
E-MuLA: An Ensemble Multi-Localized Attention Feature Extraction Network for Viral Protein Subcellular Localization

 , , , and
, , , and
Abstract
:1. Introduction
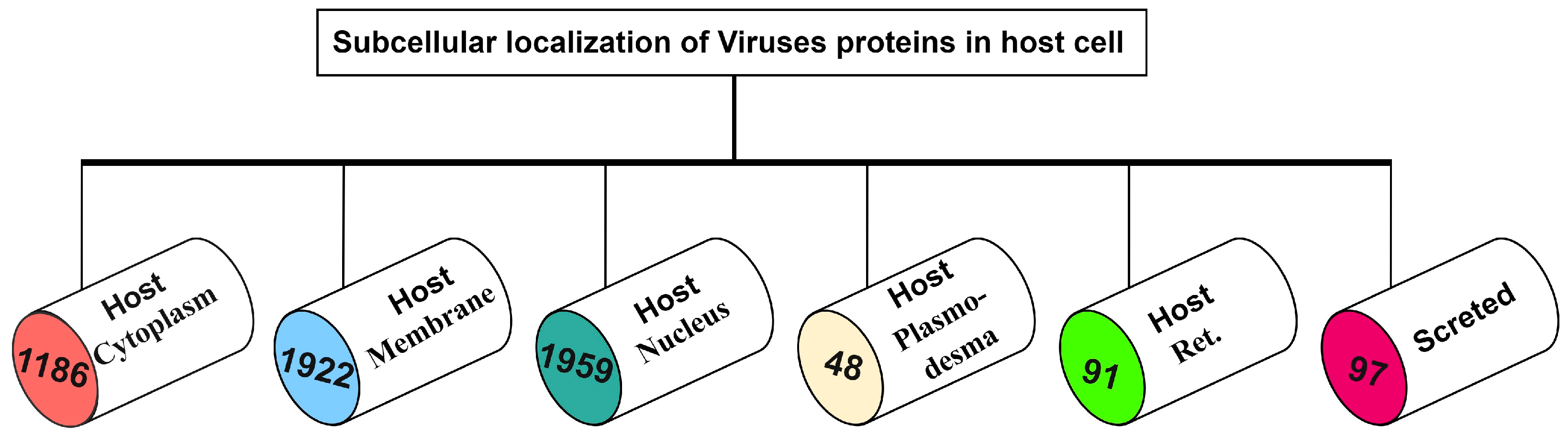
2. Materials and Methods
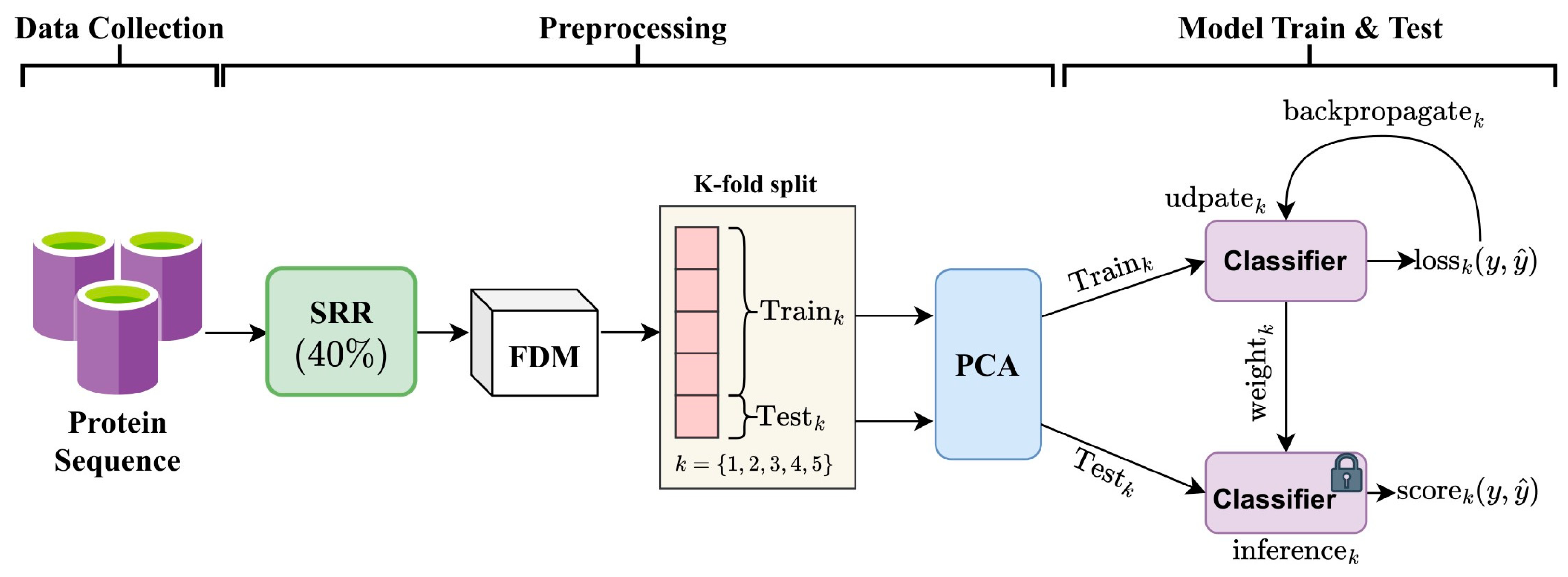
2.1. Framework
2.2. Data Transformation and Feature Extraction
2.2.1. Amino Acid Composition (AAC)
2.2.2. Pseudo-Amino Acid Composition (PseAAC)
2.2.3. Dipeptide Deviation from Expected Mean (DDE)
2.3. Principal Component Analysis (PCA)
2.4. Ensemble MuLA (E-MuLA) Network
2.4.1. Descriptor Encoder
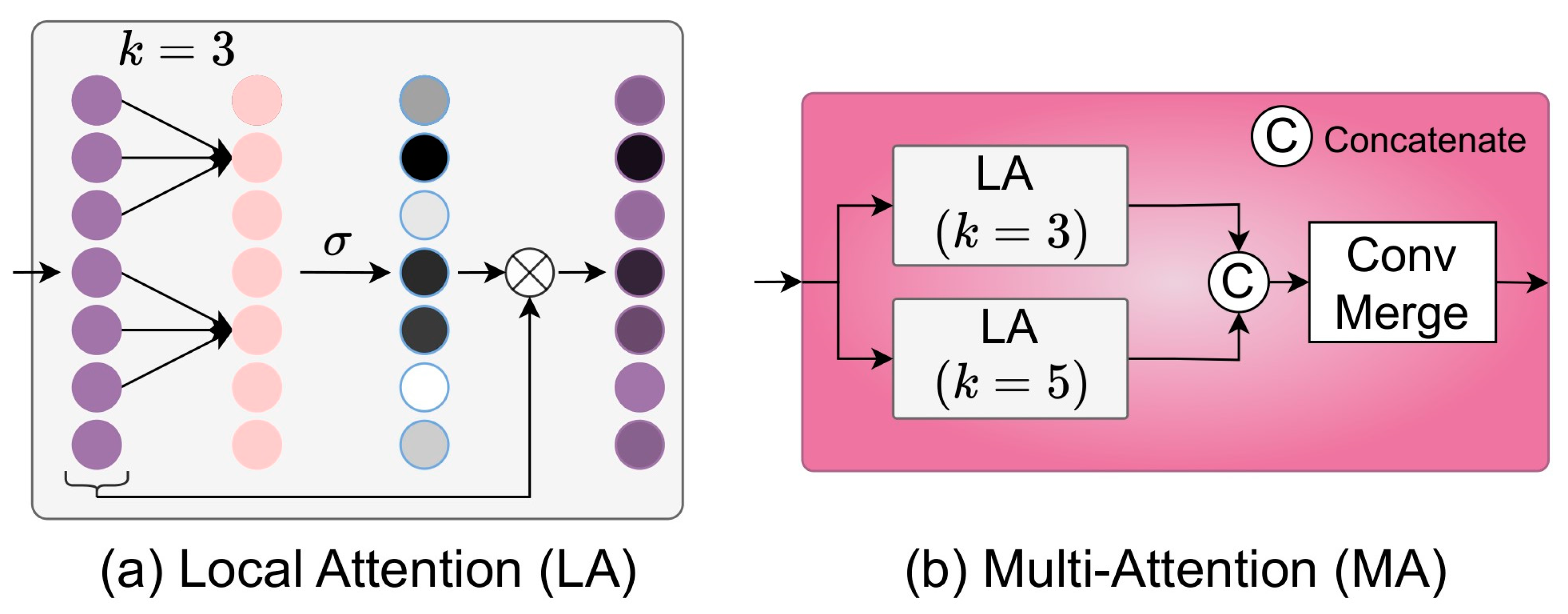
2.4.2. Multi-Attention (MA) Module
2.5. Other Algorithms
2.5.1. AdaBoost Classifier
2.5.2. Decision Trees (DTs)
2.5.3. K-Nearest Neighbors (KNNs)
2.5.4. SGD Classifier
2.5.5. Gaussian Process Classifier (GPC)
2.5.6. Linear Support Vector Machine (LSVM)
2.5.7. Long Short-Term Memory (LSTM) Classifier
2.5.8. Convolutional Neural Network (CNN) Classifier
3. Experimentation
3.1. Implementation Details
3.2. Evaluation Metrics
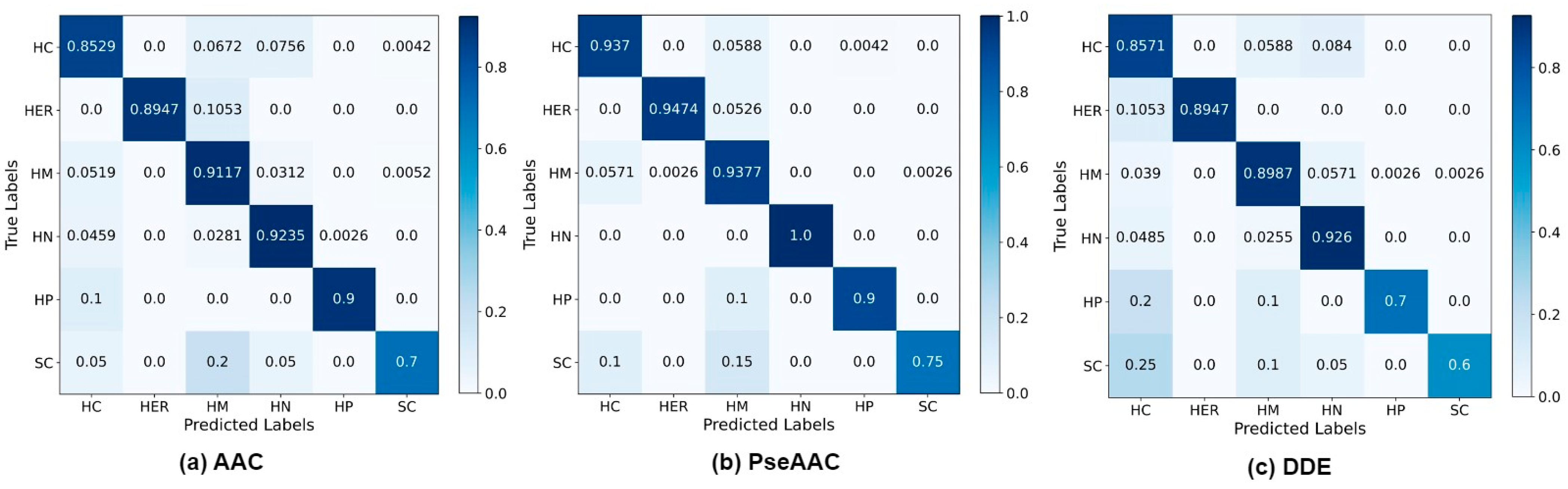
4. Results and Discussion
4.1. Investigation of PCA
4.2. Investigation of MuLA
4.3. Comparison with State-of-the-Art Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scott, M.S.; Oomen, R.; Thomas, D.Y.; Hallett, M.T. Predicting the subcellular localization of viral proteins within a mammalian host cell. Virol. J. 2006, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zou, Q.; Yuan, L. A review from biological mapping to computation-based subcellular localization. Mol. Ther. Nucleic Acid 2023, 32, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Rao, B.; Liu, L.; Cui, L.; Xiao, G.; Su, R.; Wei, L. PepFormer: End-to-End transformer-based siamese network to predict and enhance peptide detectability based on sequence only. Anal. Chem. 2021, 93, 6481–6490. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.B.; Chou, K.C. Virus-PLoc: A fusion classifier for predicting the subcellular localization of viral proteins within host and virus-infected cells. Biopolym. Orig. Res. Biomol. 2007, 85, 233–240. [Google Scholar] [CrossRef]
