
Sensing Electrochemical Signals Using a Nitrogen-Vacancy Center in Diamond


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
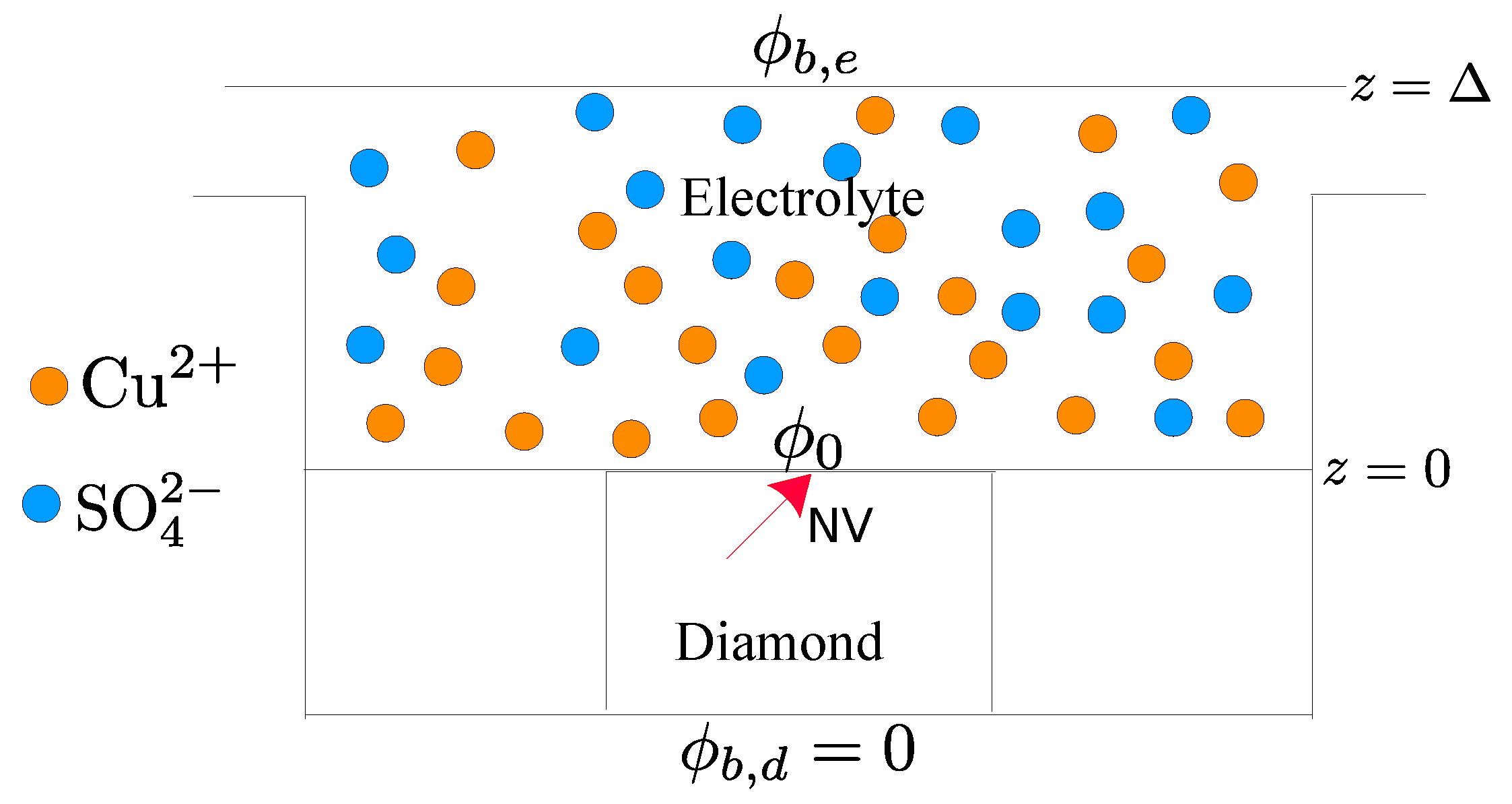
2.1. Electrolyte Solution
2.1.1. Diffusion of Ionic Species in Electrolyte Solutions
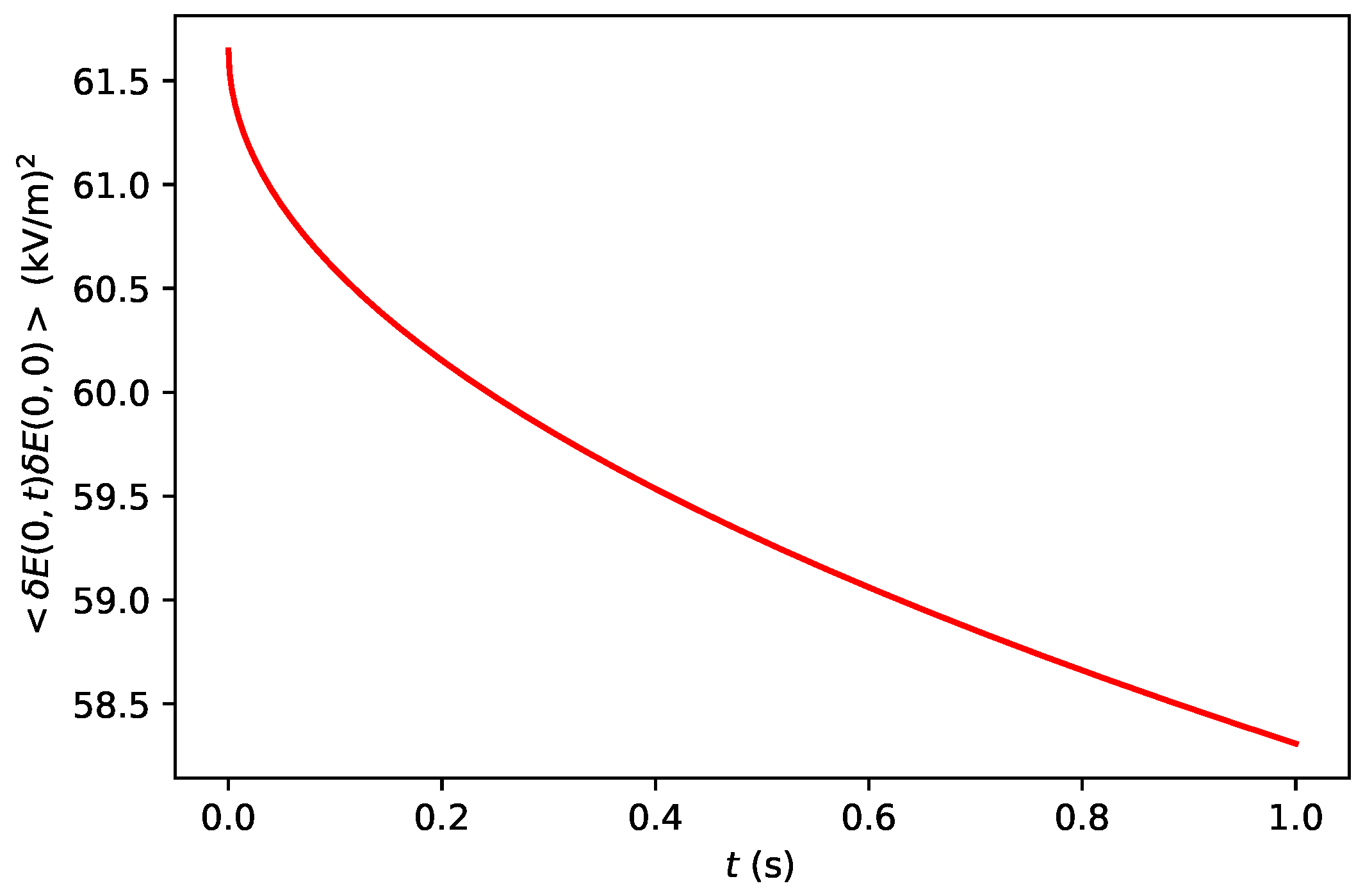
2.1.2. Electric Field Fluctuations at the Interface between the Solution and the Diamond
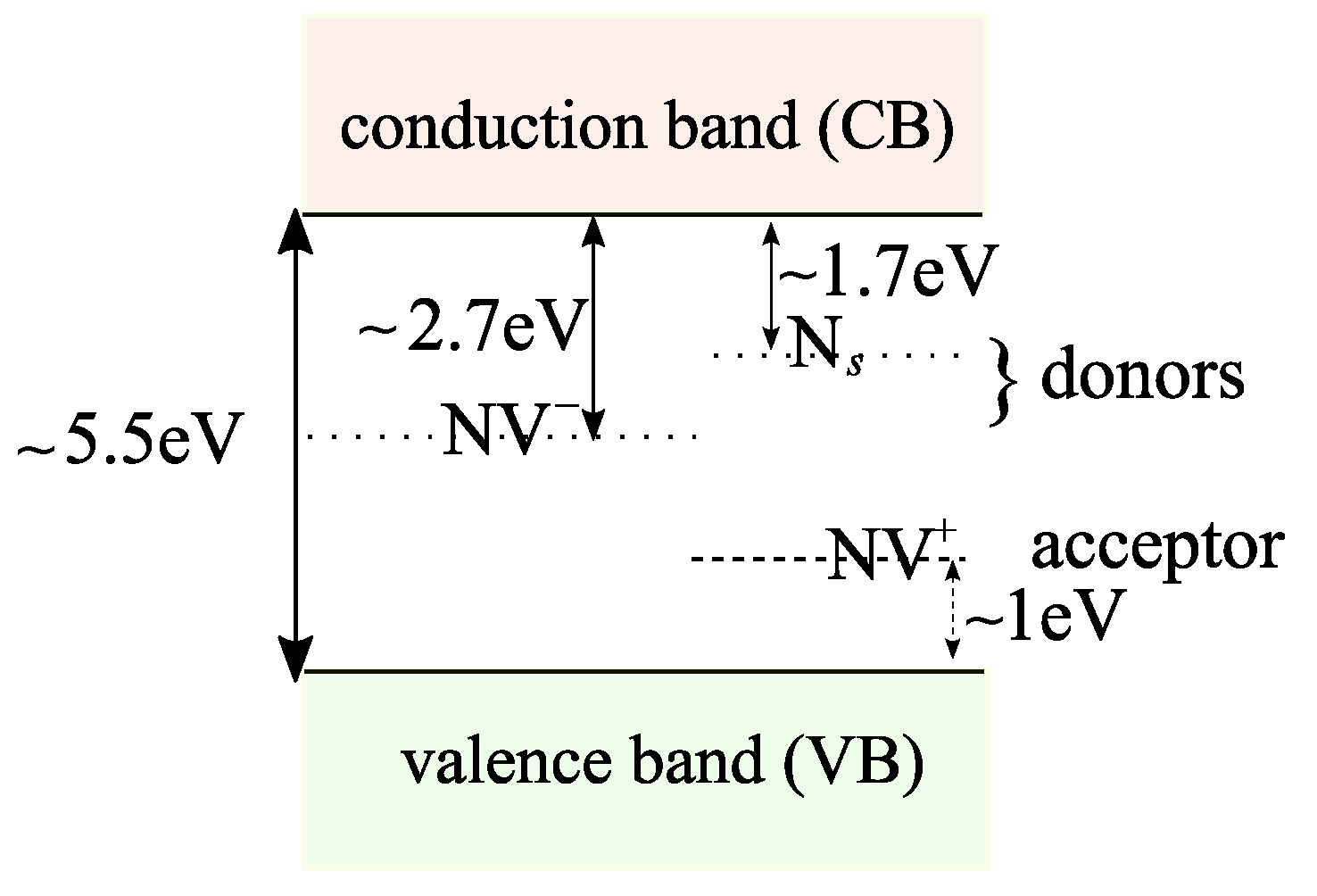
2.2. Diamond and the NV Center
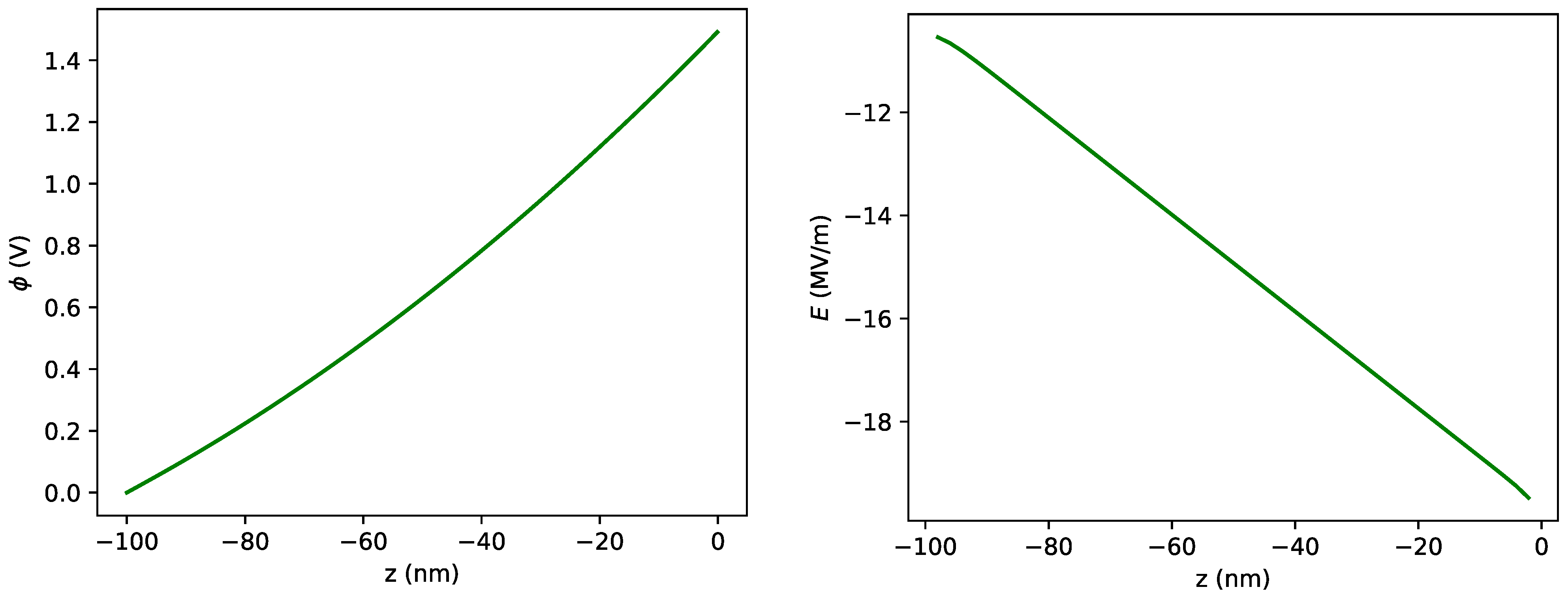
2.2.1. Potential and Electric Field Inside the Diamond
2.2.2. NV Center Inhomogenous Dephasing Rate
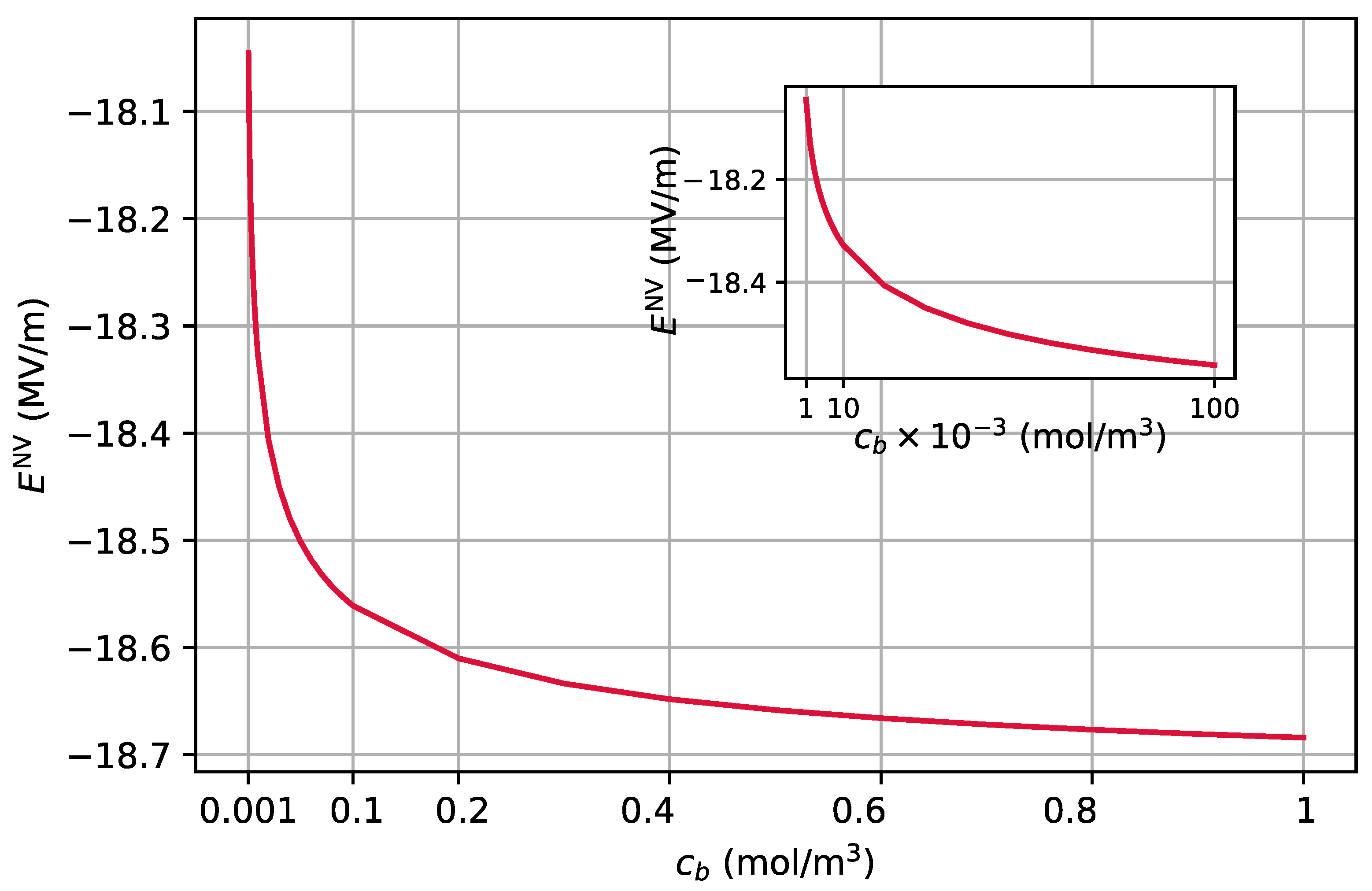
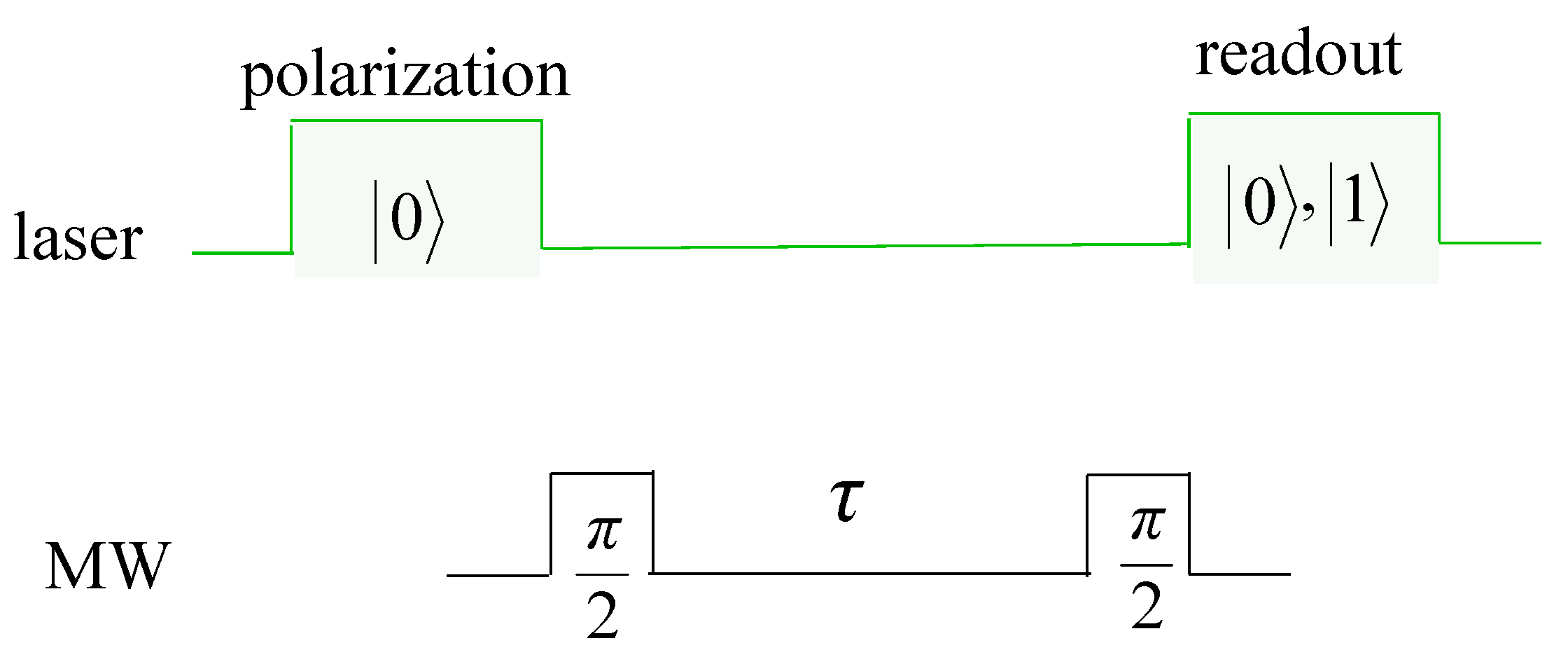
2.2.3. Electric Field Sensing
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| NV | nitrogen vacancy |
Appendix A. Electric Potential and Electric Field Inside the Electrolyte
Appendix B. Thermal Fluctuations in the Local Concentration of Chemical Species
Appendix C. Sensitivity
References
- Qureshi, A.; Kang, W.P.; Davidson, J.L.; Gurbuz, Y. Review on carbon-derived, solid-state, micro and nano sensors for electrochemical sensing applications. Diam. Relat. Mater. 2009, 18, 1401–1420. [Google Scholar] [CrossRef] [Green Version]
