Inner Ear Gene Therapies Take Off: Current Promises and Future Challenges


Abstract
:1. Introduction
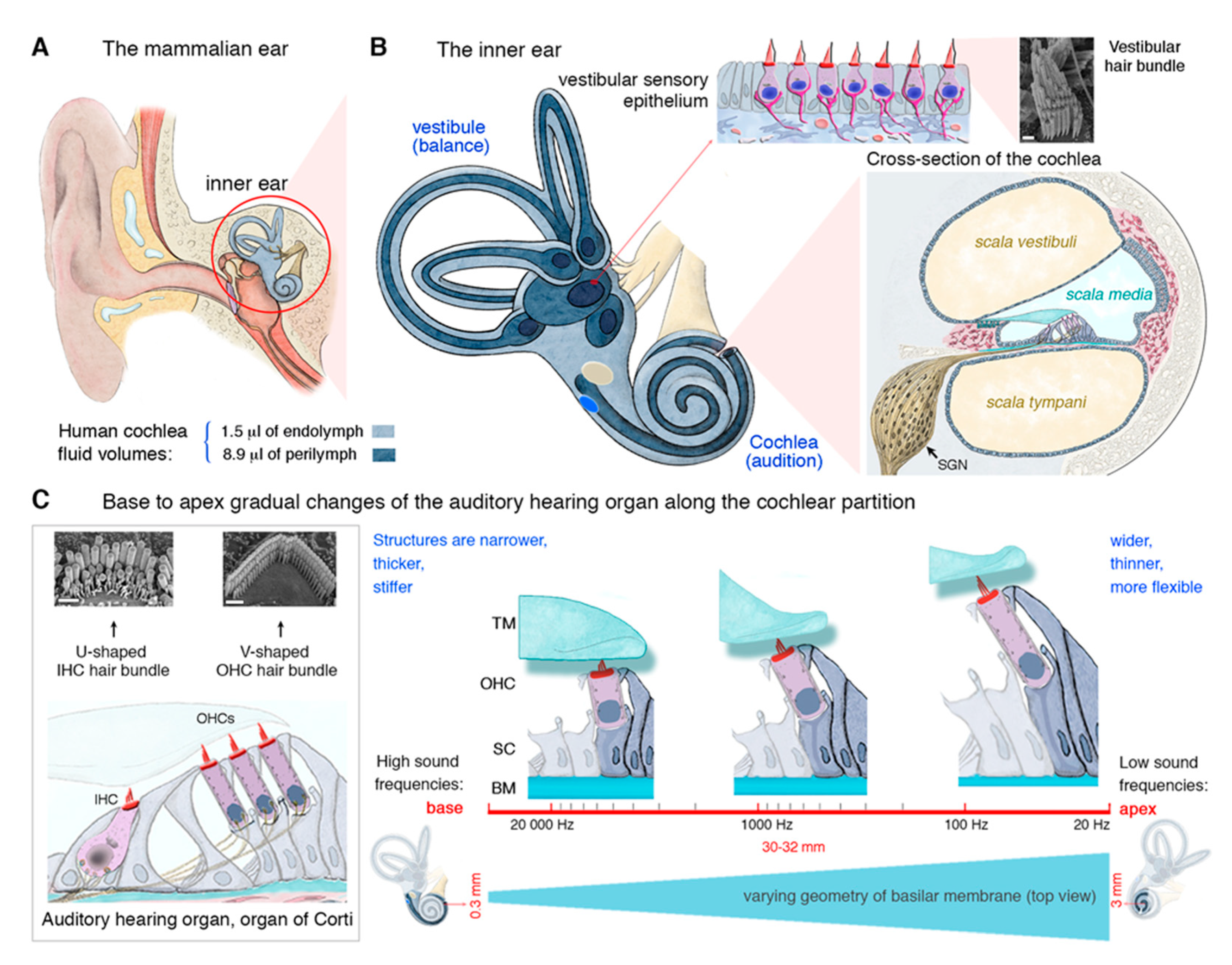
2. The Inner Ear and Its Auditory Hair Cells Specializing in Mechanoreception
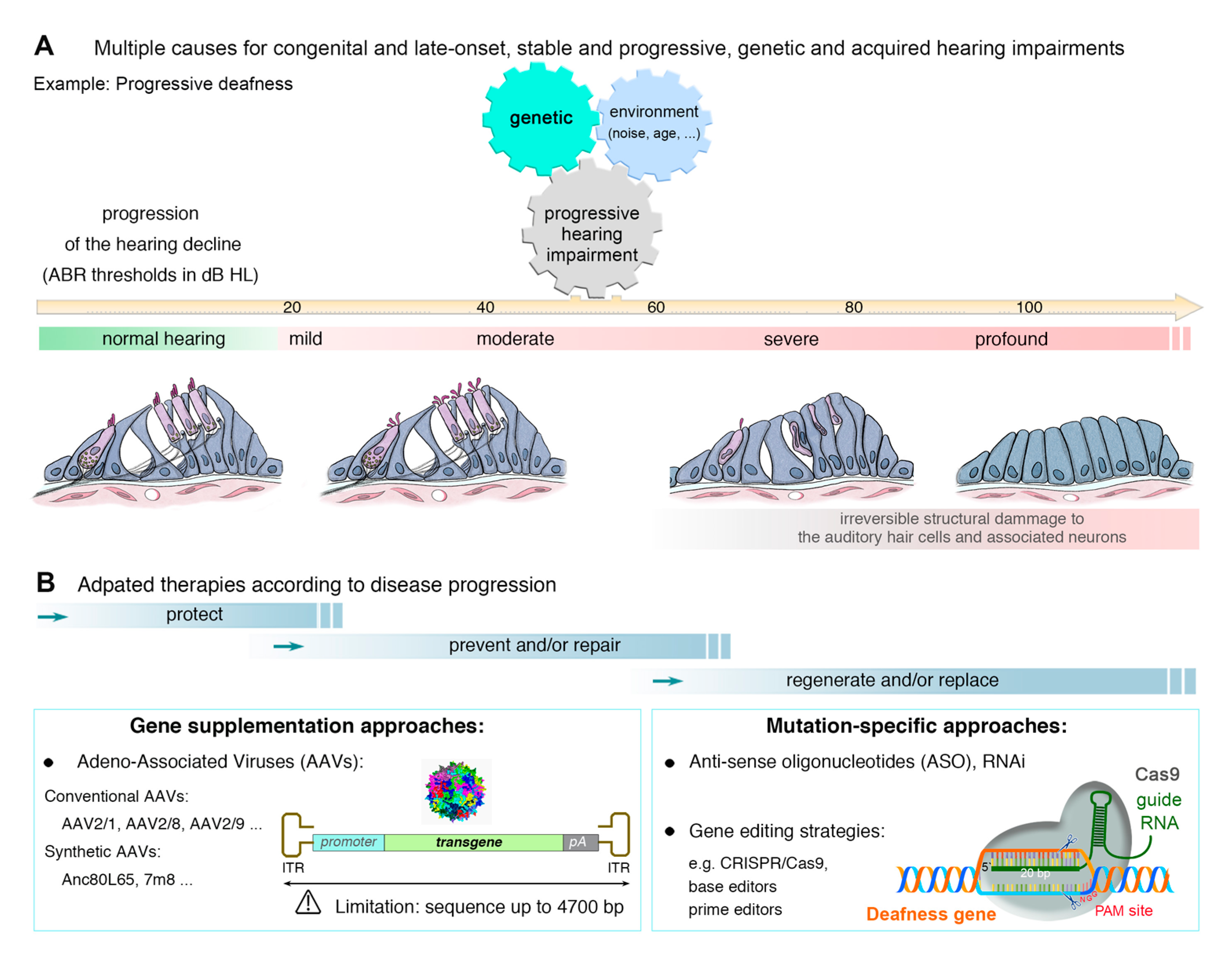
3. Genetic Hearing Impairment
4. Approaches to the Treatment of Hearing Loss
4.1. Routes for Delivery
4.2. Gene Therapy Delivery Systems
4.2.1. Viral Vectors
4.2.2. Non-Viral Delivery
4.3. Gene- and Mutation-Specific Therapies
4.3.1. Gene Replacement
4.3.2. Gene Suppression—RNA-Based Therapies
4.3.3. CRISPR/Cas9-Based Genome Editing
4.4. “Gene-Independent” Approaches—A Common Strategy for Several Forms of Deafness
4.4.1. Auditory Hair Cell Regeneration
4.4.2. Protective Local Treatments
4.5. From Animals to “One Day” in Humans: the Promises and Challenges of Preclinical Inner Ear Gene Therapy Trials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Petit, C.; El-Amraoui, A.; Avan, P. Audition: Hearing and Deafness. In Neuroscience in the 21st Century; Pfaff, D., Ed.; Springer-Science: New York, NY, USA, 2013. [Google Scholar]
- Geleoc, G.S.; Holt, J.R. Sound strategies for hearing restoration. Science 2014, 344, 1241062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberman, M.C. Noise-induced and age-related hearing loss: New perspectives and potential therapies. F1000 Res. 2017, 6, 927. [Google Scholar] [CrossRef]
- Petit, C.; Levilliers, J.; Hardelin, J.P. Molecular genetics of hearing loss. Annu. Rev. Genet. 2001, 35, 589–646. [Google Scholar] [CrossRef] [PubMed]
- Cryns, K.; Van Camp, G. Deafness genes and their diagnostic applications. Audiol. Neurootol. 2004, 9, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Raviv, D.; Dror, A.A.; Avraham, K.B. Hearing loss: A common disorder caused by many rare alleles. Ann. N. Y. Acad. Sci. 2010, 1214, 168–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dror, A.A.; Avraham, K.B. Hearing impairment: A panoply of genes and functions. Neuron 2010, 68, 293–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, G.P.; de Monvel, J.B.; Petit, C. How the genetics of deafness illuminates auditory physiology. Annu. Rev. Physiol. 2011, 73, 311–334. [Google Scholar] [CrossRef]
- Guilford, P.; Ben Arab, S.; Blanchard, S.; Levilliers, J.; Weissenbach, J.; Belkahia, A.; Petit, C. A non-syndrome form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q. Nat. Genet. 1994, 6, 24–28. [Google Scholar] [CrossRef]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef]
- Ingham, N.J.; Pearson, S.A.; Vancollie, V.E.; Rook, V.; Lewis, M.A.; Chen, J.; Buniello, A.; Martelletti, E.; Preite, L.; Lam, C.C.; et al. Mouse screen reveals multiple new genes underlying mouse and human hearing loss. PLoS Biol. 2019, 17, e3000194. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, K.; Michel, V.; Giraudet, F.; Riederer, B.; Foucher, I.; Papal, S.; Perfettini, I.; Le Gal, S.; Verpy, E.; Xia, W.; et al. An unusually powerful mode of low-frequency sound interference due to defective hair bundles of the auditory outer hair cells. Proc. Natl. Acad. Sci. USA 2014, 111, 9307–9312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, P.K.; Bowl, M.R.; Jeyarajan, P.; Wisby, L.; Blease, A.; Goldsworthy, M.E.; Simon, M.M.; Greenaway, S.; Michel, V.; Barnard, A.; et al. Novel gene function revealed by mouse mutagenesis screens for models of age-related disease. Nat. Commun. 2016, 7, 12444. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, L.A.; Patni, P.; Aguilar, C.; Mburu, P.; Corns, L.; Wells, H.R.; Delmaghani, S.; Parker, A.; Johnson, S.; Williams, D.; et al. Clarin-2 is essential for hearing by maintaining stereocilia integrity and function. EMBO Mol. Med. 2019, 11, e10288. [Google Scholar] [CrossRef] [PubMed]
