1. Introduction
Confined molecular water in nanocavities shows intriguing and unexpected behavior. The dynamic evolution of confined molecular water swings between bulk response, molecular collective actions and interface binding reactions [
1]. Translational and rotational motions of confined water point to different stretching dynamics from its bulk counterpart [
2]. It is also known that confined water builds tight hydrogen-bonded (H-bonded) networks, and its flow response is diverging by orders of magnitude from macroscopic hydrodynamics [
3]. Possible lack of H-bonding of water molecules in small volumes counts for de-wetting, cavity expulsion [
4], water self-dissociation [
5] and a diverging dielectric constant [
6]. It is plausible; therefore, that diverging behaviors of the biological and geological evolution of molecular enclosures in small systems [
7,
8,
9,
10,
11,
12,
13] also imply a nanothermodynamic approach [
14,
15].
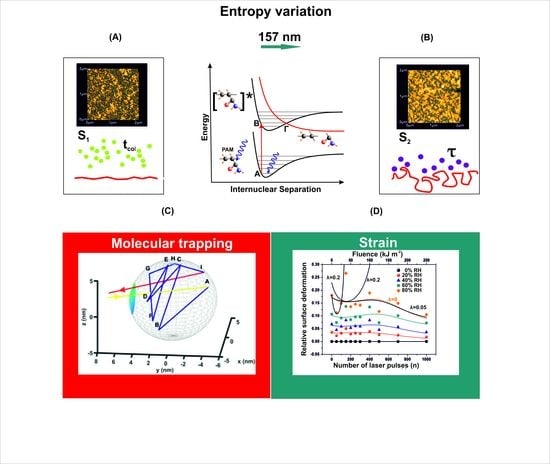
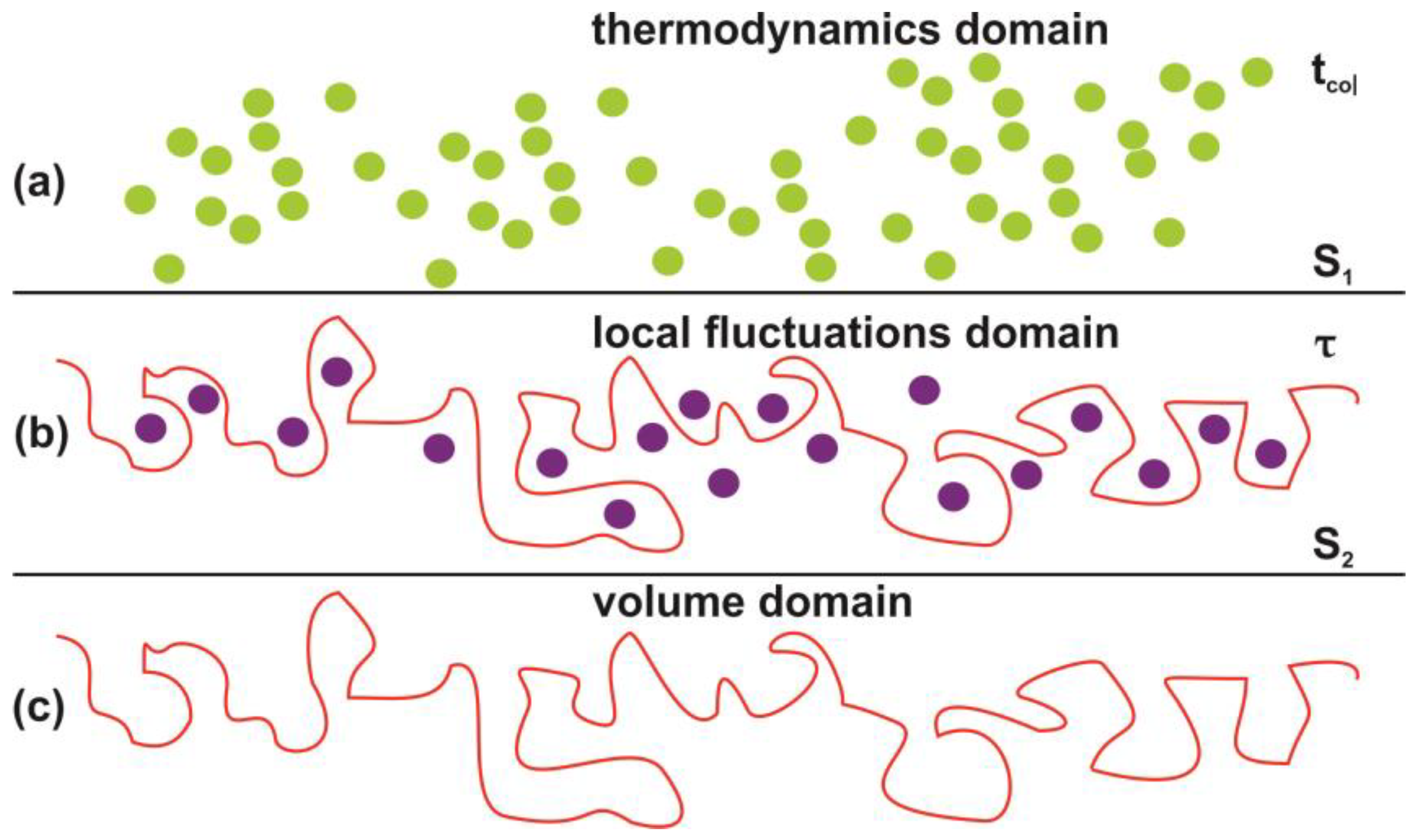
The central element of any thermodynamic theory of small systems is based on the hypothesis that nanometer-sized configurations pullout an additional physical component to the free energy of the associated macroscopic system from interactions among nanostructure entities. Moreover, the confinement of a relatively large number of molecules in nanocavities, restraints the molecular degrees of freedom (translational, vibration or rotational), and finally the system evolves through different entropic states before equilibration. Most interesting, the confinement of a small number of molecules in a large number of distinguishable tiny spaces might well indicate a thermodynamic entropic collective behavior [
13], space and time local heterogeneities, not-extensive fluctuations and intriguing surface-boundary effects. The reduction of the translational degrees of freedom of molecules in tiny spaces and the deviation of the molecular trapping time inside a cavity from the mean molecular collision time outside, highlight the presence of an entropic barrier that separates the molecular motions inside and outside the cavities.
Today, both theoretical [
14,
15,
16,
17,
18,
19] and experimental advancements [
20,
21] gradually disclose the intriguing issues of thermodynamics of small systems, with major impacts on colloids, liquids, surfaces, interphases, chemical sensors, micro/nanofluidics, nanoporous media, proteins and DNA folding [
10,
22,
23,
24,
25,
26,
27]. In cell biology, the presence of different nano-sized molecular scaffolds in the extracellular matrix environment implies a vast diversity of cellular activities and responses, including uncorrelated diverging drug delivery efficiencies [
28].
Because thermodynamic potential variations and fluctuations allow for volume and surface stressing, any experimental verification of local volume and surface stress might well point to entropic fluctuations during molecular confinement [
13,
29]. Commonly, bulk and surface stressing go along with self-assembled structures, translational symmetry breaking, non-linearity, bifurcations, chaos, instability and morphological and shape nano configurations [
30,
31]. In the non-equilibrium state, rapidly changing thermodynamic potentials across phase boundaries usually force tiny systems to pass from different morphological progressions and physical states by tracing minimum energy and maximum entropy production pathways. This universal principle appears everywhere in Nature; from self-assembled bio and macromolecular structures and folding of large protein molecules [
32] to nano/micro flower-like artificial structures [
33,
34].
The confinement of molecules within nano-size cavitations, usually on the surface of a matrix, is linked to system’s entropy diversity before and after trapping [
13,
27,
35,
36]. It is also known that for the same translational entropy, any confined molecular state attains a small variation of its rotational entropy compared to the non-confined molecular state. Likewise, rotational restriction affects surface molecular bonding and sorption/desorption kinetics [
35]. Specific response of nanoentropic potentials from molecular confinement within photon-induced nanocavitations in PDMS matrixes underlines an inherent correlation between internal stressing and 2D entropy diversion [
13].