- Schirhagl, R.; Chang, K.; Loretz, M.; Degen, C.L. Nitrogen-Vacancy Centers in Diamond: Nanoscale Sensors for Physics and Biology. Annu. Rev. Phys. Chem. 2014, 65, 83–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarracinoa, P.; Gattia, T.; Canevera, N.; Abdu-Aguyeb, M.; Loib, M.A.; Mennaa, E.; Franco, L. Probing photoinduced electron-transfer in graphene-dye hybrid materials for DSSC. Phys. Chem. Chem. Phys. 2017, 19, 27716–27724. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Lamberti, F.; Franco, L.; Collini, E.; Fortunati, I.; Bottaro, G.; Daniel, G.; Sorrentino, R.; Minotto, A.; Kukovecz, A.; et al. A film-forming graphene/diketopyrrolopyrrole covalent hybrid with far-red optical features: Evidence of photo-stability. Synth. Met. 2019, 258, 116201–116210. [Google Scholar] [CrossRef]
- Doherty, M.W.; Manson, N.B.; Delaney, P.; Jelezko, F.; Wrachtrupe, J.; Hollenberg, L.C.L. The nitrogen-vacancy color center in diamond. Phys. Rep. 2013, 528, 1–45. [Google Scholar] [CrossRef] [Green Version]
- Doherty, M.W.; Struzhkin, V.V.; Simpson, D.A.; McGuinness, L.P.; Meng, Y.; Stacey, A.; Karle, T.J.; Hemley, R.J.; Manson, N.B.; Hollenberg, L.C.L.; et al. Electronic Properties and Metrology Applications of the Diamond NV− Center under Pressure. Phys. Rev. Lett. 2014, 112, 047601. [Google Scholar] [CrossRef] [Green Version]