- Lelli, A.; Michel, V.; Boutet de Monvel, J.; Cortese, M.; Bosch-Grau, M.; Aghaie, A.; Perfettini, I.; Dupont, T.; Avan, P.; El-Amraoui, A.; et al. Class III myosins shape the auditory hair bundles by limiting microvilli and stereocilia growth. J. Cell Biol. 2016, 212, 231–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, V.; Booth, K.T.; Patni, P.; Cortese, M.; Azaiez, H.; Bahloul, A.; Kahrizi, K.; Labbe, M.; Emptoz, A.; Lelli, A.; et al. CIB2, defective in isolated deafness, is key for auditory hair cell mechanotransduction and survival. EMBO Mol. Med. 2017, 9, 1711–1731. [Google Scholar] [CrossRef]
- Geleoc, G.G.S.; El-Amraoui, A. Disease mechanisms and gene therapy for Usher syndrome. Hear. Res. 2020, 107932. [Google Scholar] [CrossRef]
- Muller, U.; Barr-Gillespie, P.G. New treatment options for hearing loss. Nat. Rev. Drug Discov. 2015, 14, 346–365. [Google Scholar] [CrossRef]
- Ahmed, H.; Shubina-Oleinik, O.; Holt, J.R. Emerging Gene Therapies for Genetic Hearing Loss. J. Assoc. Res. Otolaryngol. 2017, 18, 649–670. [Google Scholar] [CrossRef]
- Akil, O.; Seal, R.P.; Burke, K.; Wang, C.; Alemi, A.; During, M.; Edwards, R.H.; Lustig, L.R. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron 2012, 75, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Delmaghani, S.; Defourny, J.; Aghaie, A.; Beurg, M.; Dulon, D.; Thelen, N.; Perfettini, I.; Zelles, T.; Aller, M.; Meyer, A.; et al. Hypervulnerability to Sound Exposure through Impaired Adaptive Proliferation of Peroxisomes. Cell 2015, 163, 894–906. [Google Scholar] [CrossRef] [Green Version]
- Askew, C.; Rochat, C.; Pan, B.; Asai, Y.; Ahmed, H.; Child, E.; Schneider, B.L.; Aebischer, P.; Holt, J.R. Tmc gene therapy restores auditory function in deaf mice. Sci. Transl. Med. 2015, 7, 295ra108. [Google Scholar] [CrossRef] [PubMed]
- Emptoz, A.; Michel, V.; Lelli, A.; Akil, O.; Boutet de Monvel, J.; Lahlou, G.; Meyer, A.; Dupont, T.; Nouaille, S.; Ey, E.; et al. Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G. Proc. Natl. Acad. Sci. USA 2017, 114, 9695–9700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landegger, L.D.; Pan, B.; Askew, C.; Wassmer, S.J.; Gluck, S.D.; Galvin, A.; Taylor, R.; Forge, A.; Stankovic, K.M.; Holt, J.R.; et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotehnol. 2017, 35, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, B.; Askew, C.; Galvin, A.; Heman-Ackah, S.; Asai, Y.; Indzhykulian, A.A.; Jodelka, F.M.; Hastings, M.L.; Lentz, J.J.; Vandenberghe, L.H.; et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 2017, 35, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Dulon, D.; Papal, S.; Patni, P.; Cortese, M.; Vincent, P.F.; Tertrais, M.; Emptoz, A.; Tlili, A.; Bouleau, Y.; Michel, V.; et al. Clarin-1 gene transfer rescues auditory synaptopathy in model of Usher syndrome. J. Clin. Investig. 2018, 128, 3382–3401. