
Role of the Lactide:Glycolide Ratio in PLGA Nanoparticle Stability and Release under Lysosomal Conditions for Enzyme Replacement Therapy of Lysosomal Storage Disorders
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Labeling with 125Iodine
2.3. NP Preparation
2.4. NP Coating with Targeting Antibody
2.5. NP Characterization
2.6. Encapsulation Efficiency
2.7. NP Stability and Enzyme Release under Lysosomal Conditions
2.8. Released Enzyme Activity
2.9. Statistics
3. Results and Discussion
3.1. Characterization of PLGA NPs Encapsulating HAse
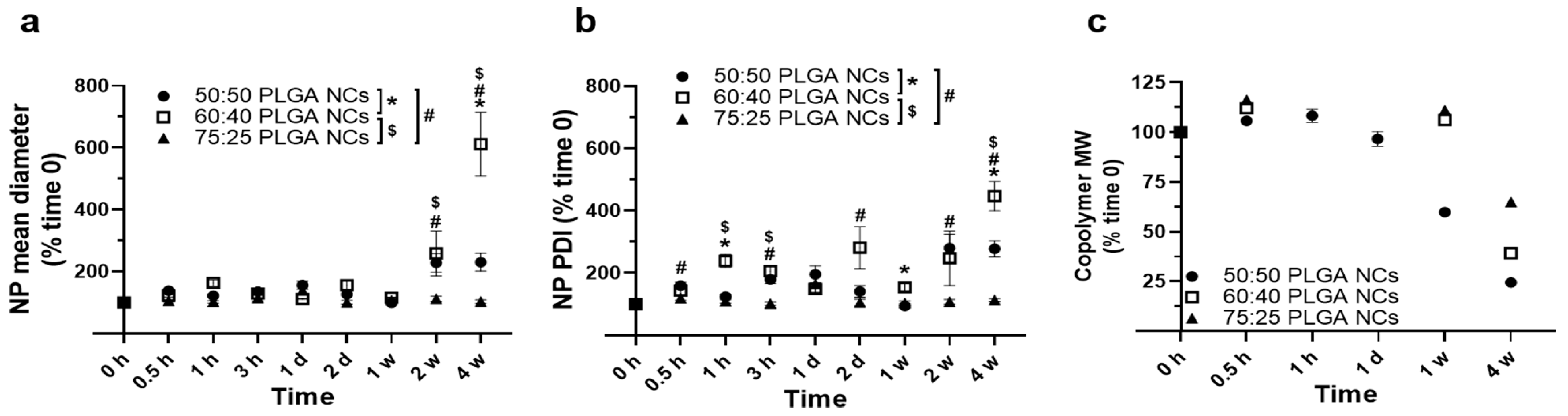
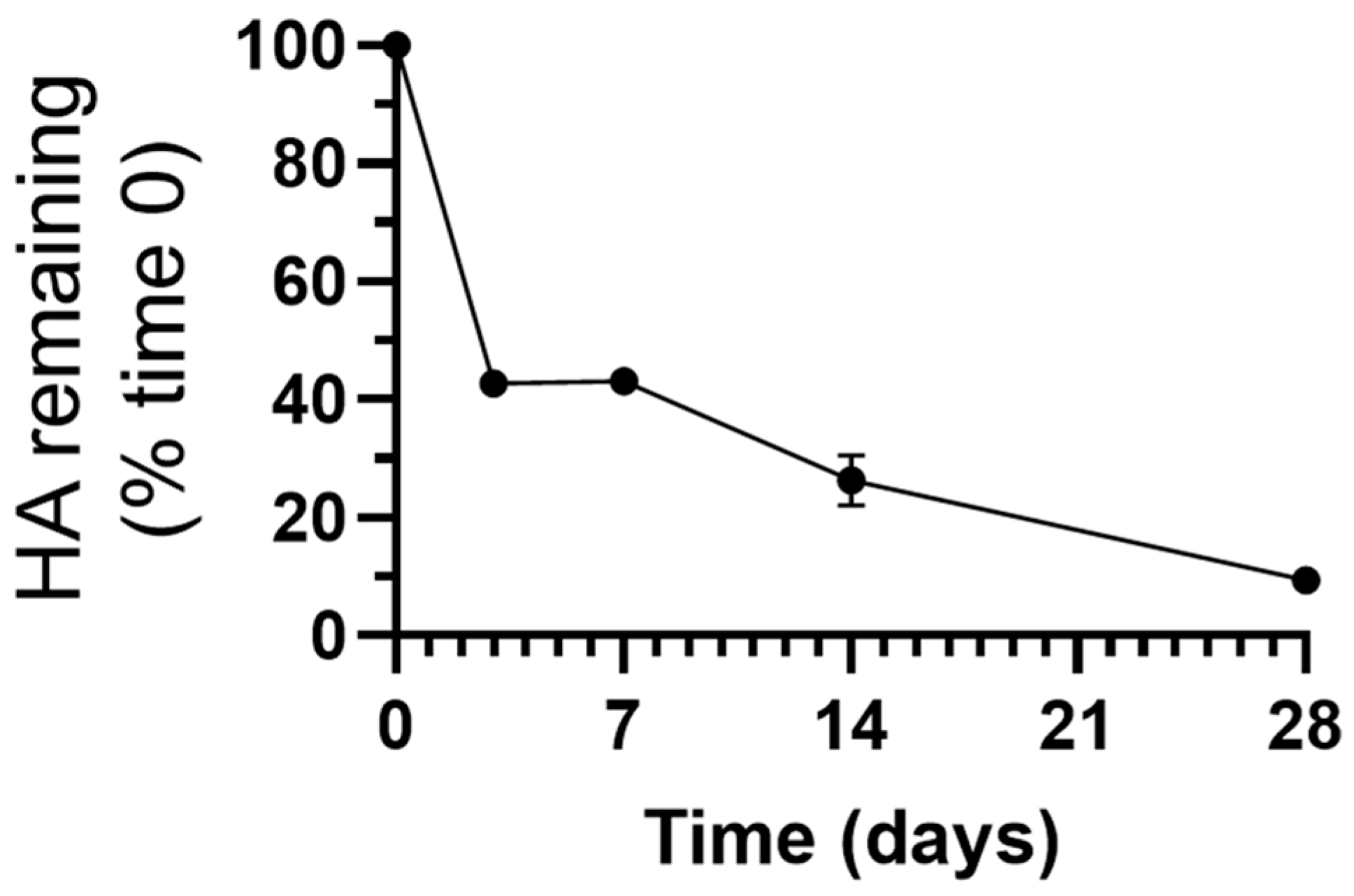
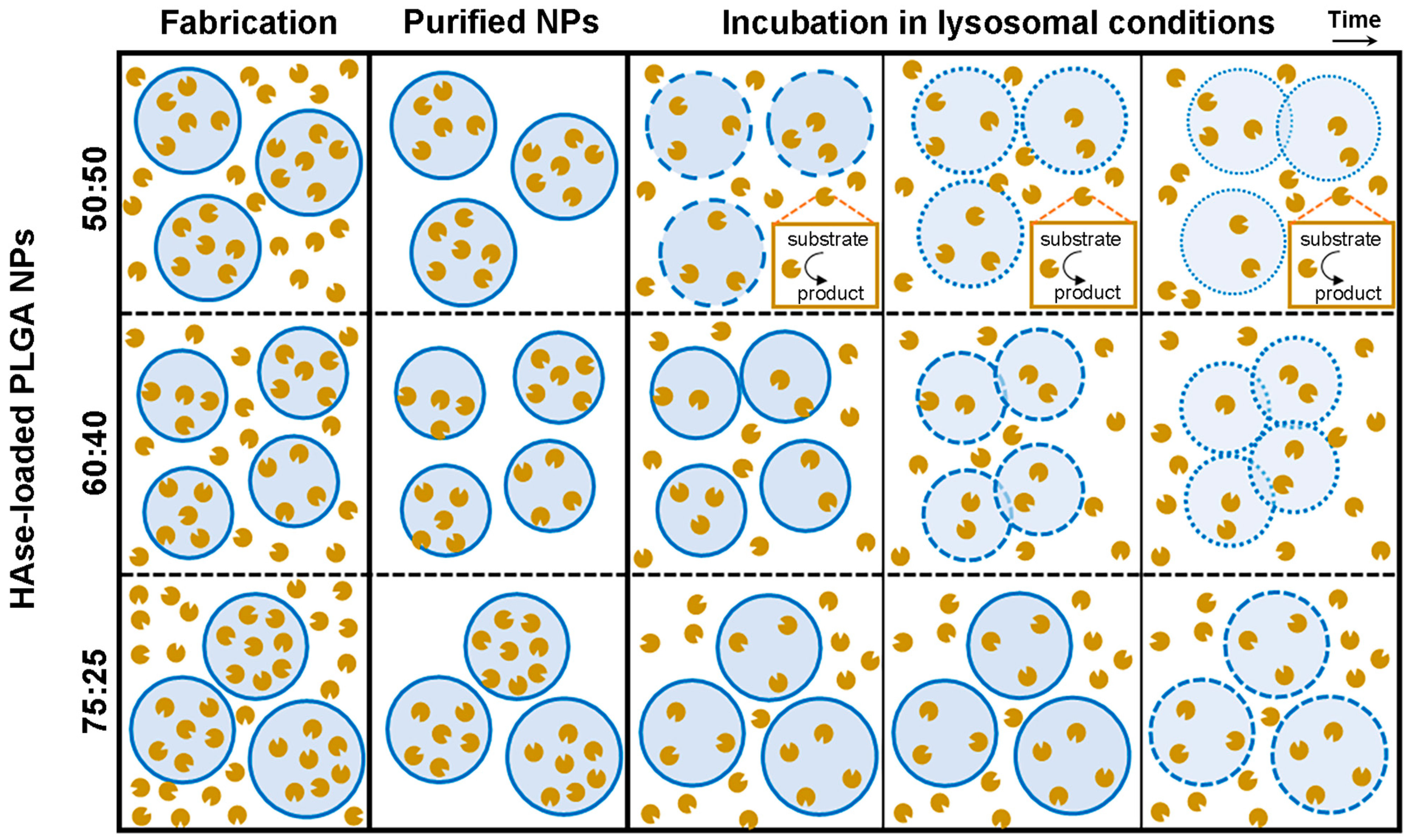
3.2. Stability under Lysosomal Conditions of PLGA NPs Encapsulating HAse
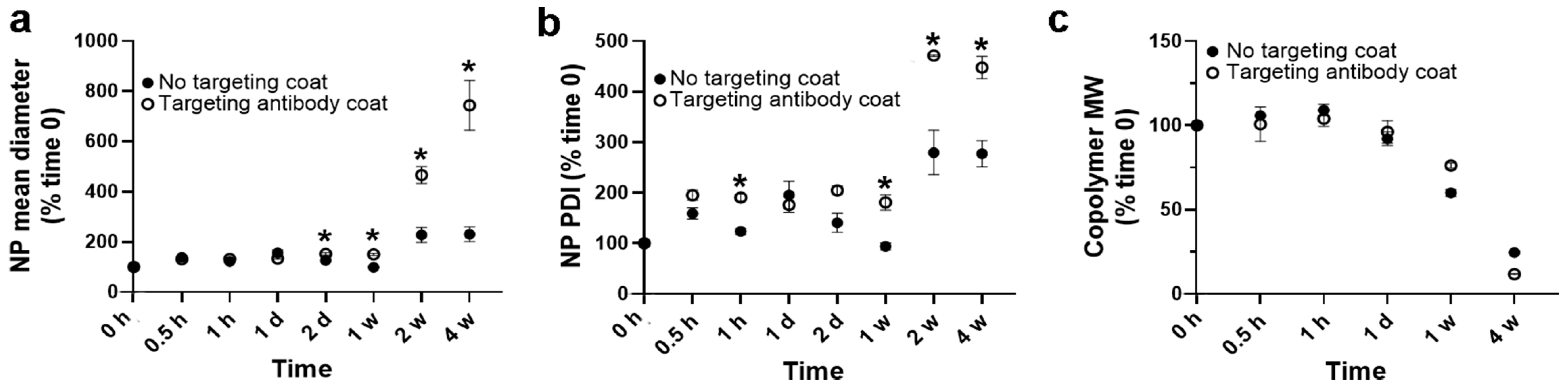
3.3. Role of a Targeting Antibody Coat on Stability under Lysosomal Conditions of PLGA NPs Encapsulating HAse
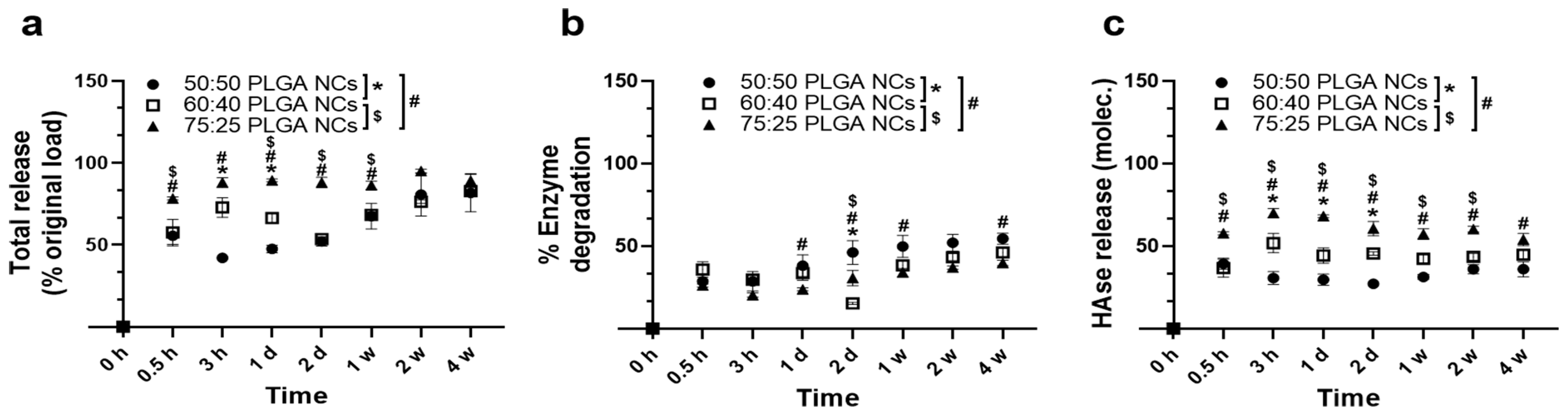
3.4. Enzyme Release under Lysosomal Conditions from PLGA NPs Encapsulating HAse
3.5. Catalytic Activity of PLGA NPs Encapsulating HAse
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Lamming, D.W.; Bar-Peled, L. Lysosome: The metabolic signaling hub. Traffic 2019, 20, 27–38. [Google Scholar] [CrossRef]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef]
- Zhang, Z.; Yue, P.; Lu, T.; Wang, Y.; Wei, Y.; Wei, X. Role of lysosomes in physiological activities, diseases, and therapy. J. Hematol. Oncol. 2021, 14, 79. [Google Scholar] [CrossRef]
- Cao, M.; Luo, X.; Wu, K.; He, X. Targeting lysosomes in human disease: From basic research to clinical applications. Signal Transduct. Target Ther. 2021, 6, 379. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef]
- Futerman, A.H.; van Meer, G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage disorders. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef]
- Aerts, J.M.F.G.; Van Weely, S.; Boot, R.; Hollak, C.E.M.; Tager, J.M. Pathogenesis of lysosomal storage disorders as illustrated by Gaucher disease. J. Inherit. Metab. Dis. 1993, 16, 288–291. [Google Scholar] [CrossRef]
- Beck, M. Variable clinical presentation in lysosomal storage disorders. J. Inherit. Metab. Dis. 2001, 24 (Suppl. 2), 47–51. [Google Scholar] [CrossRef]
- Muro, S. Strategies for delivery of therapeutics into the central nervous system for treatment of lysosomal storage disorders. Drug Deliv. Transl. Res. 2012, 2, 169–186. [Google Scholar] [CrossRef]