- Cao, C.; Shao, M.; Zuo, C.; Kwok, D.; Liu, L.; Ge, Y.; Zhang, Z.; Cui, F.; Chen, M.; Fan, R.; et al. RAVAR: A curated repository for rare variant-trait associations. Nucleic Acids Res. 2024, 52, D990–D997. [Google Scholar] [CrossRef]
- Ning, L.; Liu, M.; Gou, Y.; Yang, Y.; He, B.; Huang, J. Development and application of ribonucleic acid therapy strategies against COVID-19. Int. J. Biol. Sci. 2022, 18, 5070–5085. [Google Scholar]
- Ren, L.; Ning, L.; Yang, Y.; Yang, T.; Li, X.; Tan, S.; Ge, P.; Li, S.; Luo, N.; Tao, P.; et al. MetaboliteCOVID: A manually curated database of metabolite markers for COVID-19. Comput. Biol. Med. 2023, 167, 107661. [Google Scholar] [CrossRef]
- Shen, H.-B.; Chou, K.-C. Virus-mPLoc: A fusion classifier for viral protein subcellular location prediction by incorporating multiple sites. J. Biomol. Struct. Dyn. 2010, 28, 175–186. [Google Scholar] [CrossRef]
- Ren, L.; Xu, Y.; Ning, L.; Pan, X.; Li, Y.; Zhao, Q.; Pang, B.; Huang, J.; Deng, K.; Zhang, Y. TCM2COVID: A resource of anti-COVID-19 traditional Chinese medicine with effects and mechanisms. iMETA 2022, 1, e42. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wu, Z.-C.; Chou, K.-C. iLoc-Virus: A multi-label learning classifier for identifying the subcellular localization of virus proteins with both single and multiple sites. J. Theor. Biol. 2011, 284, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, L.C.; He, S.D.; Guo, F.; Zou, Q. SubLocEP: A novel ensemble predictor of subcellular localization of eukaryotic mRNA based on machine learning. Brief. Bioinform. 2021, 22, bbaa401. [Google Scholar] [CrossRef]