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Meijer, E.J.; Ivanchenko, M.V.; Tenneson, K.; Emond, F.; Hanlon, K.S.; Indzhykulian, A.A.; Volak, A.; Karavitaki, K.D.; Tamvakologos, P.I.; et al. Gene Transfer with AAV9-PHP.B Rescues Hearing in a Mouse Model of Usher Syndrome 3A and Transduces Hair Cells in a Non-human Primate. Mol. Ther. Methods Clin. Dev. 2019, 13, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, J.; Hashimoto, K.; Xiao, R.; Vandenberghe, L.H.; Liberman, M.C. Cochlear gene therapy with ancestral AAV in adult mice: Complete transduction of inner hair cells without cochlear dysfunction. Sci. Rep. 2017, 7, 45524. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Huang, M.; Shu, Y.; Ruprecht, A.; Wang, H.; Tang, Y.; Vandenberghe, L.H.; Wang, Q.; Gao, G.; Kong, W.J.; et al. Delivery of Adeno-Associated Virus Vectors in Adult Mammalian Inner-Ear Cell Subtypes Without Auditory Dysfunction. Hum. Gene Ther. 2018, 29, 492–506. [Google Scholar] [CrossRef]
- Guo, J.Y.; He, L.; Qu, T.F.; Liu, Y.Y.; Liu, K.; Wang, G.P.; Gong, S.S. Canalostomy As a Surgical Approach to Local Drug Delivery into the Inner Ears of Adult and Neonatal Mice. J. Vis. Exp. 2018, 135, e57351. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Smith, R.J.H. Enhanced viral—Mediated cochlear gene delivery in adult mice by combining canal fenestration with round window membrane inoculation. Sci. Rep. 2018, 8, 2980. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Nist-Lund, C.; Solanes, P.; Goldberg, H.; Wu, J.; Pan, B.; Schneider, B.L.; Holt, J.R. Efficient viral transduction in mouse inner ear hair cells with utricle injection and AAV9-PHP.B. Hear. Res. 2020, 1, 107882. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.B.; Di Pasquale, G.; Cortez, S.R.; Chiorini, J.A.; Raphael, Y. Gene transfer using bovine adeno-associated virus in the guinea pig cochlea. Gene Ther. 2009, 16, 990–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpatrick, L.A.; Li, Q.; Yang, J.; Goddard, J.C.; Fekete, D.M.; Lang, H. Adeno-associated virus-mediated gene delivery into the scala media of the normal and deafened adult mouse ear. Gene Ther. 2011, 18, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Wang, Y.; Chang, Q.; Wang, J.; Gong, S.; Li, H.; Lin, X. Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther. 2014, 21, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorgy, B.; Sage, C.; Indzhykulian, A.A.; Scheffer, D.I.; Brisson, A.R.; Tan, S.; Wu, X.; Volak, A.; Mu, D.; Tamvakologos, P.I.; et al. Rescue of Hearing by Gene Delivery to Inner-Ear Hair Cells Using Exosome-Associated AAV. Mol. Ther. 2017, 25, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Chai, R.; Guo, L.; Dong, B.; Li, W.; Shu, Y.; Huang, X.; Li, H. Transduction of Adeno-Associated Virus Vectors Targeting Hair Cells and Supporting Cells in the Neonatal Mouse Cochlea. Front. Cell Neurosci. 2019, 13, 8. [Google Scholar] [CrossRef]
- Chien, W.W.; McDougald, D.S.; Roy, S.; Fitzgerald, T.S.; Cunningham, L.L. Cochlear gene transfer mediated by adeno-associated virus: Comparison of two surgical approaches. Laryngoscope 2015, 125, 2557–2564. [Google Scholar] [CrossRef]
- Chang, Q.; Wang, J.; Li, Q.; Kim, Y.; Zhou, B.; Wang, Y.; Li, H.; Lin, X. Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol. Med. 2015, 7, 1077–1086. [Google Scholar] [CrossRef]