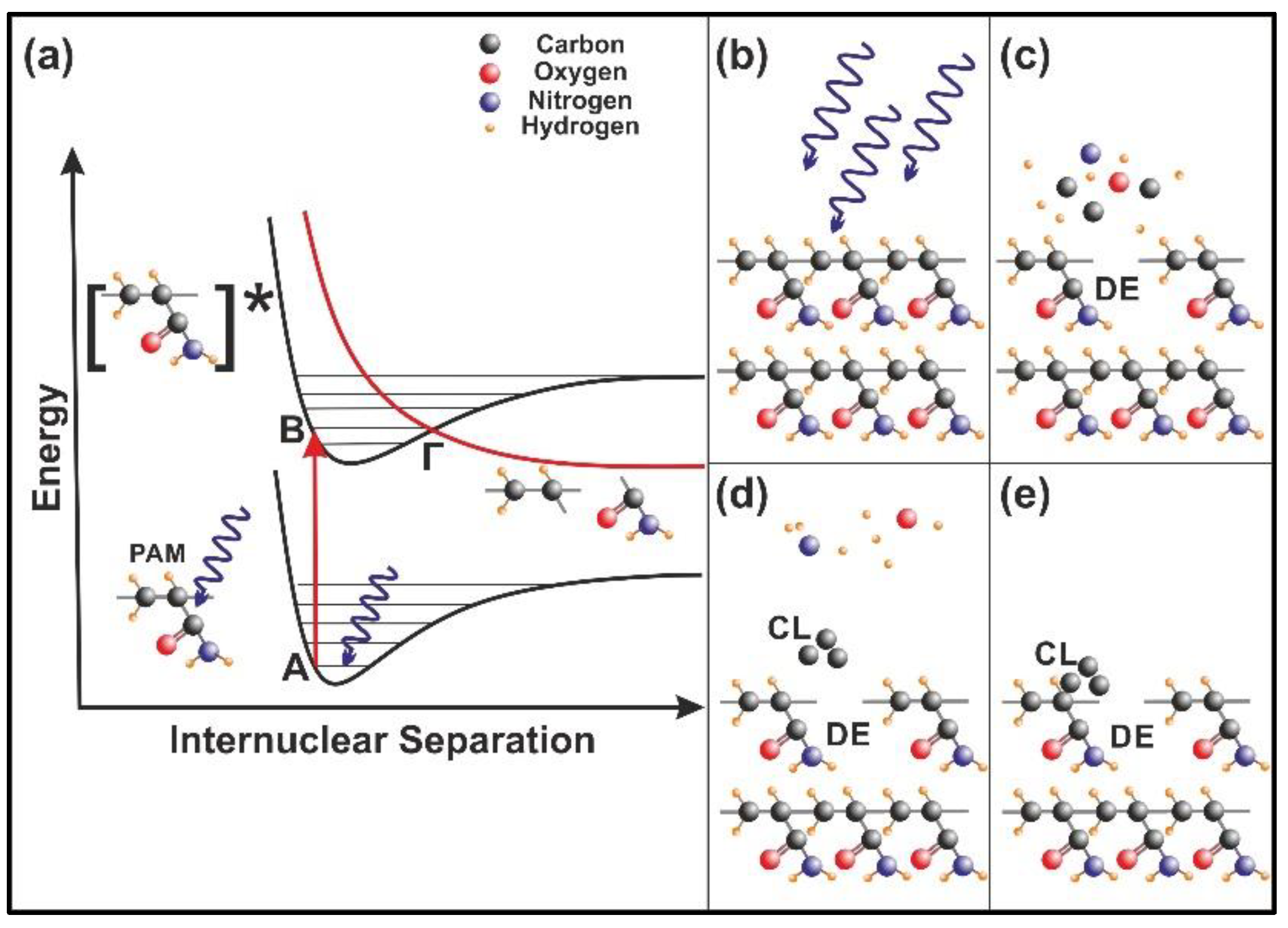
Commonly, photon-processing of surfaces reconfigures their physicochemical properties, including thermodynamic potentials [
37,
38,
39,
40]. Irradiation of a polymeric matrix with vacuum ultraviolet (VUV) light in the spectral range from 110 to 180 nm entails an extensive modification of topological and thus of physical features, because of bond breaking and formation of new bonds. Any 2D topological transform is accompanied by a diversion of surface characteristics, such as porosity, sensing efficiency, chemical stability and extensive nanocavitation [
41,
42,
43,
44,
45,
46]. The adsorption of various molecules on 2D nanostructured surfaces [
47,
48,
49], might well boost a plethora of surfactant effects along with molecular sensing [
43,
50], gas separation and storage [
51,
52,
53,
54], and also applications with particular emphasis on nanomedicine [
55], bio-engineering [
56,
57] and drug delivery systems [
58,
59]. Among other polymeric matrixes, polyacrylamide (PAM) is a hydrophilic low toxic, biocompatible, water-soluble, synthetic linear or cross-linked molecule, modified accordingly for a wide range of applications, including oil recuperation, wastewater treatment, soil conditioner, cosmetics food and biomedical industries [
60,
61,
62]. A diverging number of physical and chemical methods are currently applied to optimize the biocompatibility level of different polymers (e.g., PDMS, PET, PTFEMA, PEG), for biomedical applications, biosensors, tissue engineering and artificial organs [
46,
63,
64]. Well established methods of surface functionalization through photon irradiation with UV, VUV and EUV (extreme ultraviolet) light sources and plasma treatment at various wavelengths and electron energies, aim to optimize chemical instability and surface modification for controlling a plethora of surface functionalities [
65].
Today, several methods exist to improve the strength and the physicochemical properties of PAM matrixes by blending the matrix with chitosan, starch or other polymers [
66]. While functionalization of pure PAM polymeric surfaces is mostly done via sunlight exposure at standard environmental conditions, a limited number of studies include plasma processing [