- Thakur, A.; Rajput, A.; Kumar, M. MSLVP: Prediction of multiple subcellular localization of viral proteins using a support vector machine. Mol. BioSyst. 2016, 12, 2572–2586. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Cheng, X.; Chen, G.; Mao, Q.; Chou, K.-C. pLoc_bal-mVirus: Predict subcellular localization of multi-label virus proteins by Chou’s general PseAAC and IHTS treatment to balance training dataset. Med. Chem. 2019, 15, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Chou, K.-C. pLoc_Deep-mVirus: A CNN model for predicting subcellular localization of virus proteins by deep learning. Nat. Sci. 2020, 12, 388–399. [Google Scholar] [CrossRef]
- Ding, Y.J.; Tiwari, P.; Guo, F.; Zou, Q. Shared subspace-based radial basis function neural network for identifying ncRNAs subcellular localization. Neural Netw. 2022, 156, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jiang, Y.; Jin, J.; Yin, C.; Yu, H.; Wang, F.; Feng, J.; Su, R.; Nakai, K.; Zou, Q. DeepBIO: An automated and interpretable deep-learning platform for high-throughput biological sequence prediction, functional annotation and visualization analysis. Nucleic Acids Res. 2023, 51, 3017–3029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pan, X.; Shi, T.; Gu, Z.; Yang, Z.; Liu, M.; Xu, Y.; Yang, Y.; Ren, L.; Song, X.; et al. P450Rdb: A manually curated database of reactions catalyzed by cytochrome P450 enzymes. J. Adv. Res. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Feng, J.; Liu, C.; Wu, H.; Qiu, Z.; Ge, J.; Sun, S.; Hong, X.; Li, Y.; Wang, X.; et al. Machine learning aided construction of the quorum sensing communication network for human gut microbiota. Nat. Commun. 2022, 13, 3079. [Google Scholar] [CrossRef]
- Tang, Y.; Pang, Y.; Liu, B. IDP-Seq2Seq: Identification of intrinsically disordered regions based on sequence to sequence learning. Bioinformatics 2021, 36, 5177–5186. [Google Scholar] [CrossRef]