- Beck, M. Therapy for lysosomal storage disorders. IUBMB Life 2010, 62, 33–40. [Google Scholar] [CrossRef]
- Wraith, J.E. Limitations of enzyme replacement therapy: Current and future. J. Inherit. Metab. Dis. 2006, 29, 442–447. [Google Scholar] [CrossRef]
- Begley, D.J. Delivery of therapeutic agents to the central nervous system: The problems and the possibilities. Pharmacol. Ther. 2004, 104, 29–45. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier delivery for lysosomal storage disorders with IgG-lysosomal enzyme fusion proteins. Adv. Drug Deliv. Rev. 2022, 184, 114234. [Google Scholar] [CrossRef]
- Del Grosso, A.; Parlanti, G.; Mezzena, R.; Cecchini, M. Current treatment options and novel nanotechnology-driven enzyme replacement strategies for lysosomal storage disorders. Adv. Drug Deliv. Rev. 2022, 188, 114464. [Google Scholar] [CrossRef]
- Lu, B.; Ku, J.; Flojo, R.; Olson, C.; Bengford, D.; Marriott, G. Exosome- and extracellular vesicle-based approaches for the treatment of lysosomal storage disorders. Adv. Drug Deliv. Rev. 2022, 188, 114465. [Google Scholar] [CrossRef]
- Tomsen-Melero, J.; Merlo-Mas, J.; Carreño, A.; Sala, S.; Córdoba, A.; Veciana, J.; González-Mira, E.; Ventosa, N. Liposomal formulations for treating lysosomal storage disorders. Adv. Drug Deliv. Rev. 2022, 190, 114531. [Google Scholar] [CrossRef]
- Placci, M.; Giannotti, M.I.; Muro, S. Polymer-based drug delivery systems under investigation for enzyme replacement and other therapies of lysosomal storage disorders. Adv. Drug Deliv. Rev. 2023, 197, 114683. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Cui, J.; van Koeverden, M.P.; Müllner, M.; Kempe, K.; Caruso, F. Emerging methods for the fabrication of polymer capsules. Adv. Colloid. Interf. Sci. 2014, 207, 14–31. [Google Scholar] [CrossRef]
- Jain, R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar] [CrossRef]
- Astete, C.E.; Sabliov, C.M. Synthesis and characterization of PLGA nanoparticles. J. Biomater. Sci. Polym. Ed. 2006, 17, 247–289. [Google Scholar] [CrossRef]