- Shu, Y.; Tao, Y.; Wang, Z.; Tang, Y.; Li, H.; Dai, P.; Gao, G.; Chen, Z.Y. Identification of Adeno-Associated Viral Vectors That Target Neonatal and Adult Mammalian Inner Ear Cell Subtypes. Hum. Gene Ther. 2016, 27, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Han, J.J.; Mhatre, A.N.; Wareing, M.; Pettis, R.; Gao, W.Q.; Zufferey, R.N.; Trono, D.; Lalwani, A.K. Transgene expression in the guinea pig cochlea mediated by a lentivirus-derived gene transfer vector. Hum. Gene Ther. 1999, 10, 1867–1873. [Google Scholar] [CrossRef]
- Bedrosian, J.C.; Gratton, M.A.; Brigande, J.V.; Tang, W.; Landau, J.; Bennett, J. In vivo delivery of recombinant viruses to the fetal murine cochlea: Transduction characteristics and long-term effects on auditory function. Mol. Ther. 2006, 14, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Pietola, L.; Aarnisalo, A.A.; Joensuu, J.; Pellinen, R.; Wahlfors, J.; Jero, J. HOX-GFP and WOX-GFP lentivirus vectors for inner ear gene transfer. Acta Otolaryngol. 2008, 128, 613–620. [Google Scholar] [CrossRef]
- Wei, Y.; Fu, Y.; Liu, S.; Xia, G.; Pan, S. Effect of lentiviruses carrying enhanced green fluorescent protein injected into the scala media through a cochleostomy in rats. Am. J. Otolaryngol. 2013, 34, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Y.; Chang, Q.; Ahmad, S.; Zhou, B.; Kim, Y.; Li, H.; Lin, X. Early postnatal virus inoculation into the scala media achieved extensive expression of exogenous green fluorescent protein in the inner ear and preserved auditory brainstem response thresholds. J. Gene Med. 2013, 15, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Dazert, S.; Battaglia, A.; Ryan, A.F. Transfection of neonatal rat cochlear cells in vitro with an adenovirus vector. Int. J. Dev. Neurosci. 1997, 15, 595–600. [Google Scholar] [CrossRef]
- Suzuki, M.; Yamasoba, T.; Suzukawa, K.; Kaga, K. Adenoviral vector gene delivery via the round window membrane in guinea pigs. Neuroreport 2003, 14, 1951–1955. [Google Scholar] [CrossRef]
- Yang, J.; Cong, N.; Han, Z.; Huang, Y.; Chi, F. Ectopic hair cell-like cell induction by Math1 mainly involves direct transdifferentiation in neonatal mammalian cochlea. Neurosci. Lett. 2013, 549, 7–11. [Google Scholar] [CrossRef]
- Chien, W.W.; Monzack, E.L.; McDougald, D.S.; Cunningham, L.L. Gene therapy for sensorineural hearing loss. Ear Hear. 2015, 36, 1–7. [Google Scholar] [CrossRef]
- Husseman, J.; Raphael, Y. Gene therapy in the inner ear using adenovirus vectors. Adv. Otorhinolaryngol. 2009, 66, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Kesser, B.W.; Hashisaki, G.T.; Holt, J.R. Gene transfer in human vestibular epithelia and the prospects for inner ear gene therapy. Laryngoscope 2008, 118, 821–831. [Google Scholar] [CrossRef] [Green Version]
- Li Duan, M.; Bordet, T.; Mezzina, M.; Kahn, A.; Ulfendahl, M. Adenoviral and adeno-associated viral vector mediated gene transfer in the guinea pig cochlea. Neuroreport 2002, 13, 1295–1299. [Google Scholar] [CrossRef] [PubMed]
- Sacheli, R.; Delacroix, L.; Vandenackerveken, P.; Nguyen, L.; Malgrange, B. Gene transfer in inner ear cells: A challenging race. Gene Ther. 2013, 20, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, Y.; Takada, T.; Lee, M.Y.; Swiderski, D.L.; Kabara, L.L.; Dolan, D.F.; Raphael, Y. Ototoxicity-induced loss of hearing and inner hair cells is attenuated by HSP70 gene transfer. Mol. Ther. Methods Clin. Dev. 2015, 2, 15019. [Google Scholar] [CrossRef] [PubMed]