- Pham, N.T.; Phan, L.T.; Seo, J.; Kim, Y.; Song, M.; Lee, S.; Jeon, Y.J.; Manavalan, B. Advancing the accuracy of SARS-CoV-2 phosphorylation site detection via meta-learning approach. Brief. Bioinform. 2023, 25, bbad433. [Google Scholar] [CrossRef]
- Pham, N.T.; Rakkiyapan, R.; Park, J.; Malik, A.; Manavalan, B. H2Opred: A robust and efficient hybrid deep learning model for predicting 2’-O-methylation sites in human RNA. Brief. Bioinform. 2023, 25, bbad476. [Google Scholar] [CrossRef]
- Zhu, W.; Yuan, S.S.; Li, J.; Huang, C.B.; Lin, H.; Liao, B. A First Computational Frame for Recognizing Heparin-Binding Protein. Diagnostics 2023, 13, 2465. [Google Scholar] [CrossRef]
- Zou, X.; Ren, L.; Cai, P.; Zhang, Y.; Ding, H.; Deng, K.; Yu, X.; Lin, H.; Huang, C. Accurately identifying hemagglutinin using sequence information and machine learning methods. Front. Med. 2023, 10, 1281880. [Google Scholar] [CrossRef]
- Li, H.; Pang, Y.; Liu, B. BioSeq-BLM: A platform for analyzing DNA, RNA, and protein sequences based on biological language models. Nucleic Acids Res. 2021, 49, e129. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Chen, S.; Wang, J.; Chen, H. A robust multi-class AdaBoost algorithm for mislabeled noisy data. Knowl.-Based Syst. 2016, 102, 87–102. [Google Scholar] [CrossRef]
- Hastie, T.; Rosset, S.; Zhu, J.; Zou, H. Multi-class adaboost. Stat. Its Interface 2009, 2, 349–360. [Google Scholar] [CrossRef]
- MacKay, D.J. Introduction to Gaussian processes. NATO ASI Ser. F Comput. Syst. Sci. 1998, 168, 133–166. [Google Scholar]
- Wang, Y.; Zhai, Y.; Ding, Y.; Zou, Q. SBSM-Pro: Support Bio-sequence Machine for Proteins. arXiv 2023, arXiv:2308.10275. [Google Scholar]
- Zhang, H.Y.; Zou, Q.; Ju, Y.; Song, C.G.; Chen, D. Distance-based Support Vector Machine to Predict DNA N6-methyladenine Modification. Curr. Bioinform. 2022, 17, 473–482. [Google Scholar] [CrossRef]