- Haque, S.; Boyd, B.J.; Mcintosh, M.P.; Pouton, C.W.; Kamin-Skas, L.M.; Whittaker, M. Suggested Procedures for the Reproducible Synthesis of Poly(d,l-lactide-co-glycolide) Nanoparticles Using the Emulsification Solvent Diffusion Platform. Curr. Nanosci. 2018, 14, 448–453. [Google Scholar] [CrossRef]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, B.; Xia, G.; Choi, S.H. FDA’s poly (lactic-co-glycolic acid) research program and regulatory outcomes. AAPS J. 2021, 23, 92. [Google Scholar] [CrossRef]
- Bourdenx, M.; Daniel, J.; Genin, E.; Soria, F.N.; Blanchard-Desce, M.; Bezard, E.; Dehay, B. Nanoparticles restore lysosomal acidification defects: Implications for Parkinson and other lysosomal-related diseases. Autophagy 2016, 12, 472–483. [Google Scholar] [CrossRef]
- Prévot, G.; Soria, F.N.; Thiolat, M.L.; Daniel, J.; Verlhac, J.B.; Blanchard-Desce, M.; Bezard, E.; Barthélémy, P.; Crauste-Manciet, S.; Dehay, B. Harnessing lysosomal pH through PLGA nanoemulsion as a treatment of lysosomal-related neurodegenerative diseases. Bioconjug. Chem. 2018, 29, 4083–4089. [Google Scholar] [CrossRef]
- Zeng, J.; Martin, A.; Han, X.; Shirijai, O.S.; Grinstaff, M.W. Biodegradable PLGA Nanoparticles Restore Lysosomal Acidity and Protect Neural PC-12 Cells against Mitochondrial Toxicity. Ind. Eng. Chem. Res. 2019, 58, 13910–13917. [Google Scholar] [CrossRef]
- Manthe, R.L.; Loeck, M.; Bhowmick, T.; Solomon, M.; Muro, S. Intertwined mechanisms define transport of anti-ICAM nanocarriers across the endothelium and brain delivery of a therapeutic enzyme. J. Control. Release 2020, 324, 181–193. [Google Scholar] [CrossRef]
- Muntimadugu, E.; Silva-Abreu, M.; Vives, G.; Loeck, M.; Pham, V.; Del Moral, M.; Solomon, M.; Muro, S. Comparison between Nanoparticle Encapsulation and Surface Loading for Lysosomal Enzyme Replacement Therapy. Int. J. Mol. Sci. 2022, 23, 4034. [Google Scholar] [CrossRef]
- Solomon, M.; Loeck, M.; Silva-Abreu, M.; Moscoso, R.; Bautista, R.; Vigo, M.; Muro, S. Altered blood-brain barrier transport of nanotherapeutics in lysosomal storage diseases. J. Control. Release 2022, 349, 1031–1044. [Google Scholar] [CrossRef]
- Garnacho, C.; Dhami, R.; Solomon, M.; Schuchman, E.H.; Muro, S. Enhanced Delivery and Effects of Acid Sphingomyelinase by ICAM-1-Targeted Nanocarriers in Type B Niemann-Pick Disease Mice. Mol. Ther. 2017, 25, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Galliani, M.; Santi, M.; Del Grosso, A.; Cecchettini, A.; Santorelli, F.M.; Hofmann, S.L.; Lu, J.Y.; Angella, L.; Cecchini, M.; Signore, G. Cross-linked enzyme aggregates as versatile tool for enzyme delivery: Application to polymeric nanoparticles. Bioconjug. Chem. 2018, 29, 2225–2231. [Google Scholar] [CrossRef] [PubMed]
