
Comparative Study of Porous Iron Foams for Biodegradable Implants: Structural Analysis and In Vitro Assessment
 , , and
, , and 
Abstract
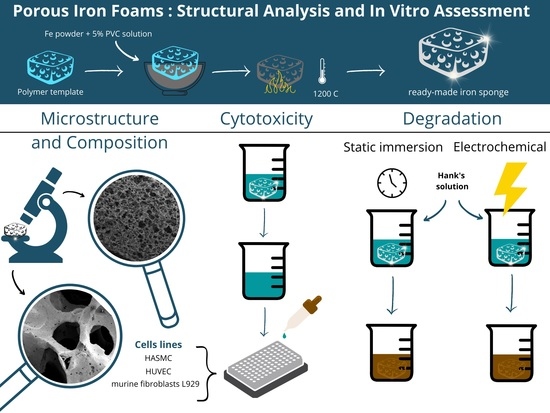
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Iron Foams
2.3. Morphology and Elemental Composition
2.4. Low-Temperature N2 Adsorption Isotherm and Pore Size Distribution
2.5. Immersion Enthalpy and Surface Energy Determination
2.6. Corrosion In Vitro
2.6.1. Static Immersion Test
2.6.2. Electrochemical Degradation
2.7. Cytotoxicity Assessment of the Foams In Vitro
2.7.1. Cell Culture
2.7.2. Preparation of Materials and Extracts
2.7.3. Cell Treatment
2.7.4. Cell Viability Evaluated Using MTT Assay
2.7.5. Cytotoxicity Assay
2.7.6. Statistical Analysis
3. Results
3.1. Morphology and Elemental Composition
3.2. Low-Temperature N2 Adsorption Isotherm and Pore Size Distribution
3.3. Immersion Enthalpy and Surface Energy Determination
3.4. Corrosion In Vitro
3.5. Electrochemical Degradation
3.6. Tested Scaffolds Induced Cytotoxic Effect on the Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mani, G.; Feldman, M.D.; Patel, D.; Agrawal, C.M. Coronary Stents: A Materials Perspective. Biomaterials 2007, 28, 1689–1710. [Google Scholar] [CrossRef] [PubMed]
- Chesta, F.; Rizvi, Z.H.; Oberoi, M.; Buttar, N. The Role of Stenting in Patients with Variceal Bleeding. Tech. Innov. Gastrointest. Endosc. 2020, 22, 205–211. [Google Scholar] [CrossRef]
- Law, M.A.; Shamszad, P.; Nugent, A.W.; Justino, H.; Breinholt, J.P.; Mullins, C.E.; Ing, F.F. Pulmonary Artery Stents: Long-Term Follow-Up. Catheter. Cardiovasc. Interv. 2010, 75, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, H. Manufacturing of Cardiovascular Stents. In Metallic Biomaterials Processing and Medical Device Manufacturing; Elsevier: Amsterdam, The Netherlands, 2020; pp. 317–340. ISBN 978-0-08-102965-7. [Google Scholar]
- Bowen, P.K.; Shearier, E.R.; Zhao, S.; Guillory, R.J.; Zhao, F.; Goldman, J.; Drelich, J.W. Biodegradable Metals for Cardiovascular Stents: From Clinical Concerns to Recent Zn-Alloys. Adv. Healthc. Mater. 2016, 5, 1121–1140. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Wang, H.; Zhu, L.; Han, Y.; Wang, K.; Li, Y.; Ma, J.; Lin, J.; Wen, C.; Huang, S. A Biodegradable in Situ Zn–Mg2Ge Composite for Bone-Implant Applications. Acta Biomater. 2022, 146, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Zartner, P.; Cesnjevar, R.; Singer, H.; Weyand, M. First Successful Implantation of a Biodegradable Metal Stent into the Left Pulmonary Artery of a Preterm Baby. Catheter. Cardiovasc. Interv. 2005, 66, 590–594. [Google Scholar] [CrossRef]
- Kaushik, V.; Nithish Kumar, B.; Sakthi Kumar, S.; Vignesh, M. Magnesium Role in Additive Manufacturing of Biomedical Implants—Challenges and Opportunities. Addit. Manuf. 2022, 55, 102802. [Google Scholar] [CrossRef]
- Gąsior, G.; Szczepański, J.; Radtke, A. Biodegradable Iron-Based Materials—What Was Done and What More Can Be Done? Materials 2021, 14, 3381. [Google Scholar] [CrossRef]