- Kucsko, G.; Maurer, P.C.; Yao, N.Y.; Kubo, M.; Noh, H.J.; Lo, P.K.; Park, H.; Lukin, M.D. Nanometre-scale thermometry in a living cell. Nature (London) 2013, 500, 54–58. [Google Scholar] [CrossRef]
- Dolde, F.; Fedder, H.; Doherty, M.W.; Nobauer, T.; Rempp, F.; Balasubramanian, G.; Wolf, T.; Reinhard, F.; Hollenberg, L.C.L.; Jelezko, F.; et al. Electric-field sensing using single diamond spins. Nat. Phys. 2011, 7, 459–463. [Google Scholar] [CrossRef]
- Rondin, L.; Tetienne, J.-P.; Hingant, T.; Roch, J.-F.; Maletinsky, P.; Jacques, V. Magnetometry with nitrogen-vacancy defects in diamond. Rep. Prog. Phys. 2014, 77, 056503–056528. [Google Scholar] [CrossRef] [Green Version]
- Maze, J.R.; Stanwix, P.L.; Hodges, J.S.; Hong, S.; Taylor, J.M.; Cappellaro, P.; Jiang, L.; Dutt, M.V.G.; Togan, E.; Zibrov, A.S.; et al. Nanoscale magnetic sensing with an individual electronic spin in diamond. Nature (London) 2008, 455, 644–647. [Google Scholar] [CrossRef]
- Bonato, C.; Blok, M.S.; Dinani, H.T.; Berry, D.W.; Markham, L.; Twichen, D.J.; Hanson, R. Optimized quantum sensing with a single electron spin using real-time adaptive measurements. Nat. Nanotechnol. 2016, 11, 247–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinani, H.T.; Berry, D.W.; Gonzalez, R.; Maze, J.R.; Bonato, C. Bayesian estimation for quantum sensing in the absence of single-shot detection. Phys. Rev. B 2019, 99, 125413. [Google Scholar] [CrossRef] [Green Version]