- Del Grosso, A.; Galliani, M.; Angella, L.; Santi, M.; Tonazzini, I.; Parlanti, G.; Signore, G.; Cecchini, M. Brain-targeted enzyme-loaded nanoparticles: A breach through the blood-brain barrier for enzyme replacement therapy in Krabbe disease. Sci. Adv. 2019, 5, eaax7462. [Google Scholar] [CrossRef]
- Costantino, L.; Gandolfi, F.; Tosi, G.; Rivasi, F.; Vandelli, M.A.; Forni, F. Peptide-derivatized biodegradable nanoparticles able to cross the blood-brain barrier. J. Control. Release 2005, 108, 84–96. [Google Scholar] [CrossRef]
- Salvalaio, M.; Rigon, L.; Belletti, D.; D’Avanzo, F.; Pederzoli, F.; Ruozi, B.; Marin, O.; Vandelli, M.A.; Forni, F.; Scarpa, M.; et al. Targeted polymeric nanoparticles for brain delivery of high molecular weight molecules in lysosomal storage disorders. PLoS ONE 2016, 11, e0156452. [Google Scholar] [CrossRef] [PubMed]
- Rigon, L.; Salvalaio, M.; Pederzoli, F.; Legnini, E.; Duskey, J.T.; D’Avanzo, F.; De Filippis, C.; Ruozi, B.; Marin, O.; Vandelli, M.A.; et al. Targeting brain disease in MPSII: Preclinical evaluation of IDS-loaded PLGA nanoparticles. Int. J. Mol. Sci. 2019, 20, 2014. [Google Scholar] [CrossRef]
- Tosi, G.; Ruozi, B.; Belletti, D.; Vilella, A.; Zoli, M.; Vandelli, M.A.; Forni, F. Brain-targeted polymeric nanoparticles: In vivo evidence of different routes of administration in rodents. Nanomedicine 2013, 8, 1373–1383. [Google Scholar] [CrossRef]
- Tancini, B.; Tosi, G.; Bortot, B.; Dolcetta, D.; Magini, A.; De Martino, E.; Urbanelli, L.; Ruozi, B.; Forni, F.; Emiliani, C.; et al. Use of Polylactide-Co-Glycolide-Nanoparticles for Lysosomal Delivery of a Therapeutic Enzyme in Glycogenosis Type II Fibroblasts. J. Nanosci. Nanotechnol. 2015, 15, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Kao, W.J. Drug Release Kinetics and Transport Mechanisms of Non-degradable and Degradable Polymeric Delivery Systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- Park, T.G. Degradation of poly(lactic-co-glycolic acid) microspheres: Effect of copolymer composition. Biomaterials 1995, 16, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Vert, M.; Mauduit, J.; Li, S. Biodegradation of PLA/GA polymers: Increasing complexity. Biomaterials 1994, 15, 1209–1213. [Google Scholar] [CrossRef] [PubMed]