- Liu, B.; Gao, X.; Zhang, H. BioSeq-Analysis2.0: An updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res. 2019, 47, e127. [Google Scholar] [CrossRef]
- Ao, C.; Ye, X.; Sakurai, T.; Zou, Q.; Yu, L. m5U-SVM: Identification of RNA 5-methyluridine modification sites based on multi-view features of physicochemical features and distributed representation. BMC Biol. 2023, 21, 93. [Google Scholar] [CrossRef] [PubMed]
- Muhuri, P.S.; Chatterjee, P.; Yuan, X.; Roy, K.; Esterline, A. Using a long short-term memory recurrent neural network (LSTM-RNN) to classify network attacks. Information 2020, 11, 243. [Google Scholar] [CrossRef]
- Chen, J.; Zou, Q.; Li, J. DeepM6ASeq-EL: Prediction of Human N6-Methyladenosine (m6A) Sites with LSTM and Ensemble Learning. Front. Comput. Sci. 2022, 16, 162302. [Google Scholar] [CrossRef]
- Tang, Y. Deep learning using linear support vector machines. arXiv 2013, arXiv:1306.0239. [Google Scholar]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene Subsequence Embedding for Prediction of Mammalian N6-Methyladenosine Sites from mRNA. RNA 2019, 25, 205–218. [Google Scholar] [CrossRef]
- Zulfiqar, H.; Guo, Z.; Ahmad, R.M.; Ahmed, Z.; Cai, P.; Chen, X.; Zhang, Y.; Lin, H.; Shi, Z. Deep-STP: A deep learning-based approach to predict snake toxin proteins by using word embeddings. Front. Med. 2024, 10, 1291352. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Hao, H.; Yu, L. Identifying disease-related microbes based on multi-scale variational graph autoencoder embedding Wasserstein distance. BMC Biol. 2023, 21, 294. [Google Scholar] [CrossRef]
- Hasan, M.M.; Tsukiyama, S.; Cho, J.Y.; Kurata, H.; Alam, M.A.; Liu, X.; Manavalan, B.; Deng, H.W. Deepm5C: A deep-learning-based hybrid framework for identifying human RNA N5-methylcytosine sites using a stacking strategy. Mol. Ther. 2022, 30, 2856–2867. [Google Scholar] [CrossRef]
