | 2.1 ± 1.4 | 0.7 ± 0.2 | |
| Unfilled P3HT | 3.35 ± 0.03 | 4.2 ± 0.3 | 3.0 ± 0.3 | 0.43 ± 0.04 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borzdun, N.; Glova, A.; Larin, S.; Lyulin, S. Influence of Asphaltene Modification on Structure of P3HT/Asphaltene Blends: Molecular Dynamics Simulations. Nanomaterials 2022, 12, 2867. https://doi.org/10.3390/nano12162867
Borzdun N, Glova A, Larin S, Lyulin S. Influence of Asphaltene Modification on Structure of P3HT/Asphaltene Blends: Molecular Dynamics Simulations. Nanomaterials. 2022; 12(16):2867. https://doi.org/10.3390/nano12162867
Chicago/Turabian StyleBorzdun, Natalia, Artyom Glova, Sergey Larin, and Sergey Lyulin. 2022. "Influence of Asphaltene Modification on Structure of P3HT/Asphaltene Blends: Molecular Dynamics Simulations" Nanomaterials 12, no. 16: 2867. https://doi.org/10.3390/nano12162867