- Salama, M.; Vaz, M.F.; Colaço, R.; Santos, C.; Carmezim, M. Biodegradable Iron and Porous Iron: Mechanical Properties, Degradation Behaviour, Manufacturing Routes and Biomedical Applications. J. Funct. Biomater. 2022, 13, 72. [Google Scholar] [CrossRef]
- Hrubovčáková, M.; Kupková, M.; Džupon, M. Fe and Fe-P Foam for Biodegradable Bone Replacement Material: Morphology, Corrosion Behaviour, and Mechanical Properties. Adv. Mater. Sci. Eng. 2016, 2016, 6257368. [Google Scholar] [CrossRef]
- Oriňaková, R.; Gorejová, R.; Králová, Z.O.; Petráková, M.; Oriňak, A. Novel Trends and Recent Progress on Preparation Methods of Biodegradable Metallic Foams for Biomedicine: A Review. J. Mater. Sci. 2021, 56, 13925–13963. [Google Scholar] [CrossRef]
- Wiśniewski, M.; Rychlicki, G.; Arcimowicz, A. Experimental and Theoretical Estimations of the Polar Force Contributions to the Heat of Immersion of Carbon Nanotubes. Chem. Phys. Lett. 2010, 485, 331–334. [Google Scholar] [CrossRef]
- Grodzicka, M.; Gąsior, G.; Wiśniewski, M.; Bartmański, M.; Radtke, A. A Simple Replica Method as the Way to Obtain a Morphologically and Mechanically Bone-like Iron-Based Biodegradable Material. Materials 2022, 15, 4552. [Google Scholar] [CrossRef]
- Douillard, J.-M.; Salles, F.; Henry, M.; Malandrini, H.; Clauss, F. Surface Energy of Talc and Chlorite: Comparison between Electronegativity Calculation and Immersion Results. J. Colloid Interface Sci. 2007, 305, 352–360. [Google Scholar] [CrossRef]
- ISO 10993-5; Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity. ISO: Geneva, Switzerland, 2009.
- ISO 10993-12; Biological Evaluation of Medical Devices—Part 12: Sample Preparation and Reference Materials. ISO: Geneva, Switzerland, 2021.
- Kubásek, J.; Vojtěch, D.; Jablonská, E.; Pospíšilová, I.; Lipov, J.; Ruml, T. Structure, Mechanical Characteristics and in Vitro Degradation, Cytotoxicity, Genotoxicity and Mutagenicity of Novel Biodegradable Zn–Mg Alloys. Mater. Sci. Eng. C 2016, 58, 24–35. [Google Scholar] [CrossRef]
- Lowell, S.; Shields, J.E. Adsorption Isotherms. In Powder Surface Area and Porosity; Springer: Dordrecht, The Netherlands, 1991; pp. 11–13. ISBN 978-90-481-4005-3. [Google Scholar]
- Birbilis, N.; Holloway, L.J. Use of the Time Constant to Detect Corrosion Speed in Reinforced Concrete Structures. Cem. Concr. Compos. 2007, 29, 330–336. [Google Scholar] [CrossRef]
- Sriramadasu, R.C.; Lu, Y.; Banerjee, S.; Sriramula, S. Advances in Corrosion Monitoring of Reinforced Concrete Using Active and Passive Sensing Approaches. In The Rise of Smart Cities; Elsevier: Amsterdam, The Netherlands, 2022; pp. 407–429. ISBN 978-0-12-817784-6. [Google Scholar]
- Han, Y.; Choi, J.; Kim, H.-S.; Kim, H.; Park, J. Control of Pore and Window Size of Ceramic Foams with Tri-Modal Pore Structure: Influence of Agar Concentration. Mater. Lett. 2013, 110, 256–259. [Google Scholar] [CrossRef]
- Karageorgiou, V.; Kaplan, D. Porosity of 3D Biomaterial Scaffolds and Osteogenesis. Biomaterials 2005, 26, 5474–5491. [Google Scholar] [CrossRef]
- Murphy, C.M.; Haugh, M.G.; O’Brien, F.J. The Effect of Mean Pore Size on Cell Attachment, Proliferation and Migration in Collagen–Glycosaminoglycan Scaffolds for Bone Tissue Engineering. Biomaterials 2010, 31, 461–466. [Google Scholar] [CrossRef]