- Newell, A.N.; Dowdell, D.A.; Santamore, D.H. Surface effects on nitrogen vacancy centers neutralization in diamond. J. Appl. Phys. 2016, 120, 185104. [Google Scholar] [CrossRef] [Green Version]
- Rendler, T.; Neburkova, J.; Zemek, O.; Kotek, J.; Zappe, A.; Chu, Z.; Cigler, P.; Wrachtrup, J. Optical imaging of localized chemical events using programmable diamond quantum nanosensors. Nat. Comm. 2017, 8, 14701–14709. [Google Scholar] [CrossRef]
- Luan, L.; Grinolds, M.S.; Hong, S.; Maletinsky, P.; Walsworth, R.L.; Yacoby, A. Decoherence imaging of spin ensembles using a scanning single-electron spin in diamond. Sci. Rep. 2015, 5, 8119–8123. [Google Scholar] [CrossRef] [Green Version]
- Staudacher, T.; Raatz, N.; Pezzagna, S.; Meijer, J.; Reinhard, F.; Meriles, C.A.; Wrachtrup, J. Probing molecular dynamics at the nanoscale via an individual paramagnetic centre. Nat. Comm. 2015, 6, 8527–8533. [Google Scholar] [CrossRef]
- Cohen, D.; Nigmatullin, R.; Kenneth, O.; Jelezko, F.; Khodas, M.; Retzker, A. Utilising NV based quantum sensing for velocimetry at the nanoscale. Sci. Rep. 2020, 10, 5298–5310. [Google Scholar] [CrossRef] [Green Version]