- Venail, F.; Wang, J.; Ruel, J.; Ballana, E.; Rebillard, G.; Eybalin, M.; Arbones, M.; Bosch, A.; Puel, J.L. Coxsackie adenovirus receptor and alpha nu beta3/alpha nu beta5 integrins in adenovirus gene transfer of rat cochlea. Gene Ther. 2007, 14, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Bergelson, J.M. Adenovirus receptors. J. Virol. 2005, 79, 12125–12131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Frisina, R.D.; Bowers, W.J.; Frisina, D.R.; Federoff, H.J. HSV amplicon-mediated neurotrophin-3 expression protects murine spiral ganglion neurons from cisplatin-induced damage. Mol. Ther. 2001, 3, 958–963. [Google Scholar] [CrossRef]
- Derby, M.L.; Sena-Esteves, M.; Breakefield, X.O.; Corey, D.P. Gene transfer into the mammalian inner ear using HSV-1 and vaccinia virus vectors. Hear. Res. 1999, 134, 1–8. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [Green Version]
- Carter, B.J. Adeno-associated virus and the development of adeno-associated virus vectors: A historical perspective. Mol. Ther. 2004, 10, 981–989. [Google Scholar] [CrossRef]
- Choi, V.W.; McCarty, D.M.; Samulski, R.J. AAV hybrid serotypes: Improved vectors for gene delivery. Curr Gene Ther. 2005, 5, 299–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Vandenberghe, L.H.; Wilson, J.M. New recombinant serotypes of AAV vectors. Curr. Gene Ther. 2005, 5, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Voutetakis, A.; Afione, S.; Zheng, C.; Mandikian, D.; Chiorini, J.A. Adeno-associated virus type 12 (AAV12): A novel AAV serotype with sialic acid—And heparan sulfate proteoglycan-independent transduction activity. J. Virol. 2008, 82, 1399–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Okada, T.; Sheykholeslami, K.; Shimazaki, K.; Nomoto, T.; Muramatsu, S.; Kanazawa, T.; Takeuchi, K.; Ajalli, R.; Mizukami, H.; et al. Specific and efficient transduction of Cochlear inner hair cells with recombinant adeno-associated virus type 3 vector. Mol. Ther. 2005, 12, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Ballana, E.; Wang, J.; Venail, F.; Estivill, X.; Puel, J.L.; Arbones, M.L.; Bosch, A. Efficient and specific transduction of cochlear supporting cells by adeno-associated virus serotype. Neurosci. Lett. 2008, 442, 134–139. [Google Scholar] [CrossRef]
- Konishi, M.; Kawamoto, K.; Izumikawa, M.; Kuriyama, H.; Yamashita, T. Gene transfer into guinea pig cochlea using adeno-associated virus vectors. J. Gene Med. 2008, 10, 610–618. [Google Scholar] [CrossRef]
- Stone, I.M.; Lurie, D.I.; Kelley, M.W.; Poulsen, D.J. Adeno-associated virus-mediated gene transfer to hair cells and support cells of the murine cochlea. Mol. Ther. 2005, 11, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, A.N.; Couchman, J.R.; Whiteford, J.R. The CMV early enhancer/chicken beta actin (CAG) promoter can be used to drive transgene expression during the differentiation of murine embryonic stem cells into vascular progenitors. BMC Cell Biol. 2008, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Okada, T.; Nomoto, T.; Ke, X.; Kume, A.; Ozawa, K.; Xiao, S. Promoter effects of adeno-associated viral vector for transgene expression in the cochlea in vivo. Exp. Mol. Med. 2007, 39, 170–175. [Google Scholar] [CrossRef]