- Keles, H.; Naylor, A.; Clegg, F.; Sammon, C. Investigation of factors influencing the hydrolytic degradation of single PLGA microparticles. Polym. Deg. Stab. 2015, 119, 228–241. [Google Scholar] [CrossRef]
- Romero, G.; Echeverría, M.; Qiu, Y.; Murray, R.A.; Moya, S.E. A novel approach to monitor intracellular degradation kinetics of poly(lactide-co-glycolide) nanoparticles by means of flow cytometry. J. Mater. Chem. B 2014, 2, 826–833. [Google Scholar] [CrossRef]
- Xu, Y.; Kim, C.S.; Saylor, D.M.; Koo, D. Polymer degradation and drug delivery in PLGA-based drug-polymer applications: A review of experiments and theories. J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 1692–1716. [Google Scholar] [CrossRef]
- Natowicz, M.R.; Short, M.P.; Wang, Y.; Dickersin, G.R.; Gebhardt, M.C.; Rosenthal, D.I.; Sims, K.B.; Rosenberg, A.E. Clinical and biochemical manifestations of hyaluronidase deficiency. N. Engl. J. Med. 1996, 335, 029–1033. [Google Scholar] [CrossRef]
- Imundo, L.; LeDuc, C.A.; Guha, S.; Brown, M.; Perino, G.; Gushulak, L.; Triggs-Raine, B.; Chung, W.K. A complete deficiency of Hyaluronoglucosaminidase 1(HYAL1) presenting as familial juvenile idiopathic arthritis. J. Inherit. Metab. Dis. 2011, 34, 1013–1022. [Google Scholar] [CrossRef]
- Triggs-Raine, B.; Salo, T.J.; Zhang, H.; Wicklow, B.A.; Natowicz, M.R. Mutations in HYAL1, a member of a tandemly distributed multigene family encoding disparate hyaluronidase activities, cause a newly described lysosomal disorder, mucopolysaccharidosis IX. Proc. Natl. Acad. Sci. USA 1999, 96, 6296–6300. [Google Scholar] [CrossRef]
- Wiseman, M.E.; Frank, C.W. Antibody adsorption and orientation on hydrophobic surfaces. Langmuir 2012, 28, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Pochapski, D.J.; Carvalho Dos Santos, C.; Leite, G.W.; Pulcinelli, S.H.; Santilli, C.V. Zeta Potential and Colloidal Stability Predictions for Inorganic Nanoparticle Dispersions: Effects of Experimental Conditions and Electrokinetic Models on the Interpretation of Results. Langmuir 2021, 37, 13379–13389. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Toghani, D.; Wong, B.; Nance, E. Colloidal stability as a determinant of nanoparticle behavior in the brain. Colloids Surf. B Biointerfaces 2018, 170, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Muro, S.; Wiewrodt, R.; Thomas, A.; Koniaris, L.; Albelda, S.M.; Muzykantov, V.R.; Koval, M. A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J. Cell. Sci. 2003, 116, 1599–1609. [Google Scholar] [CrossRef]