- Hall, L.T.; Hill, C.D.; Cole, J.H.; Stadler, B.; Caruso, F.; Mulvaney, P.; Wrachtrup, J.; Hollenberg, L.C.L. Monitoring ion-channel function in real time through quantum decoherence. Proc. Natl. Acad. Sci. USA 2010, 107, 18777–18782. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, M.; Tsukahara, R.; Sera, Y.; Yukawa, H.; Baba, Y.; Shikatab, S.; Hashimoto, H. Monitoring spin coherence of single nitrogen-vacancy centers in nanodiamonds during pH changes in aqueous buffer solutions. RSC Adv. 2019, 9, 12606–12614. [Google Scholar] [CrossRef] [Green Version]
- Kirby, B.J. Micro-and Nanoscale Fluid Mechanics: Transport in Microfluidic Devices; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Gavish, N.; Promislow, K. Dependence of the dielectric constant of electrolyte solutions on ionic concentration: A microfield approach. Phys. Rev. E 2016, 94, 012611. [Google Scholar] [CrossRef] [Green Version]
- Broadway, D.A.; Dontschuk, N.; Tsai, A.; Lillie, S.E.; Lew, C.T.-K.; McCallum, J.C.; Johnson, B.C.; Doherty, M.W.; Stacey, A.; Hollenberg, L.C.L.; et al. Spatial mapping of band bending in semiconductor devices using in situ quantum sensors. Nat. Electron. 2018, 1, 502–507. [Google Scholar] [CrossRef]
- Deák, P.; Aradi, B.; Kaviani, M.; Frauenheim, T.; Gali, A. Formation of NV centers in diamond: A theoretical study based on calculated transitions and migration of nitrogen and vacancy related defects. Phys. Rev. B 2014, 89, 075203. [Google Scholar]