- Sharma, P.; Jain, K.G.; Pandey, P.M.; Mohanty, S. In Vitro Degradation Behaviour, Cytocompatibility and Hemocompatibility of Topologically Ordered Porous Iron Scaffold Prepared Using 3D Printing and Pressureless Microwave Sintering. Mater. Sci. Eng. C 2020, 106, 110247. [Google Scholar] [CrossRef]
- Li, Z.; Deng, L.; Kinloch, I.A.; Young, R.J. Raman Spectroscopy of Carbon Materials and Their Composites: Graphene, Nanotubes and Fibres. Prog. Mater. Sci. 2023, 135, 101089. [Google Scholar] [CrossRef]
- Puech, P.; Kandara, M.; Paredes, G.; Moulin, L.; Weiss-Hortala, E.; Kundu, A.; Ratel-Ramond, N.; Plewa, J.-M.; Pellenq, R.; Monthioux, M. Analyzing the Raman Spectra of Graphenic Carbon Materials from Kerogens to Nanotubes: What Type of Information Can Be Extracted from Defect Bands? C—J. Carbon Res. 2019, 5, 69. [Google Scholar] [CrossRef]
- Sharma, P.; Pandey, P.M. Rapid Manufacturing of Biodegradable Pure Iron Scaffold Using Amalgamation of Three-Dimensional Printing and Pressureless Microwave Sintering. Proc. Inst. Mech. Eng. J. Mech. Eng. Sci. 2019, 233, 1876–1895. [Google Scholar] [CrossRef]
- Haverová, L.; Oriňaková, R.; Oriňak, A.; Gorejová, R.; Baláž, M.; Vanýsek, P.; Kupková, M.; Hrubovčáková, M.; Mudroň, P.; Radoňák, J.; et al. An In Vitro Corrosion Study of Open Cell Iron Structures with PEG Coating for Bone Replacement Applications. Metals 2018, 8, 499. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, Y.F. Effects of Alloying Elements (Mn, Co, Al, W, Sn, B, C and S) on Biodegradability and in Vitro Biocompatibility of Pure Iron. Acta Biomater. 2011, 7, 1407–1420. [Google Scholar] [CrossRef]
- Huang, T.; Zheng, Y.; Han, Y. Accelerating Degradation Rate of Pure Iron by Zinc Ion Implantation. Regen. Biomater. 2016, 3, 205–215. [Google Scholar] [CrossRef]
- Huang, S.; Ulloa, A.; Nauman, E.; Stanciu, L. Collagen Coating Effects on Fe–Mn Bioresorbable Alloys. J. Orthop. Res. 2020, 38, 523–535. [Google Scholar] [CrossRef]
- Moravej, M.; Mantovani, D. Biodegradable Metals for Cardiovascular Stent Application: Interests and New Opportunities. Int. J. Mol. Sci. 2011, 12, 4250–4270. [Google Scholar] [CrossRef]
- Scarcello, E.; Lison, D. Are Fe-Based Stenting Materials Biocompatible? A Critical Review of In Vitro and In Vivo Studies. J. Funct. Biomater. 2019, 11, 2. [Google Scholar] [CrossRef]
- Zhao, Y.; Zang, G.; Yin, T.; Ma, X.; Zhou, L.; Wu, L.; Daniel, R.; Wang, Y.; Qiu, J.; Wang, G. A Novel Mechanism of Inhibiting In-Stent Restenosis with Arsenic Trioxide Drug-Eluting Stent: Enhancing Contractile Phenotype of Vascular Smooth Muscle Cells via YAP Pathway. Bioact. Mater. 2021, 6, 375–385. [Google Scholar] [CrossRef]
- Ceylan, H.; Tekinay, A.B.; Guler, M.O. Selective Adhesion and Growth of Vascular Endothelial Cells on Bioactive Peptide Nanofiber Functionalized Stainless Steel Surface. Biomaterials 2011, 32, 8797–8805. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial Cell Control of Thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef]