- Ghosh, A.; Yue, Y.; Lai, Y.; Duan, D. A hybrid vector system expands adeno-associated viral vector packaging capacity in a transgene-independent manner. Mol. Ther. 2008, 16, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Akil, O.; Dyka, F.; Calvet, C.; Emptoz, A.; Lahlou, G.; Nouaille, S.; Boutet de Monvel, J.; Hardelin, J.P.; Hauswirth, W.W.; Avan, P.; et al. Dual AAV—Mediated gene therapy restores hearing in a DFNB9 mouse model. Proc. Natl. Acad. Sci. USA 2019, 116, 4496–4501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Moyed, H.; Cepeda, A.P.; Jung, S.; Moser, T.; Kugler, S.; Reisinger, E. A dual—AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Nist-Lund, C.A.; Pan, B.; Patterson, A.; Asai, Y.; Chen, T.; Zhou, W.; Zhu, H.; Romero, S.; Resnik, J.; Polley, D.B.; et al. Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat. Commun. 2019, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Isgrig, K.; McDougald, D.S.; Zhu, J.; Wang, H.J.; Bennett, J.; Chien, W.W. AAV2.7m8 is a powerful viral vector for inner ear gene therapy. Nat. Commun. 2019, 10, 427. [Google Scholar] [CrossRef] [Green Version]
- Zinn, E.; Pacouret, S.; Khaychuk, V.; Turunen, H.T.; Carvalho, L.S.; Andres-Mateos, E.; Shah, S.; Shelke, R.; Maurer, A.C.; Plovie, E.; et al. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015, 12, 1056–1068. [Google Scholar] [CrossRef] [Green Version]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-Dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef]
- Ivanchenko, M.V.; Hanlon, K.S.; Devine, M.K.; Tenneson, K.; Emond, F.; Lafond, J.F.; Kenna, M.A.; Corey, D.P.; Maguire, C.A. Preclinical testing of AAV9-PHP.B for transgene expression in the non-human primate cochlea. Hear. Res. 2020, 107930. [Google Scholar] [CrossRef]
- Dalkara, D.; Byrne, L.C.; Klimczak, R.R.; Visel, M.; Yin, L.; Merigan, W.H.; Flannery, J.G.; Schaffer, D.V. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 2013, 5, 189ra176. [Google Scholar] [CrossRef]
- Chai, R.; Kuo, B.; Wang, T.; Liaw, E.J.; Xia, A.; Jan, T.A.; Liu, Z.; Taketo, M.M.; Oghalai, J.S.; Nusse, R.; et al. Wnt signaling induces proliferation of sensory precursors in the postnatal mouse cochlea. Proc. Natl. Acad. Sci. USA 2012, 109, 8167–8172. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.; Hu, L.; Edge, A.S. Generation of hair cells in neonatal mice by Beta—Catenin overexpression in Lgr5-positive cochlear progenitors. Proc. Natl. Acad. Sci. USA 2013, 110, 13851–13856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell. Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Hung, M.E.; Breakefield, X.O.; Leonard, J.N. Therapeutic applications of extracellular vesicles: Clinical promise and open questions. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 439–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, Z.; Gyorgy, B.; Skog, J.; Maguire, C.A. Extracellular vesicles as enhancers of virus vector-mediated gene delivery. Hum. Gene Ther. 2014, 25, 785–786. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, T.; Kamiya, K.; Gotoh, S.; Sugitani, Y.; Suzuki, M.; Noda, T.; Minowa, O.; Ikeda, K. Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Hum. Mol. Genet. 2015, 24, 3651–3661. [Google Scholar] [CrossRef] [Green Version]