- Ashcroft, N.W.; Mermin, N.D. Solid State Physics; Saunders College: Philadelphia, PA, USA, 1976. [Google Scholar]
- Norambuena, A.; Muñoz, E.; Dinani, H.T.; Jarmola, A.; Maletinsky, P.; Budker, D.; Maze, J.R. Spin-lattice relaxation of individual solid-state spins. Phys. Rev. B 2018, 97, 094304. [Google Scholar] [CrossRef] [Green Version]
- Udvarhelyi, P.; Shkolnikov, V.O.; Gali, A.; Burkard, G.; Pályi, A. Spin-strain interaction in nitrogen-vacancy centers in diamond. Phys. Rev. B 2018, 98, 075201. [Google Scholar] [CrossRef] [Green Version]
- Doherty, M.W.; Dolde, F.; Fedder, H.; Jelezko, F.; Wrachtrup, J.; Manson, N.B.; Hollenberg, L.C.L. Theory of the ground-state spin of the NV− center in diamond. Phys. Rev. B 2012, 85, 205203. [Google Scholar] [CrossRef] [Green Version]
- Jamonneau, P.; Lesik, M.; Tetienne, J.P.; Alvizu, I.; Mayer, L.; Dréau, A.; Kosen, S.; Roch, J.-F.; Pezzagna, S.; Meijer, J.; et al. Competition between electric field and magnetic field noise in the decoherence of a single spin in diamond. Phys. Rev. B 2016, 93, 024305. [Google Scholar] [CrossRef] [Green Version]