- Thrivikraman, G.; Madras, G.; Basu, B. In Vitro/In Vivo Assessment and Mechanisms of Toxicity of Bioceramic Materials and Its Wear Particulates. RSC Adv. 2014, 4, 12763. [Google Scholar] [CrossRef]
- Furuhashi, A.; Ayukawa, Y.; Atsuta, I.; Okawachi, H.; Koyano, K. The Difference of Fibroblast Behavior on Titanium Substrata with Different Surface Characteristics. Odontology 2012, 100, 199–205. [Google Scholar] [CrossRef]
- Bae, W.-J.; Chang, S.-W.; Lee, S.-I.; Kum, K.-Y.; Bae, K.-S.; Kim, E.-C. Human Periodontal Ligament Cell Response to a Newly Developed Calcium Phosphate–Based Root Canal Sealer. J. Endod. 2010, 36, 1658–1663. [Google Scholar] [CrossRef]
- Gomes, M.E.; Reis, R.L.; Cunha, A.M.; Blitterswijk, C.A.; de Bruijn, J.D. Cytocompatibility and Response of Osteoblastic-like Cells to Starch-Based Polymers: Effect of Several Additives and Processing Conditions. Biomaterials 2001, 22, 1911–1917. [Google Scholar] [CrossRef]
- He, H.; Qiao, Y.; Zhou, Q.; Wang, Z.; Chen, X.; Liu, D.; Yin, D.; He, M. Iron Overload Damages the Endothelial Mitochondria via the ROS/ADMA/DDAHII/ENOS/NO Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 2340392. [Google Scholar] [CrossRef]
- Zhu, S.; Huang, N.; Xu, L.; Zhang, Y.; Liu, H.; Sun, H.; Leng, Y. Biocompatibility of Pure Iron: In Vitro Assessment of Degradation Kinetics and Cytotoxicity on Endothelial Cells. Mater. Sci. Eng. C 2009, 29, 1589–1592. [Google Scholar] [CrossRef]
- Moravej, M.; Purnama, A.; Fiset, M.; Couet, J.; Mantovani, D. Electroformed Pure Iron as a New Biomaterial for Degradable Stents: In Vitro Degradation and Preliminary Cell Viability Studies. Acta Biomater. 2010, 6, 1843–1851. [Google Scholar] [CrossRef]
- Igarashi, K.; Shoji, Y.; Sekine-Suzuki, E.; Ueno, M.; Matsumoto, K.; Nakanishi, I.; Fukui, K. Importance of Locations of Iron Ions to Elicit Cytotoxicity Induced by a Fenton-Type Reaction. Cancers 2022, 14, 3642. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hwater [J/m2] | hn-heptane [J/m2] | hformamide [J/m2] | [J/m2] | [J/m2] | [J/m2] | [J/m2] | [J/m2] |
|---|---|---|---|---|---|---|---|
| −1.763 (0.21) | −0.352 (0.0201) | −1.951 (0.141) | 0.760 (0.263) | 13.669 (0.382) | 0.530 (0.089) | 3.453 (0.029) | 2.123 (0.042) |
| E corr [mV] | I corr [μA] | |
|---|---|---|
| Fe01 | 705,739 | 16,951 |
| Fe02 | 542,397 | 80,490 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gąsior, G.; Grodzicka, M.; Jędrzejewski, T.; Wiśniewski, M.; Radtke, A. Comparative Study of Porous Iron Foams for Biodegradable Implants: Structural Analysis and In Vitro Assessment. J. Funct. Biomater. 2023, 14, 293. https://doi.org/10.3390/jfb14060293
Gąsior G, Grodzicka M, Jędrzejewski T, Wiśniewski M, Radtke A. Comparative Study of Porous Iron Foams for Biodegradable Implants: Structural Analysis and In Vitro Assessment. Journal of Functional Biomaterials. 2023; 14(6):293. https://doi.org/10.3390/jfb14060293
Chicago/Turabian StyleGąsior, Gabriela, Marlena Grodzicka, Tomasz Jędrzejewski, Marek Wiśniewski, and Aleksandra Radtke. 2023. "Comparative Study of Porous Iron Foams for Biodegradable Implants: Structural Analysis and In Vitro Assessment" Journal of Functional Biomaterials 14, no. 6: 293. https://doi.org/10.3390/jfb14060293