67,
68,
69,
70,
71]. However, no data exist for VUV processing of PAM surfaces, preventing thus precise tailoring of PAM’s physicochemical surface characteristics (surface roughness, structure size, elasticity, chemical composition, etc.) and the formation of controlled micro/nanopatterns and cavitations for different applications [
37,
42,
43,
63,
64].
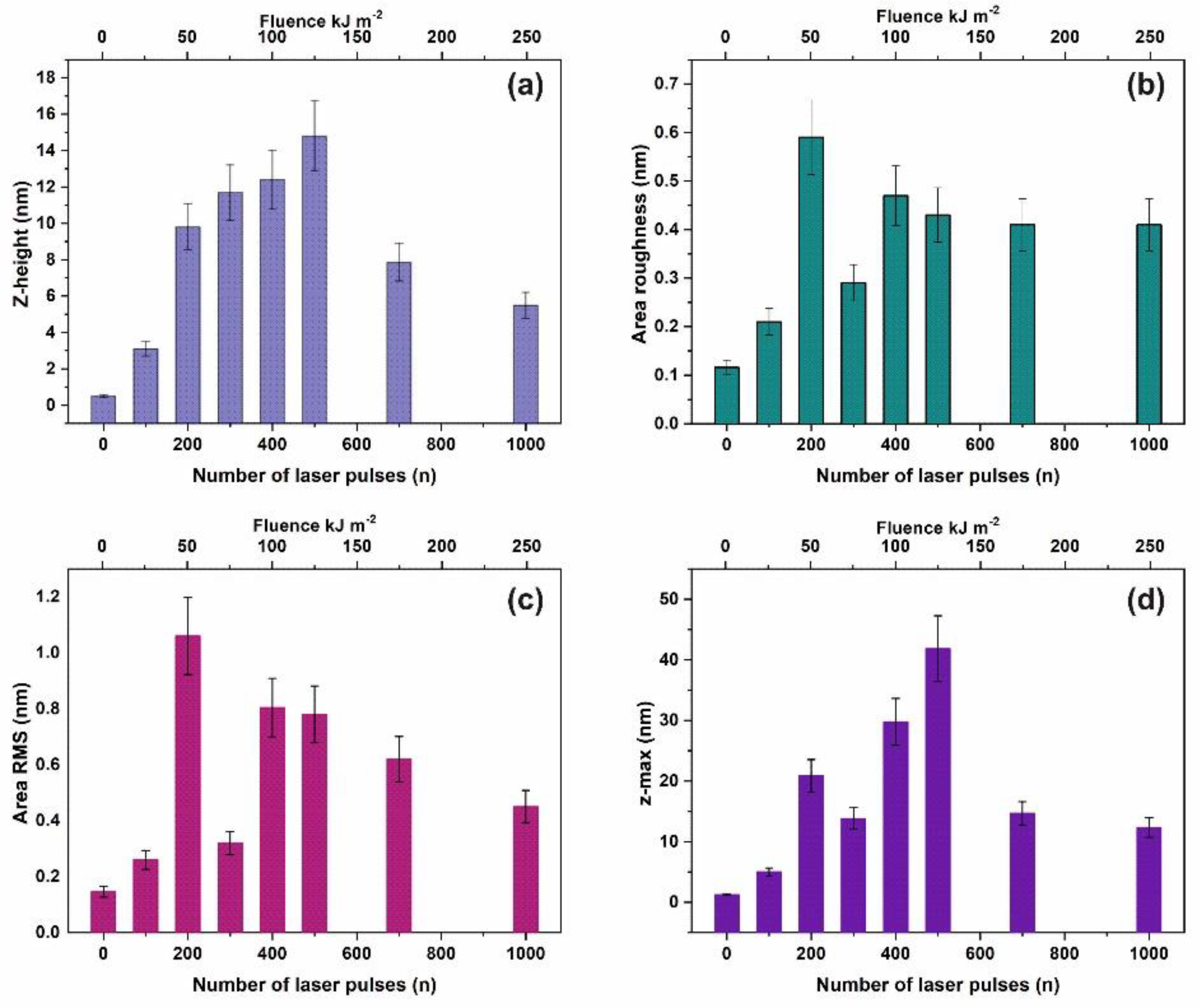
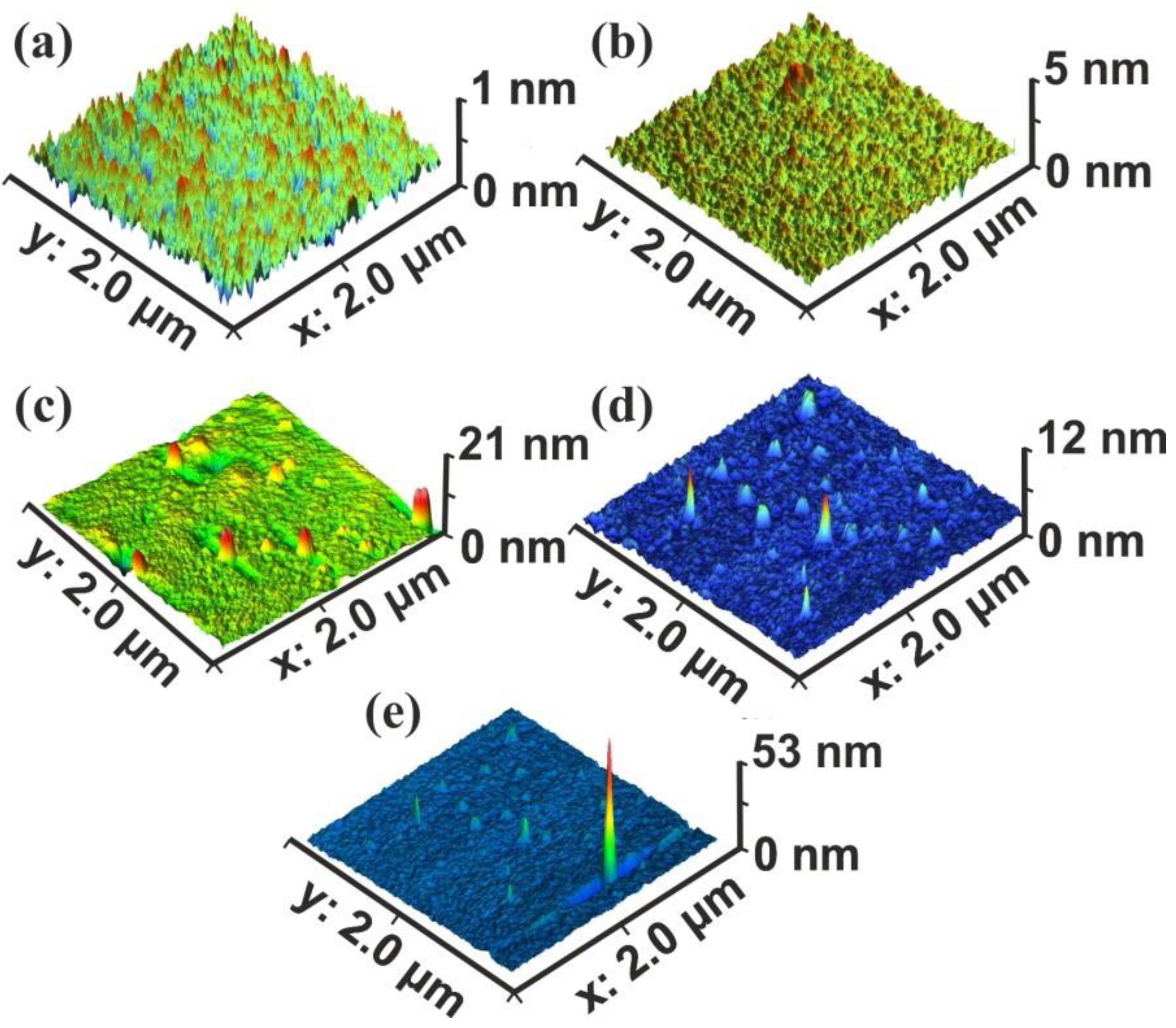
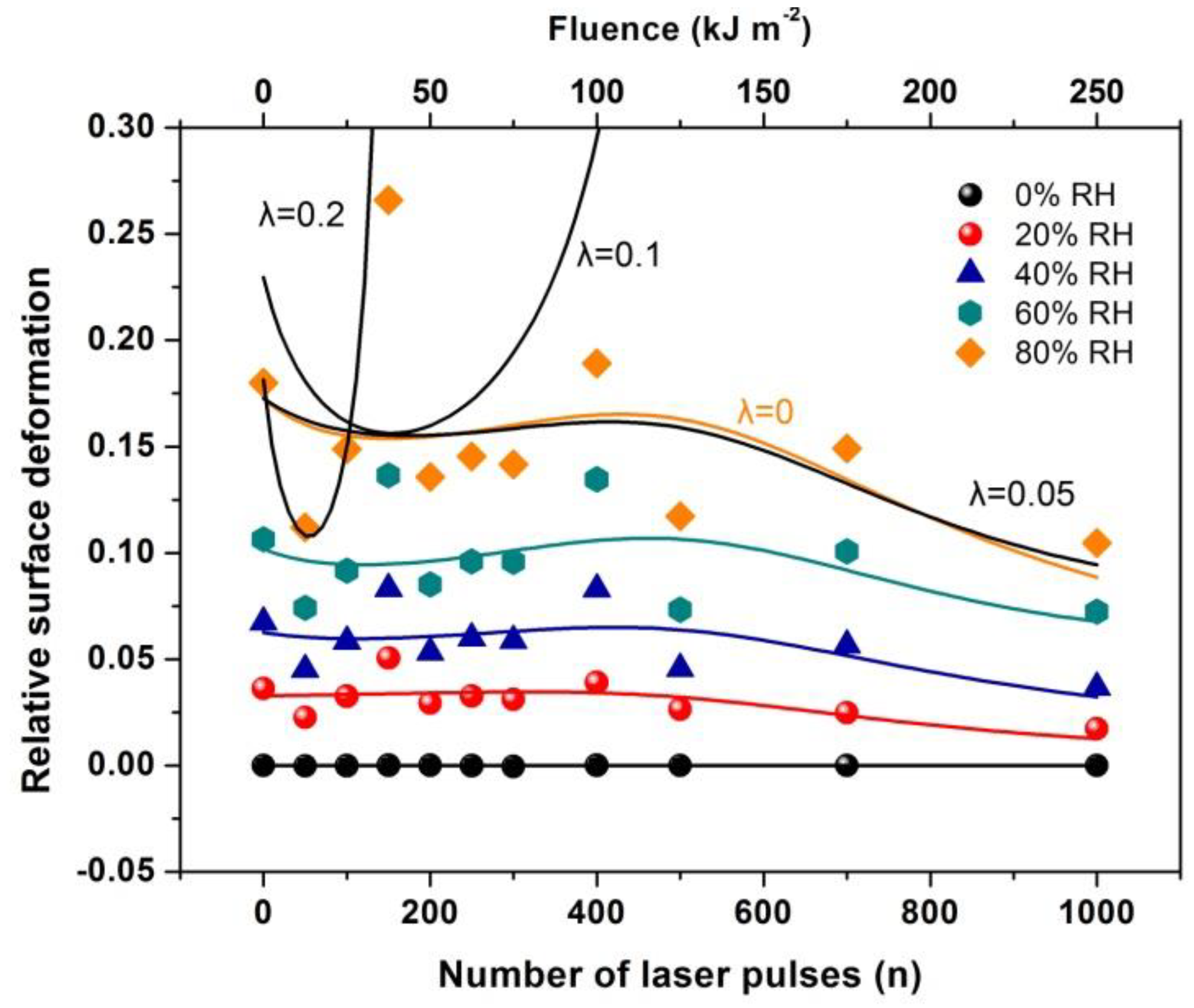
The current work establishes the link between entropy variation and molecular water confinement in small nanocavities fabricated by 157 nm laser photons in polymeric PAM matrixes. The work follows a line of a rational evolution. First, the correlation between 157 nm molecular photodissociation (laser fluence or a number of laser pulses) and surface topological features, including nanocavitations, is established from fractal and surface analysis by using atomic force microscopy (AFM). Next, the correlation between surface strain and 157 nm molecular photodissociation is revealed by applying AFM nanoindentation (AFM-NI), contact angle (CA) wetting and white light reflection spectroscopy (WLRS). Random walk simulations of water molecules inside cavitations differentiate the escape time of confined molecular water and the mean collision time of water molecules near the PAM surface. The different time scales inside and outside the nanocavities point to an additional state of ordered arrangements between nanocavities and the molecular water ensembles of fixed molecular length near the surface. The configured number of microstates properly counts for the experimental surface entropy deviation during molecular water confinement, in agreement with the experimental results. Finally, the mean time distribution for a small number of water molecules for different runs reveals a non-equilibrium state inside tiny cavities. The experimental method has the potential to identify confined water molecules in nanocavities via entropy variation. The proposed roadmap of analysis may be used in applications related to life science.