- Maze, J.R.; Dréau, A.; Waselowski, V.; Duarte, H.; Roch, J.-F.; Jacques, V. Free induction decay of single spins in diamond. New J. Phys. 2012, 14, 103041–103058. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-H.; Takahashi, S. Spin decoherence and electron spin bath noise of a nitrogen-vacancy center in diamond. Phys. Rev. B 2013, 87, 115122. [Google Scholar] [CrossRef] [Green Version]
- Maurer, P.C.; Kucsko, G.; Latta, C.; Jiang, L.; Yao, N.Y.; Bennett, S.D.; Pastawski, F.; Hunger, D.; Chisholm, N.; Markham, M.; et al. Room-temperature quantum bit memory exceeding one second. Science 2012, 336, 1283–1286. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Fu, K.-M.C.; Santori, C.; Acosta, V.M.; Beausoleil, R.G.; Watanabe, H.; Shikata, S.; Itoh, K.M. Optical and spin coherence properties of nitrogen vacancy centers placed in a 100 nm thick isotopically purified diamond layer. Nano Lett. 2012, 12, 2083–2087. [Google Scholar] [CrossRef] [Green Version]
- Michl, J.; Steiner, J.; Denisenko, A.; Bulau, A.; Zimmermann, A.; Nakamura, K.; Sumiya, H.; Onoda, S.; Neumann, P.; Isoya, J.; et al. Robust and Accurate Electric Field Sensing with Solid State Spin Ensembles. Nano Lett. 2019, 19, 4904–4910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kampen, N.G. Stochastic Processes in Physics and Chemistry; Elsevier: North Holand, The Netherlands, 1992; Chapter XIV. [Google Scholar]