- Muro, S.; Garnacho, C.; Champion, J.A.; Leferovich, J.; Gajewski, C.; Schuchman, E.H.; Mitragotri, S.; Muzykantov, V.R. Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol. Ther. 2008, 16, 1450–1458. [Google Scholar] [CrossRef]
- Serrano, D.; Bhowmick, T.; Chadha, R.; Garnacho, C.; Muro, S. Intercellular adhesion molecule 1 engagement modulates sphingomyelinase and ceramide, supporting uptake of drug carriers by the vascular endothelium. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1178–1185. [Google Scholar] [CrossRef]
- Ansar, M.; Serrano, D.; Papademetriou, I.; Bhowmick, T.K.; Muro, S. Biological functionalization of drug delivery carriers to bypass size restrictions of receptor-mediated endocytosis independently from receptor targeting. ACS Nano 2013, 7, 10597–10611. [Google Scholar] [CrossRef]
- Schnitzer, J.E. Caveolae: From basic trafficking mechanisms to targeting transcytosis for tissue-specific drug and gene delivery in vivo. Adv. Drug. Deliv. Rev. 2001, 49, 265–280. [Google Scholar] [CrossRef]
- Shuvaev, V.V.; Kiseleva, R.Y.; Arguiri, E.; Villa, C.H.; Muro, S.; Christofidou-Solomidou, M.; Stan, R.V.; Muzykantov, V.R. Targeting superoxide dismutase to endothelial caveolae profoundly alleviates inflammation caused by endotoxin. J. Control. Release 2018, 272, 1–8. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, H.; Fontana, F.; Hirvonen, J.T.; Santos, H.A. Current developments and applications of microfluidic technology toward clinical translation of nanomedicines. Adv. Drug Deliv. Rev. 2018, 128, 54–83. [Google Scholar] [CrossRef]
- Lim, J.M.; Swami, A.; Gilson, L.M.; Chopra, S.; Choi, S.; Wu, J.; Langer, R.; Karnik, R.; Farokhzad, O.C. Ultra-high throughput synthesis of nanoparticles with homogeneous size distribution using a coaxial turbulent jet mixer. ACS Nano 2014, 8, 6056–6065. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Kim, J.M.; Seo, K.S.; Jeong, Y.K.; Lee, H.B.; Khang, G. Characterization of degradation behavior for PLGA in various pH condition by simple liquid chromatography method. Biomed. Mater. Eng. 2005, 15, 279–288. [Google Scholar] [PubMed]