2. Materials and Methods
2.1. Materials
PAM (typical Mn = 150 K, Mw 400 K) purchased from Sigma-Aldrich (St. Louis, MO, USA) used to prepare solution 5% w/w in water. Thin layers (426 ± 1 nm) on Si wafer substrates were made by spin-coating for 60 s at 2500 rpm, and finally, cured at 110 °C for 15 min at a temperature rate of 0.37 °C s−1 and then left to cool at room temperature. WLRS measures the thickness of PAM films coated on Si wafers.
2.2. 157 nm Laser
PAM layers irradiated with a high power pulse discharged molecular fluorine laser at 157 nm (Lambda Physik 250 (LPFTM 200), Lambda Physik AG (Coherent), Göttingen, Germany), under continuous nitrogen flow (99.999%) at 105 Pa and room temperature. The layers were mounted into a computer-controlled X-Y-Z-θ translation-rotation motorized stage, placed inside a 316 stainless-steel chamber. The laser temporal pulse duration at FWHM, the energy of an unfocused laser beam per laser pulse, the photon fluence per laser pulse and laser repetition rate were set up at 15 ns, 28 mJ, 250 Jm−2 and 10 Hz. For dipping the amount of oxygen inside the stainless-steel chamber, nitrogen purging of the chamber was applied for 10 min before the irradiating stage.
2.3. AFM Imaging and AFM-NI
An AFM system (diInnova, Veeco Instruments Inc. (SPM Bruker), Santa Barbara, CA, USA) used for surface imaging of exposed/non exposed areas and the AFM-NI measurements. The imaging carried out in a tapping mode at a scanning rate of 0.5 Hz, using phosphorus-(n)-doped silicon cantilever (MPP-11123-10), having a spring constant of 40 nN nm−1 and tip radius of 8 nm, operating at a resonance frequency of 300 kHz at ambient conditions. The surface parameters of the samples were also evaluated.
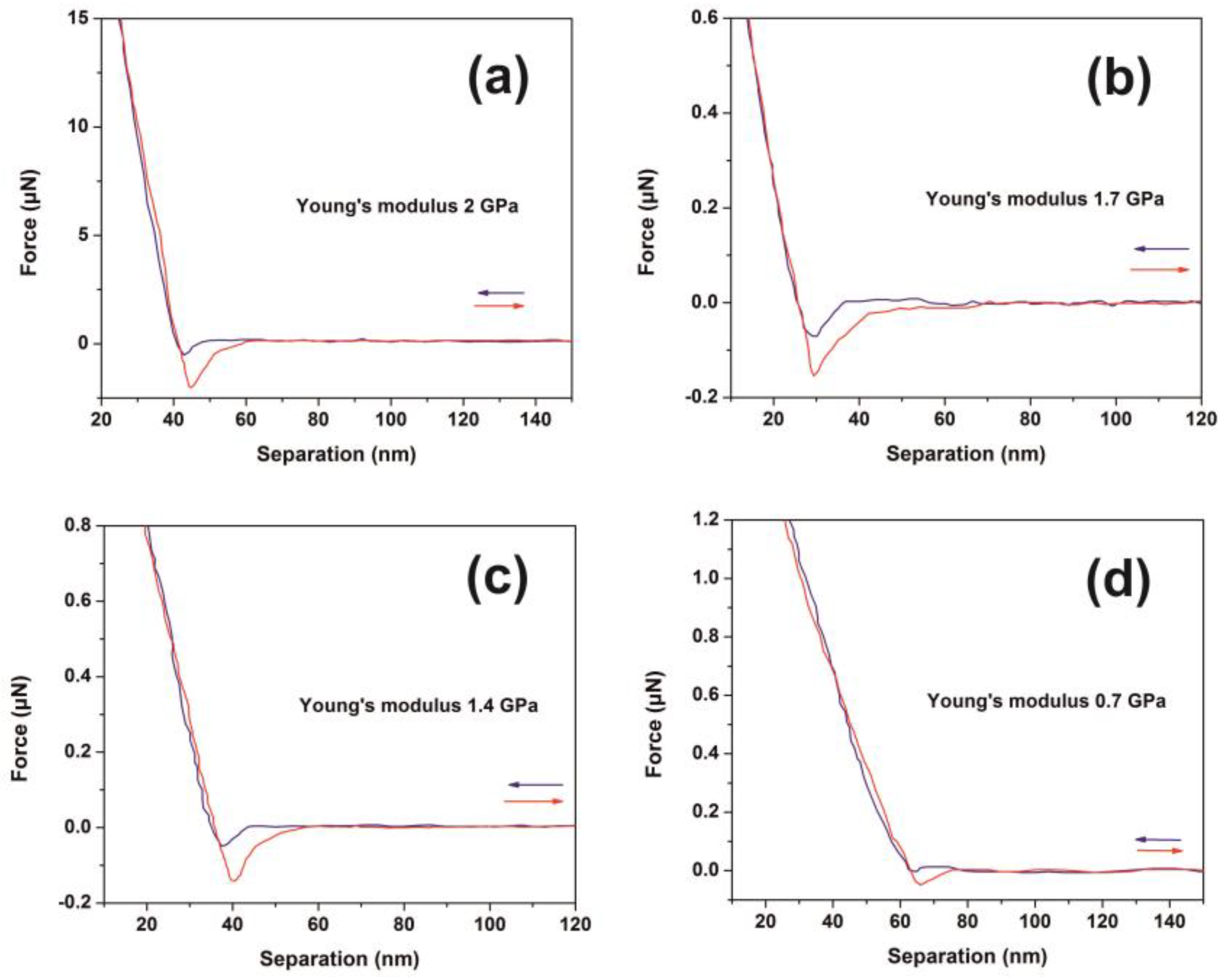
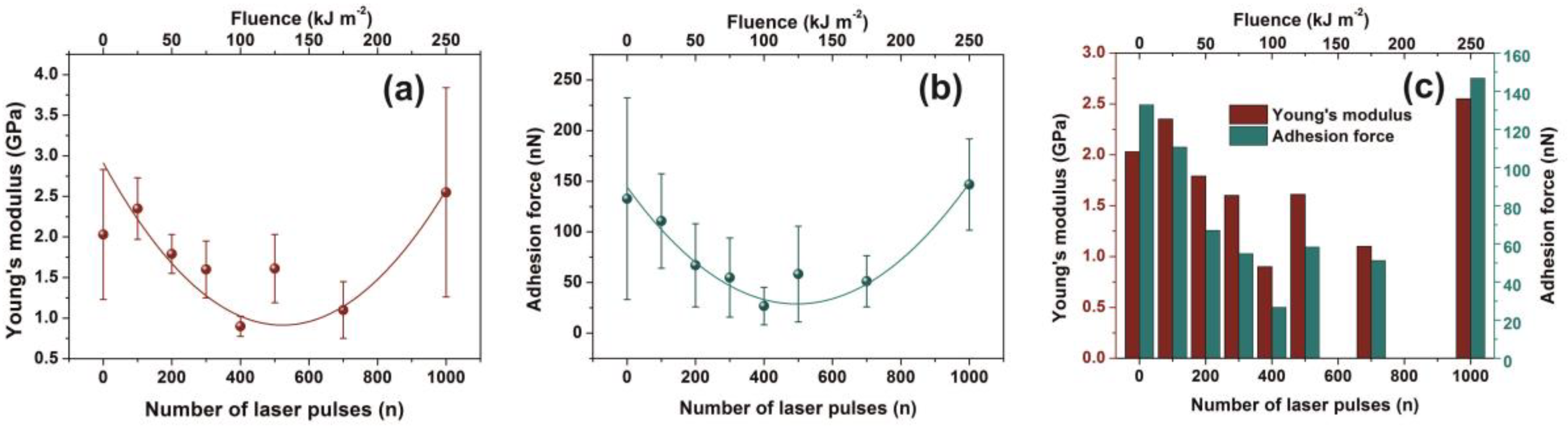
The force versus distance (F-D) response from ten different points on each non-exposed and exposed areas was also recorded with the same cantilever. The elastic modulus (Young’s modulus) was calculated using the SPIP force curve analysis software by fitting a Hertz model to the force-distance curve. The hysteresis between approach and retract curves were corrected by the same software. Calculations performed with a Poisson’s ratio value of 0.3 [
72].