- Meriles, C.A.; Jiang, L.; Goldstein, G.; Hodges, J.S.; Maze, J.; Lukin, M.D.; Cappellaro, P. Imaging mesoscopic nuclear spin noise with a diamond magnetometer. J. Chem. Phys. 2010, 133, 124105–124113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babinec, T.; Hausmann, B.J.M.; Khan, M.; Zhang, Y.; Maze, J.; Hemmer, P.R.; Loncar, M. A diamond nanowire single-photon source. Nat. Nanotechnol. 2010, 5, 195–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, B.J.; Unterreithmeier, Q.P.; de Leon, N.P.; Park, H.; Lukin, M.D. Efficient readout of a single spin state in diamond via spin-to-charge conversion. Phys. Rev. Lett. 2015, 114, 136402–136406. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinani, H.T.; Muñoz, E.; Maze, J.R. Sensing Electrochemical Signals Using a Nitrogen-Vacancy Center in Diamond. Nanomaterials 2021, 11, 358. https://doi.org/10.3390/nano11020358
Dinani HT, Muñoz E, Maze JR. Sensing Electrochemical Signals Using a Nitrogen-Vacancy Center in Diamond. Nanomaterials. 2021; 11(2):358. https://doi.org/10.3390/nano11020358
Chicago/Turabian StyleDinani, Hossein T., Enrique Muñoz, and Jeronimo R. Maze. 2021. "Sensing Electrochemical Signals Using a Nitrogen-Vacancy Center in Diamond" Nanomaterials 11, no. 2: 358. https://doi.org/10.3390/nano11020358