- Chien, W.W.; Isgrig, K.; Roy, S.; Belyantseva, I.A.; Drummond, M.C.; May, L.A.; Fitzgerald, T.S.; Friedman, T.B.; Cunningham, L.L. Gene Therapy Restores Hair Cell Stereocilia Morphology in Inner Ears of Deaf Whirler Mice. Mol. Ther. 2016, 24, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Isgrig, K.; Shteamer, J.W.; Belyantseva, I.A.; Drummond, M.C.; Fitzgerald, T.S.; Vijayakumar, S.; Jones, S.M.; Griffith, A.J.; Friedman, T.B.; Cunningham, L.L.; et al. Gene Therapy Restores Balance and Auditory Functions in a Mouse Model of Usher Syndrome. Mol. Ther. 2017, 25, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.A.; Cho, H.J.; Bae, S.H.; Lee, B.; Oh, S.K.; Kwon, T.J.; Ryoo, Z.Y.; Kim, H.Y.; Cho, J.H.; Kim, U.K.; et al. Methionine Sulfoxide Reductase B3-Targeted In Utero Gene Therapy Rescues Hearing Function in a Mouse Model of Congenital Sensorineural Hearing Loss. Antioxid. Redox Signal. 2016, 24, 590–602. [Google Scholar] [CrossRef] [Green Version]
- Geng, R.; Omar, A.; Gopal, S.R.; Chen, D.H.; Stepanyan, R.; Basch, M.L.; Dinculescu, A.; Furness, D.N.; Saperstein, D.; Hauswirth, W.; et al. Modeling and Preventing Progressive Hearing Loss in Usher Syndrome III. Sci. Rep. 2017, 7, 13480. [Google Scholar] [CrossRef]
- Kim, M.A.; Kim, S.H.; Ryu, N.; Ma, J.H.; Kim, Y.R.; Jung, J.; Hsu, C.J.; Choi, J.Y.; Lee, K.Y.; Wangemann, P.; et al. Gene therapy for hereditary hearing loss by SLC26A4 mutations in mice reveals distinct functional roles of pendrin in normal hearing. Theranostics 2019, 9, 7184–7199. [Google Scholar] [CrossRef]
- Staecker, H.; Li, D.; O’Malley, B.W., Jr.; Van De Water, T.R. Gene expression in the mammalian cochlea: A study of multiple vector systems. Acta. Otolaryngol. 2001, 121, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.; Sood, R.; Ranjan, S.; Surovtseva, E.; Ahmad, A.; Kinnunen, P.K.; Pyykko, I.; Zou, J. Visualization of intracellular trafficking of Math1 protein in different cell types with a newly-constructed nonviral gene delivery plasmid. J. Gene Med. 2011, 13, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Wareing, M.; Mhatre, A.N.; Pettis, R.; Han, J.J.; Haut, T.; Pfister, M.H.; Hong, K.; Zheng, W.W.; Lalwani, A.K. Cationic liposome mediated transgene expression in the guinea pig cochlea. Hear. Res. 1999, 128, 61–69. [Google Scholar] [CrossRef]
- Jero, J.; Mhatre, A.N.; Tseng, C.J.; Stern, R.E.; Coling, D.E.; Goldstein, J.A.; Hong, K.; Zheng, W.W.; Hoque, A.T.; Lalwani, A.K. Cochlear gene delivery through an intact round window membrane in mouse. Hum. Gene Ther. 2001, 12, 539–548. [Google Scholar] [CrossRef]
- Jero, J.; Tseng, C.J.; Mhatre, A.N.; Lalwani, A.K. A surgical approach appropriate for targeted cochlear gene therapy in the mouse. Hear. Res. 2001, 151, 106–114. [Google Scholar] [CrossRef]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.H.; Pan, B.; Hu, Y.J.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 2018, 553, 217–221. [Google Scholar] [CrossRef]
- Gao, X.; Kim, K.S.; Liu, D. Nonviral gene delivery: What we know and what is next. AAPS J. 2007, 9, E92–E104. [Google Scholar] [CrossRef]
- Tamura, T.; Kita, T.; Nakagawa, T.; Endo, T.; Kim, T.S.; Ishihara, T.; Mizushima, Y.; Higaki, M.; Ito, J. Drug delivery to the cochlea using PLGA nanoparticles. Laryngoscope 2005, 115, 2000–2005. [Google Scholar] [CrossRef]