- Bupi, N.; Sangaraju, V.K.; Phan, L.T.; Lal, A.; Vo, T.T.B.; Ho, P.T.; Qureshi, M.A.; Tabassum, M.; Lee, S.; Manavalan, B. An Effective Integrated Machine Learning Framework for Identifying Severity of Tomato Yellow Leaf Curl Virus and Their Experimental Validation. Research 2023, 6, 0016. [Google Scholar] [CrossRef]
- Manavalan, B.; Patra, M.C. MLCPP 2.0: An Updated Cell-penetrating Peptides and Their Uptake Efficiency Predictor. J. Mol. Biol. 2022, 434, 167604. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Basith, S.; Pitti, T.; Lee, G.; Manavalan, B. THRONE: A New Approach for Accurate Prediction of Human RNA N7-Methylguanosine Sites. J. Mol. Biol. 2022, 434, 167549. [Google Scholar] [CrossRef] [PubMed]
- Thi Phan, L.; Woo Park, H.; Pitti, T.; Madhavan, T.; Jeon, Y.J.; Manavalan, B. MLACP 2.0: An updated machine learning tool for anticancer peptide prediction. Comput. Struct. Biotechnol. J. 2022, 20, 4473–4480. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rat (ρ) | AAC | PseAAC | DDE | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acc | Prec | Rec | F1 | MCC | Acc | Prec | Rec | F1 | MCC | Acc | Prec | Rec | F1 | MCC | |
| 0.2 | 74.60 | 71.83 | 65.33 | 66.87 | 62.62 | 90.09 | 80.72 | 72.15 | 74.1 | 85.45 | 86.33 | 87.93 | 78.19 | 81.94 | 79.89 |
| 0.4 | 84.55 | 80.91 | 75.83 | 77.39 | 77.32 | 92.55 | 85.5 | 78.64 | 80.74 | 89.07 | 82.77 | 82.88 | 74.39 | 77.54 | 74.64 |
| 0.6 | 86.98 | 84.46 | 78.54 | 80.67 | 80.85 | 93.59 | 88.1 | 80.79 | 83.21 | 90.6 | 80.64 | 81.71 | 71.82 | 75.37 | 71.47 |
| 0.8 | 87.71 | 86.55 | 79.53 | 82.16 | 81.92 | 94.00 | 88.66 | 82.2 | 84.17 | 91.21 | 79.15 | 81.35 | 69.55 | 73.71 | 69.22 |
| 1.0 | 87.62 | 85.72 | 79.40 | 81.74 | 81.80 | 93.94 | 88.12 | 81.38 | 83.62 | 91.12 | 74.56 | 76.32 | 65.57 | 69.44 | 62.43 |
| MA | AAC | PseAAC | DDE | |||
|---|---|---|---|---|---|---|