- Baiti, R.N.; Ardhyananta, H.; El Kirat, K. Effect of acidic and basic environment to the degradation behavior of PLGA nanocapcules for biomedical application. Adv. Mater. Res. 2015, 1123, 213–216. [Google Scholar] [CrossRef]
- Moin, A.; Wani, S.U.D.; Osmani, R.A.; Abu Lila, A.S.; Khafagy, E.S.; Arab, H.H.; Gangadharappa, H.V.; Allam, A.N. Formulation, characterization, and cellular toxicity assessment of tamoxifen-loaded silk fibroin nanoparticles in breast cancer. Drug Deliv. 2021, 28, 1626–1636. [Google Scholar] [CrossRef]
- Rescignano, N.; Tarpani, L.; Romani, A.; Bicchi, I.; Mattioli, S.; Emiliani, C.; Torre, L.; Kenny, J.M.; Martino, S.; Latterini, L.; et al. In-vitro degradation of PLGA nanoparticles in aqueous medium and in stem cell cultures by monitoring the cargo fluorescence spectrum. Polym. Degr. Stab. 2016, 134, 296–304. [Google Scholar] [CrossRef]
- Mohammad, A.K.; Reineke, J.J. Quantitative detection of PLGA nanoparticle degradation in tissues following intravenous administration. Mol. Pharm. 2013, 10, 2183–2189. [Google Scholar] [CrossRef]
- Wang, T.; Xue, P.; Wang, A.; Yin, M.; Han, J.; Tang, S.; Liang, R. Pore change during degradation of octreotide acetate-loaded PLGA microspheres: The effect of polymer blends. Eur. J. Pharm. Sci. 2019, 138, 104990. [Google Scholar] [CrossRef]
- Ma, H.; Ó’Fágáin, C.; O’Kennedy, R. Antibody stability: A key to performance—Analysis, influences and improvement. Biochimie 2020, 177, 213–225. [Google Scholar] [CrossRef]
- Ejima, D.; Tsumoto, K.; Fukada, H.; Yumioka, R.; Nagase, K.; Arakawa, T.; Philo, J.S. Effects of acid exposure on the conformation, stability, and aggregation of monoclonal antibodies. Proteins 2007, 66, 954–962. [Google Scholar] [CrossRef]
- Yoo, J.; Won, Y.Y. Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef]
- Rafati, A.; Boussahel, A.; Shakesheff, K.M.; Shard, A.G.; Roberts, C.J.; Chen, X.; Scurr, D.J.; Rigby-Singleton, S.; Whiteside, P.; Alexander, M.R.; et al. Chemical and spatial analysis of protein loaded PLGA microspheres for drug delivery applications. J. Control. Release 2012, 162, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Washington, C. Drug release from microparticulate systems. In Microencapsulation Methods and Industrial Applications; Benita, S., Ed.; Taylor & Francis: New York, NY, USA, 2006; pp. 156–175. [Google Scholar]