2.4. Fractal Analysis
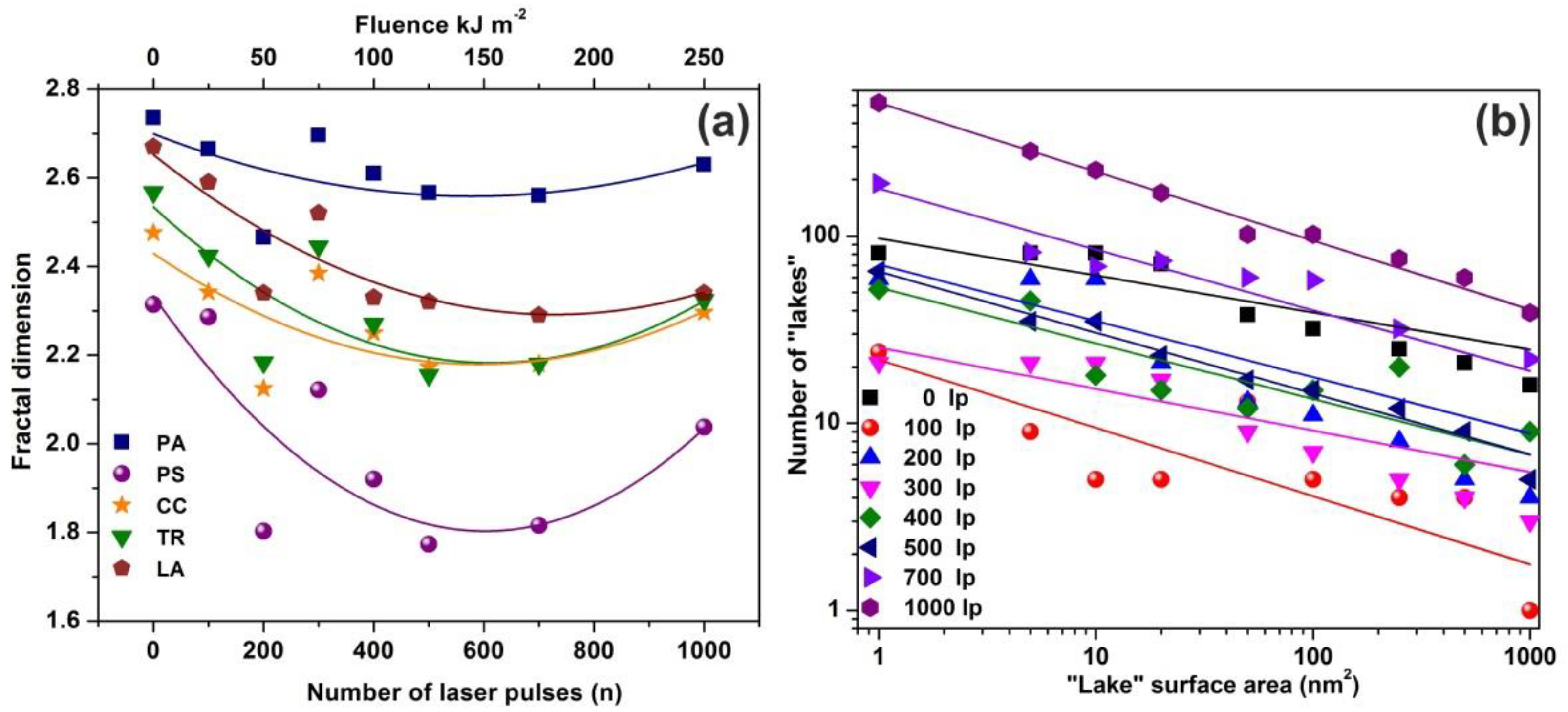
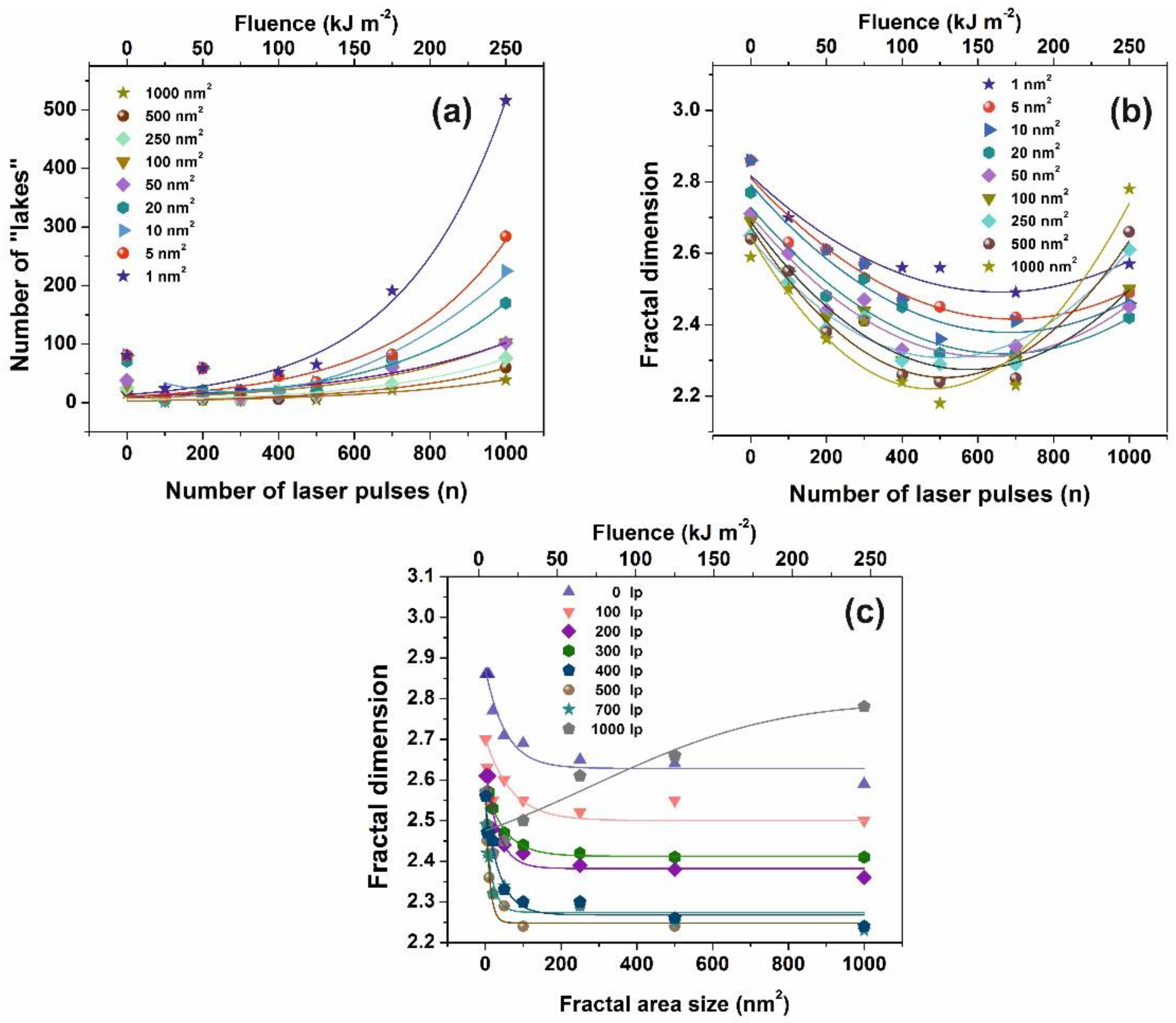
The fractal characteristics of the exposed and non-exposed areas were quantified through the fractal dimensionality D
f that describes the topology and the cavitation of a surface quantitatively. D
f was derived from AFM images by four different algorithms, the cube counting, triangulation, variance and power spectrum methods, besides an algorithm provided by the AFM’s “lake pattern” software (diSPMLab Vr.5.01). A detailed description of the concept and the specific methodologies of the different algorithms can be found in [
27]. The D
f was calculated for the four different methods using “Gwyddion, SPM data visualisation and analysis tool” [
73]. The D
f calculated with the four different algorithms follow the same trend, despite small dimensionality divergences coming up from systematic errors, because of the different converging speed of the fractal analytical approaches.