| Acc | MCC | Acc | MCC | Acc | MCC | |
| w/o | 84.13 | 79.65 | 93.01 | 90.22 | 80.74 | 73.89 |
| w (k1 = 3) | 85.43 | 80.11 | 93.74 | 90.83 | 82.63 | 74.44 |
| w (k2 = 5) | 86.72 | 80.46 | 93.84 | 90.98 | 84.47 | 77.15 |
| w (k1 = 3, k2 = 5) | 87.71 | 81.92 | 94.00 | 91.21 | 86.33 | 79.89 |
| Model | Acc | Prec | Rec | F1 | Spec | Sen | MCC |
|---|---|---|---|---|---|---|---|
| AAC (Only) | |||||||
| AdaBoost | 38.04 | 28.88 | 32.2 | 25.61 | 86.96 | 32.2 | 20.61 |
| DT | 78.49 | 68.61 | 69.75 | 68.84 | 94.82 | 69.75 | 68.48 |
| KNN | 84.16 | 81.17 | 71.35 | 73.95 | 96.21 | 71.35 | 76.83 |
| GPC | 86.7 | 84.43 | 77.25 | 80.01 | 96.49 | 77.25 | 80.85 |
| LSVM | 69.73 | 48.93 | 44.04 | 45.32 | 92.33 | 44.04 | 54.69 |
| SGD | 64.81 | 44.03 | 34.65 | 35.44 | 91.11 | 34.65 | 47.21 |
| LSTM | 63.75 | 41.93 | 40.74 | 40.57 | 90.87 | 40.74 | 45.73 |
| CNN | 62.83 | 38.66 | 36.06 | 35.84 | 90.49 | 36.06 | 44.14 |
| Virus-mPLoc [8] | 65.63 | 42.13 | 39.29 | 39.57 | 91.33 | 39.29 | 48.41 |
| MuLA | 87.71 | 86.55 | 79.53 | 82.16 | 97.0 | 79.53 | 81.92 |
| PseACC (only) | |||||||
| AdaBoost | 51.15 | 21.24 | 23.39 | 18.95 | 88.26 | 23.39 | 31.67 |
| DT | 89.31 | 73.51 | 75.65 | 73.51 | 97.6 | 75.65 | 84.36 |
| KNN | 92.74 | 84.14 | 80.1 | 80.49 | 97.36 | 80.1 | 89.4 |
| GPC | 92.85 | 87.06 | 81.19 | 82.59 | 97.8 | 81.19 | 89.88 |
| LSVM | 86.37 | 58.48 | 55.54 | 56.57 | 96.74 | 55.54 | 79.8 |
| SGD | 84.41 | 55.93 | 48.38 | 49.51 | 96.28 | 48.38 | 76.92 |
| LSTM | 80.92 | 50.31 | 48.77 | 48.55 | 95.38 | 48.77 | 72.03 |
| CNN | 82.72 | 51.65 | 48.19 | 48.34 | 95.71 | 48.19 | 74.94 |
| Virus-mPLoc [8] | 81.21 | 47.13 | 45.25 | 45.1 | 95.38 | 45.25 | 72.37 |
| MuLA | 94.0 | 88.66 | 82.2 | 84.17 | 98.61 | 82.2 | 91.21 |
| DDE (only) | |||||||
| AdaBoost | 48.06 | 40.88 | 32.84 | 31.18 | 87.06 | 32.84 | 25.24 |
| DT | 77.67 | 69.15 | 68.09 | 68.11 | 94.61 | 68.09 | 67.25 |