- Roki, N.; Solomon, M.; Casta, L.; Bowers, J.; Getts, R.C.; Muro, S. A method to improve quantitative radiotracing-based analysis of the in vivo biodistribution of drug carriers. Bioeng. Transl. Med. 2021, 6, e10208. [Google Scholar] [CrossRef] [PubMed]
- Sah, H. Protein instability toward organic solvent/water emulsification: Implications for protein microencapsulation into microspheres. P.D.A. J. Pharm. Sci. Technol. 1999, 53, 3–10. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLGA Copolymer | Diameter (nm) | PDI | ζ-Potential (mV) | %EE | HAse molec./NP | µgHAse/mg Copolymer |
|---|---|---|---|---|---|---|
| 50:50 | 167.8 ± 4.2 | 0.14 ±0.01 | −38.4 ± 0.6 | 20.4 ± 5.7 | 400.9 ± 85.1 | 1.6 ± 0.5 |
| 60:40 | 121.4 ± 2.2 * | 0.12 ± 0.02 | −34.7 ± 1.0 * | 27.9 ± 1.2 | 243.7 ± 35.0 | 2.5 ± 0.1 |
| 75:25 | 164.7 ± 13.1 $ | 0.16 ± 0.02 | −34.9 ± 1.8 | 15.0 ± 2.2 $ | 335.6 ± 63.4 | 1.2 ± 0.2 $ |
| 50:50 PLGA | Diameter (nm) | PDI | ζ-Potential (mV) | HAse molec./NP | Ab molec./NP |
|---|---|---|---|---|---|
| Non-coated | 167.8 ± 4.2 | 0.14 ±0.01 | −38.4 ± 0.6 | 400.9 ± 85.1 | ND |
| Ab-coated | 224.9x ± 26.5 * | 0.19 ± 0.02 * | −32.6 ± 2.8 * | (same as above) | 187.4 ± 13.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Moral, M.; Loeck, M.; Muntimadugu, E.; Vives, G.; Pham, V.; Pfeifer, P.; Battaglia, G.; Muro, S. Role of the Lactide:Glycolide Ratio in PLGA Nanoparticle Stability and Release under Lysosomal Conditions for Enzyme Replacement Therapy of Lysosomal Storage Disorders. J. Funct. Biomater. 2023, 14, 440. https://doi.org/10.3390/jfb14090440
del Moral M, Loeck M, Muntimadugu E, Vives G, Pham V, Pfeifer P, Battaglia G, Muro S. Role of the Lactide:Glycolide Ratio in PLGA Nanoparticle Stability and Release under Lysosomal Conditions for Enzyme Replacement Therapy of Lysosomal Storage Disorders. Journal of Functional Biomaterials. 2023; 14(9):440. https://doi.org/10.3390/jfb14090440
Chicago/Turabian Styledel Moral, Maria, Maximilian Loeck, Eameema Muntimadugu, Guillem Vives, Vy Pham, Peter Pfeifer, Giuseppe Battaglia, and Silvia Muro. 2023. "Role of the Lactide:Glycolide Ratio in PLGA Nanoparticle Stability and Release under Lysosomal Conditions for Enzyme Replacement Therapy of Lysosomal Storage Disorders" Journal of Functional Biomaterials 14, no. 9: 440. https://doi.org/10.3390/jfb14090440