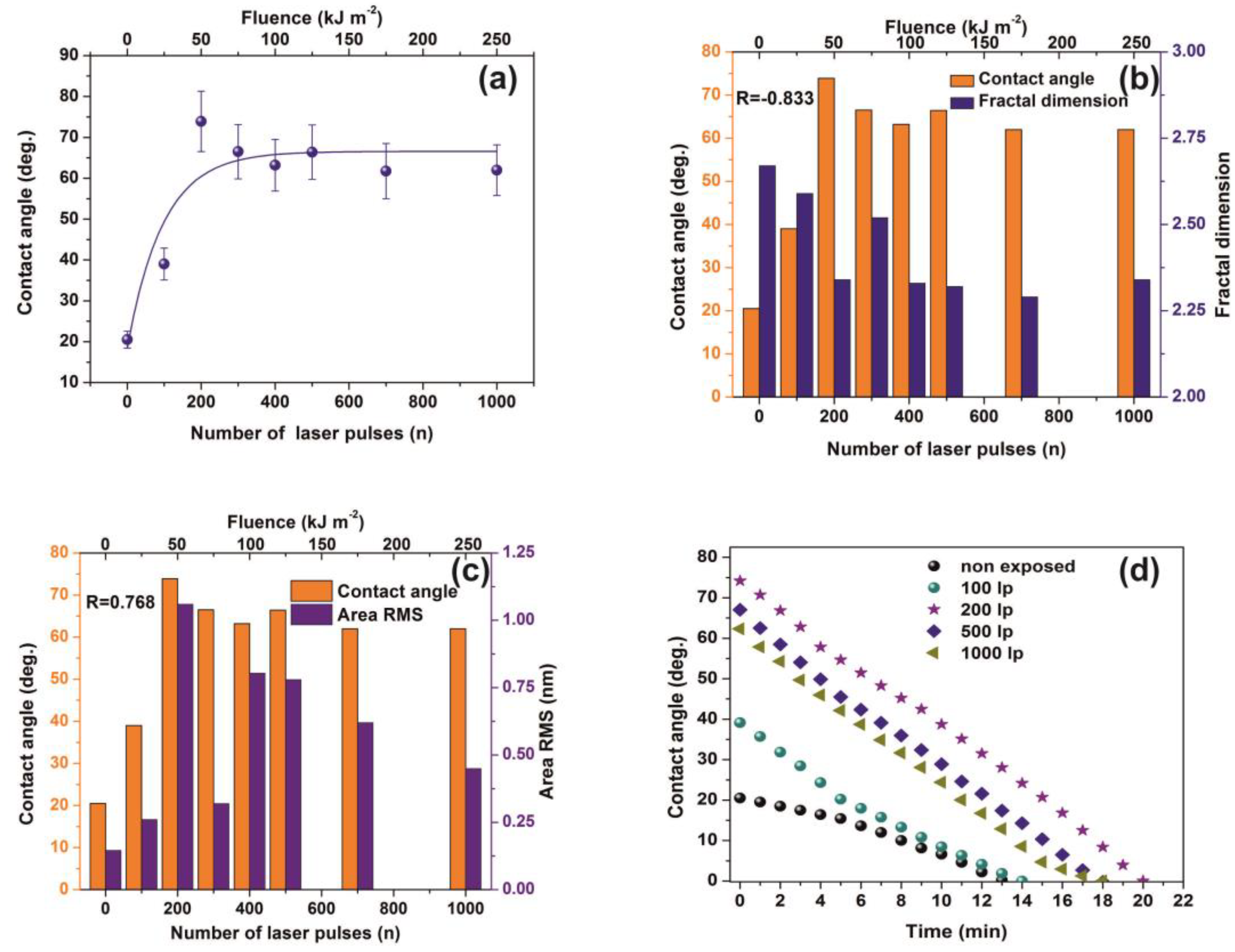
2.5. Water Contact Angle (CA)
The chemical modification of PAM surfaces following PAM surface laser irradiation was monitored by water CA surface measurements under ambient atmospheric conditions. Distilled water droplets with a volume of 0.5 μL were gently deposited onto the sample surface using a microsyringe. Water CAs on samples before and after irradiation and at different time intervals were measured using a CA measurement system (Digidrop, GBX, Romans sur Isere, Drôme, France) equipped with a CCD camera to capture lateral snapshots of a droplet deposited on top of the preselected area, suitable for both static and dynamic CA measurements. Droplet images captured at a speed of 50 frames/s. CA values were obtained via the Digidrop software analysis, approximating the tangent of the drop profile at the triple point (three-phase contact point). Three different CA measurements were taken from each sample at different sample positions to calculate the average values.
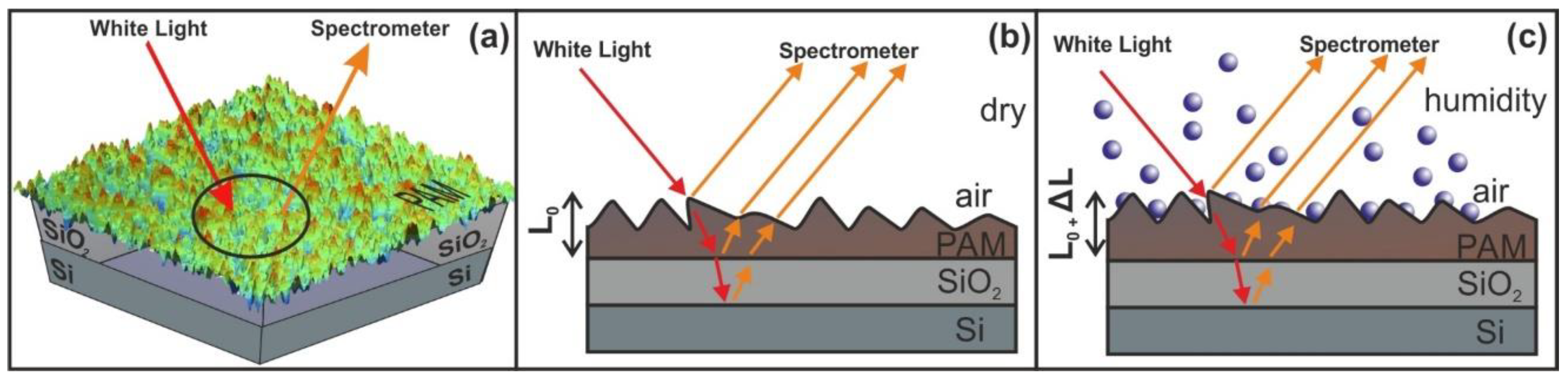
2.6. White Light Reflectance Spectroscopy (WLRS)
The WLRS measurements were performed by an FR-Basic, ThetaMetrisis™ (ThetaMetrisis SA, Athens, Greece) equipped with a VIS–NIR spectrometer (Theta Metrisis SA, Athens, Greece) having 2048 pixels detector and optical resolution of 0.35 nm. The beam of the light source comes from a white light halogen lamp, with a uniquely designed stable power supply and soft-start circuit, ensuring stable operation over time that is necessary for long time duration experiments. Software controls the instrument, performing the data acquisition and film thickness calculations. The PAM films were spin-coated on native oxide Si wafers and SiO2 layer on the top with a thickness of 2–3 nm.