| KNN | 84.86 | 84.09 | 76.62 | 79.76 | 95.36 | 76.62 | 77.78 |
| GPC | 85.85 | 86.48 | 77.36 | 80.09 | 95.99 | 77.36 | 78.98 |
| LSVM | 79.46 | 77.68 | 62.54 | 66.24 | 94.92 | 62.54 | 69.53 |
| SGD | 73.56 | 69.49 | 60.27 | 63.71 | 93.52 | 60.27 | 60.92 |
| LSTM | 60.52 | 42.1 | 38.95 | 39.22 | 90.0 | 38.95 | 40.68 |
| CNN | 58.06 | 37.68 | 33.76 | 33.76 | 89.31 | 33.76 | 37.15 |
| Virus-mPLoc [8] | 57.27 | 36.56 | 32.41 | 32.53 | 89.15 | 32.41 | 35.57 |
| MuLA | 86.33 | 87.93 | 78.18 | 81.94 | 96.65 | 78.18 | 79.89 |
| Ensemble (AAC, PseAAC, and DDE) with the ternary operators U (·) = {M, C, S} to merge the descriptors features. | |||||||
| E-MuLA (M = maximum) | 94.72 | 92.31 | 84.15 | 87.39 | 98.78 | 84.15 | 92.26 |
| E-MuLA (C = concatenate) | 94.55 | 91.56 | 83.83 | 86.84 | 98.74 | 83.83 | 92.01 |
| E-MuLA (S = summation) | 94.87 | 92.72 | 84.18 | 87.61 | 98.81 | 84.18 | 92.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakanina Kissanga, G.-M.; Zulfiqar, H.; Gao, S.; Yussif, S.B.; Momanyi, B.M.; Ning, L.; Lin, H.; Huang, C.-B. E-MuLA: An Ensemble Multi-Localized Attention Feature Extraction Network for Viral Protein Subcellular Localization. Information 2024, 15, 163. https://doi.org/10.3390/info15030163
Bakanina Kissanga G-M, Zulfiqar H, Gao S, Yussif SB, Momanyi BM, Ning L, Lin H, Huang C-B. E-MuLA: An Ensemble Multi-Localized Attention Feature Extraction Network for Viral Protein Subcellular Localization. Information. 2024; 15(3):163. https://doi.org/10.3390/info15030163
Chicago/Turabian StyleBakanina Kissanga, Grace-Mercure, Hasan Zulfiqar, Shenghan Gao, Sophyani Banaamwini Yussif, Biffon Manyura Momanyi, Lin Ning, Hao Lin, and Cheng-Bing Huang. 2024. "E-MuLA: An Ensemble Multi-Localized Attention Feature Extraction Network for Viral Protein Subcellular Localization" Information 15, no. 3: 163. https://doi.org/10.3390/info15030163