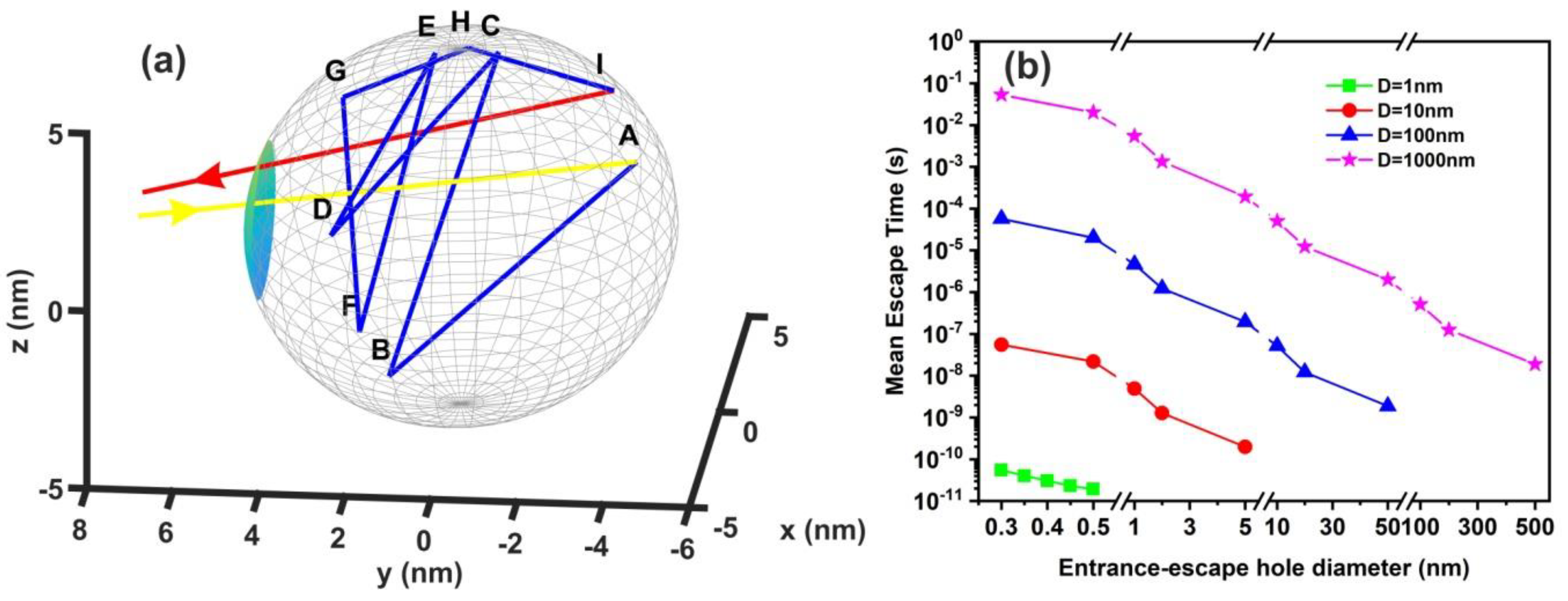
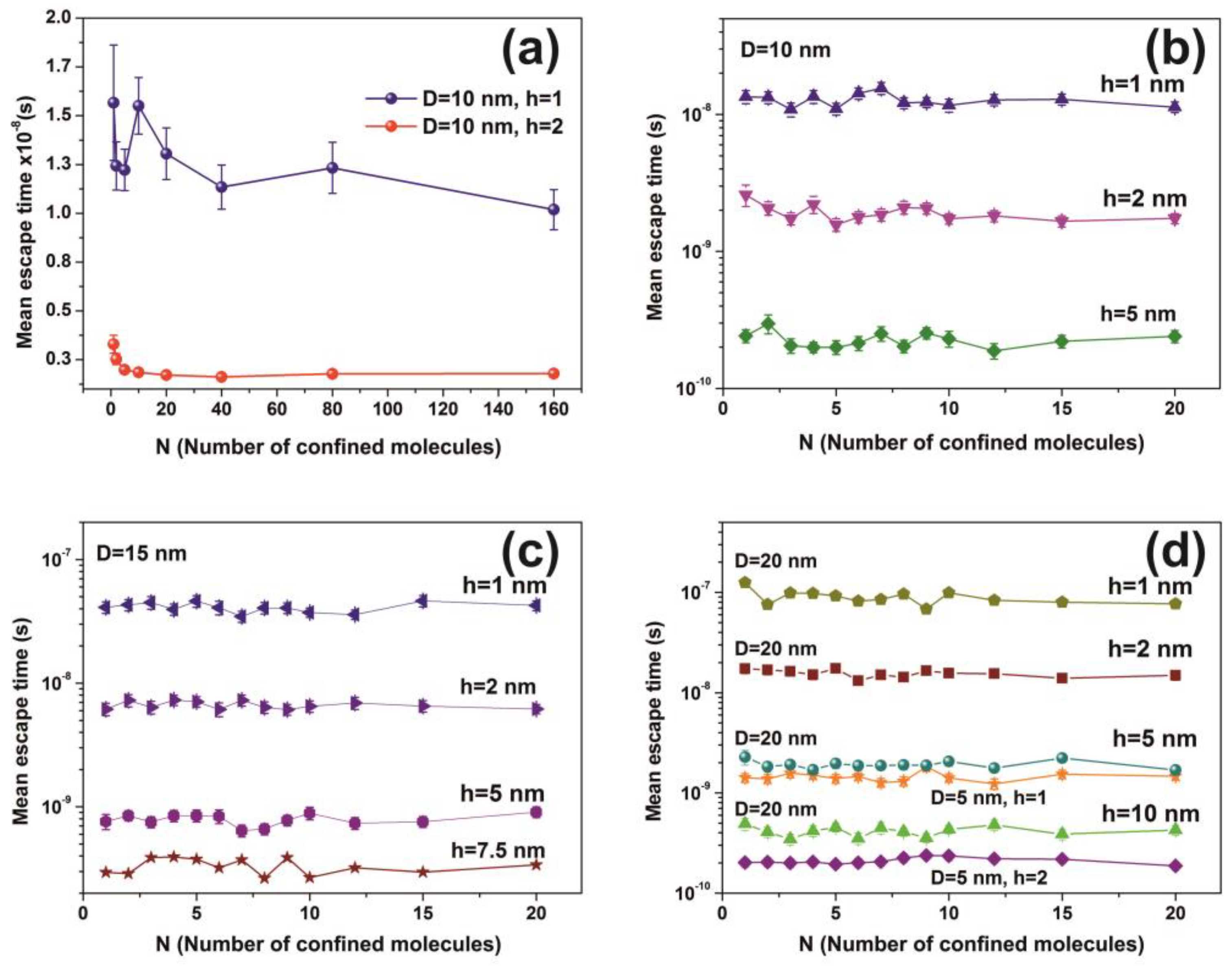
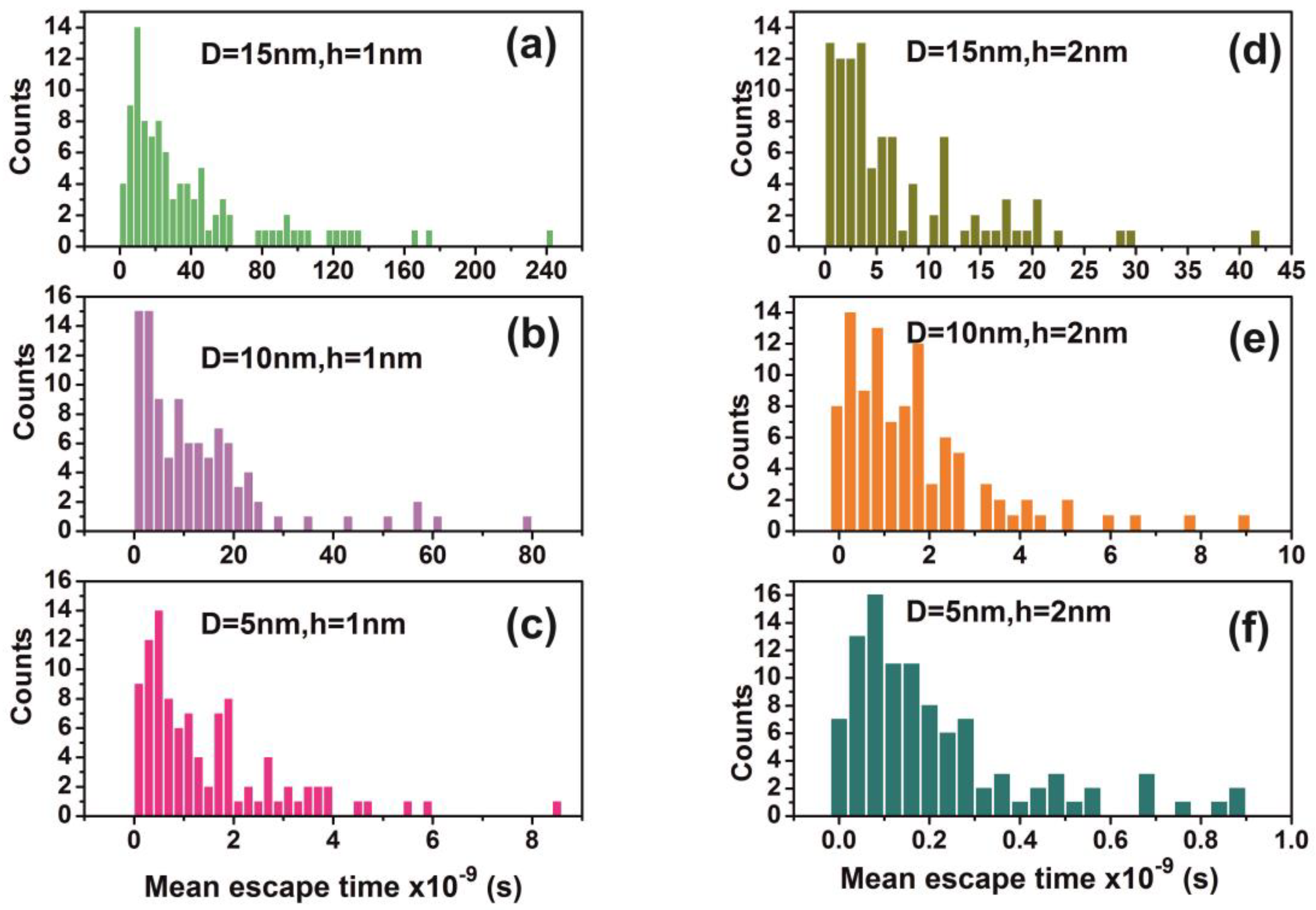
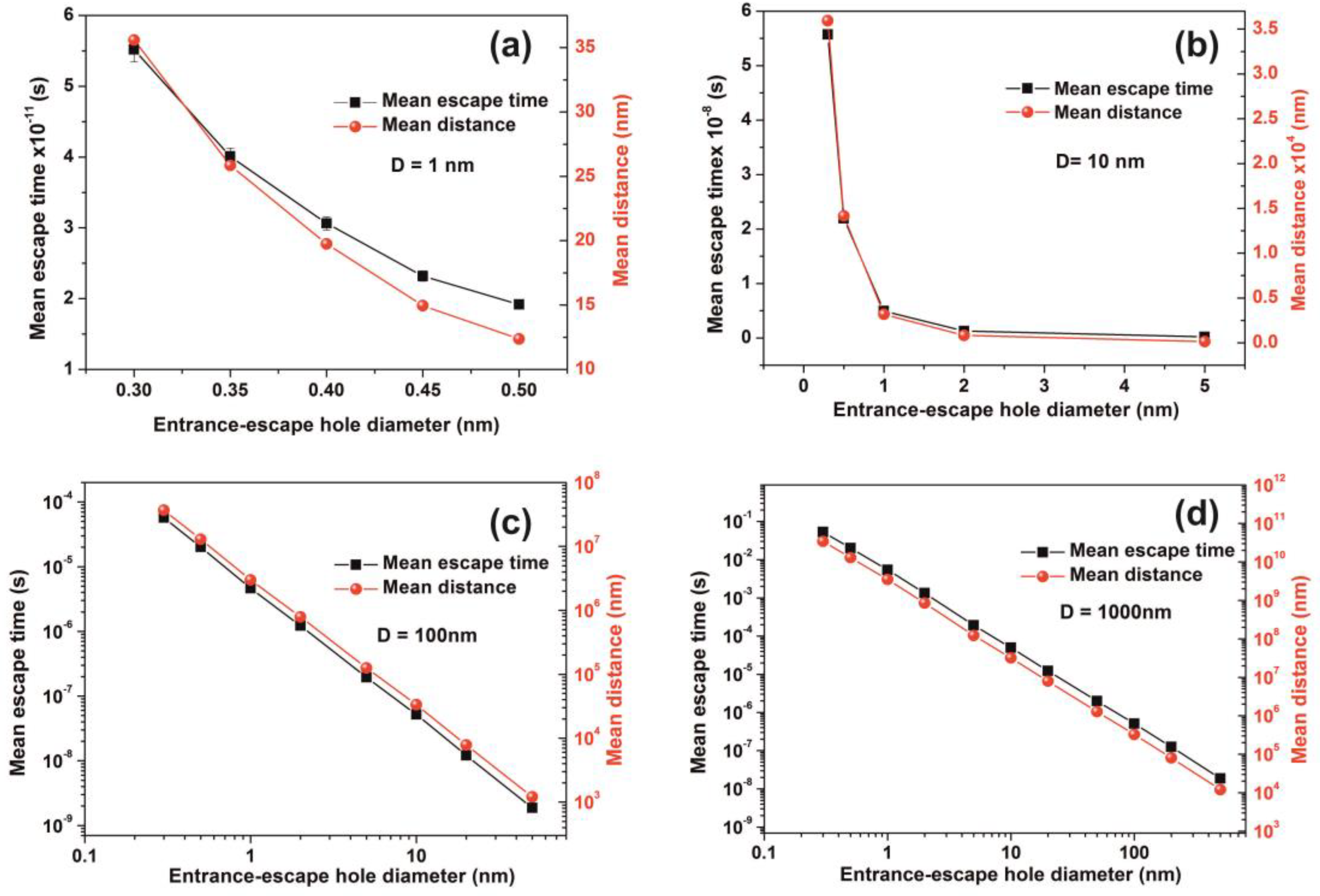
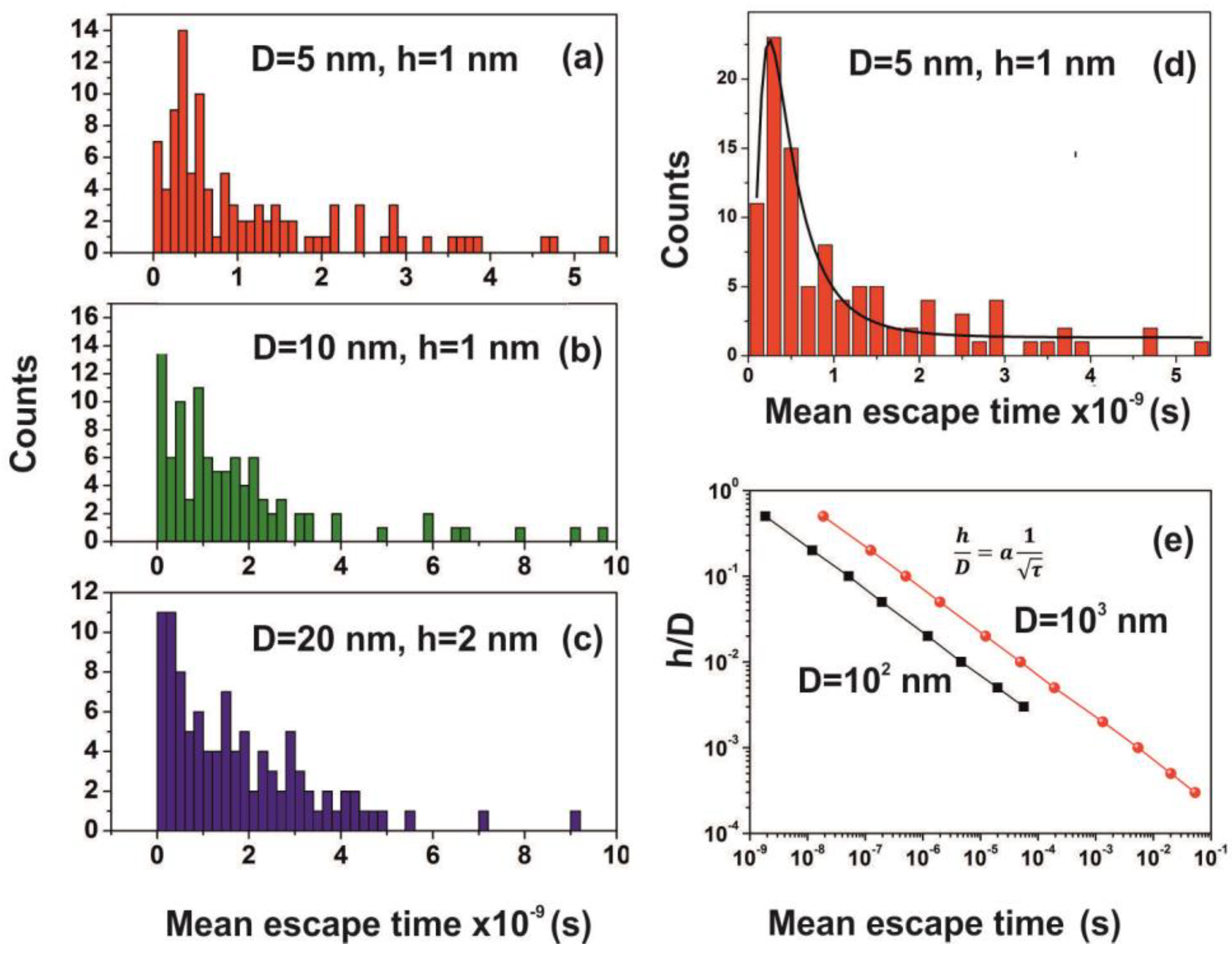
2.7. Random Walk Model
The mean escape time of a water molecule confined in nanocavities was computed by applying different 3D random walk models with diverging numbers of water molecules, variable spherical size nanocavities, and entrance-escape hole sizes. Two different models of non-interactive and interactive water molecules inside the cavities were used. The first model, the non-interactive random walk model, uses molecular masses of zero volume and elastic collisions of the water molecule with the cavity wall and it records the sequence of positions of water molecules inside the spherical cavity until it gets back to the entrance-escape hole. The collision angle was varied randomly with a uniform distribution. The model calculates the total distance that molecules travel in the cavity before they escape from the entrance-escape hole. The mean escape time was calculated by considering that the molecule attains its kinetic energy after an elastic collision with the walls of the cavity. Therefore the kinetic energy transfer from the wall to the molecule should be equal with the thermal energy of the wall , where is Boltzmann’s constant.
The escape time from the entrance-escape hole for a non-interactive water molecule in the cavity is given by the equation:
where
n is the number of collisions in each run,
R is the radius of the spherical cavity,
is the position of the molecule in the
ith collision. The entrance and the exit point in the cavity wall are given in spherical coordinates
and
, accordingly, and
is the molecular mass of water.
The interactive random walk model records the sequence of positions of a specific molecule that enters a spherical cavity through the entrance-escape hole, alongside with the locations of a variable number of neighboring molecules trapped in the cavity, until it gets back to the entrance-escape hole. At first, because of non-thermal equilibrium between water molecules within the cavity, the molecules are placed inside the cavity in random positions with random velocities of uniform distribution between 0 and . The position of each molecule was recorded every. The collision of each water molecule with the cavity wall and its neighbouring molecules is considered to be elastic. The collision angle was varied randomly with a uniform distribution. Contrary to the non-interactive model of zero-size molecules, the interactive model uses a spherical molecular diameter of 0.3 nm.
For every pair of the cavity size and entrance-escape hole, the random walk was run 102 times and the mean escape time was calculated. In addition, the mean-escape time distribution for different cavities and number of molecules was used to evaluate the thermodynamic state inside the cavity. The model was designed and run in MATLAB. 9.4.0.813654 (R2018a), The MathWorks Inc.; Natick, MA, USA.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