- Toyama, K.; Ozeki, M.; Hamajima, Y.; Lin, J. Expression of the integrin genes in the developing cochlea of rats. Hear. Res. 2005, 201, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.T.; Foong, K.H.; Lee, M.M.; Ruan, R. Polyethylenimine-mediated cochlear gene transfer in guinea pigs. Arch. Otolaryngol. Head Neck Surg. 2008, 134, 884–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhang, Y.; Lobler, M.; Schmitz, K.P.; Ahmad, A.; Pyykko, I.; Zou, J. Nuclear entry of hyperbranched polylysine nanoparticles into cochlear cells. Int. J. Nanomed. 2011, 6, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyantseva, I.A.; Boger, E.T.; Friedman, T.B. Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc. Natl. Acad. Sci. USA 2003, 100, 13958–13963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyantseva, I.A.; Boger, E.T.; Naz, S.; Frolenkov, G.I.; Sellers, J.R.; Ahmed, Z.M.; Griffith, A.J.; Friedman, T.B. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat. Cell Biol. 2005, 7, 148–156. [Google Scholar] [CrossRef]
- Woods, C.; Montcouquiol, M.; Kelley, M.W. Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat. Neurosci. 2004, 7, 1310–1318. [Google Scholar] [CrossRef]
- Zheng, J.L.; Gao, W.Q. Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat. Neurosci. 2000, 3, 580–586. [Google Scholar] [CrossRef]
- Gubbels, S.P.; Woessner, D.W.; Mitchell, J.C.; Ricci, A.J.; Brigande, J.V. Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 2008, 455, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Miwa, T.; Minoda, R.; Ise, M.; Yamada, T.; Yumoto, E. Mouse otocyst transuterine gene transfer restores hearing in mice with connexin 30 deletion-associated hearing loss. Mol. Ther. 2013, 21, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- Puligilla, C.; Dabdoub, A.; Brenowitz, S.D.; Kelley, M.W. Sox2 induces neuronal formation in the developing mammalian cochlea. J. Neurosci. 2010, 30, 714–722. [Google Scholar] [CrossRef]
- Pinyon, J.L.; Tadros, S.F.; Froud, K.E.; AC, Y.W.; Tompson, I.T.; Crawford, E.N.; Ko, M.; Morris, R.; Klugmann, M.; Housley, G.D. Close-field electroporation gene delivery using the cochlear implant electrode array enhances the bionic ear. Sci. Transl. Med. 2014, 6, 233ra254. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N., Jr.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M.; et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef] [Green Version]
- Cideciyan, A.V.; Aleman, T.S.; Boye, S.L.; Schwartz, S.B.; Kaushal, S.; Roman, A.J.; Pang, J.J.; Sumaroka, A.; Windsor, E.A.; Wilson, J.M.; et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. USA 2008, 105, 15112–15117. [Google Scholar] [CrossRef] [Green Version]
- MacLaren, R.E.; Groppe, M.; Barnard, A.R.; Cottriall, C.L.; Tolmachova, T.; Seymour, L.; Clark, K.R.; During, M.J.; Cremers, F.P.; Black, G.C.; et al. Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet 2014, 383, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Schilder, A.G.M.; Su, M.P.; Blackshaw, H.; Lustig, L.; Staecker, H.; Lenarz, T.; Safieddine, S.; Gomes-Santos, C.S.; Holme, R.; Warnecke, A. Hearing Protection, Restoration, and Regeneration: An Overview of Emerging Therapeutics for Inner Ear and Central Hearing Disorders. Otol. Neurotol. 2019, 40, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Lopes, V.S.; Williams, D.S. Gene Therapy for the Retinal Degeneration of Usher Syndrome Caused by Mutations in MYO7A. Cold Spring Harb Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta. 2015, 1852, 406–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustapha, M.; Chouery, E.; Chardenoux, S.; Naboulsi, M.; Paronnaud, J.; Lemainque, A.; Megarbane, A.; Loiselet, J.; Weil, D.; Lathrop, M.; et al. DFNB31, a recessive form of sensorineural hearing loss, maps to chromosome 9q32-34. Eur. J. Hum. Genet. 2002, 10, 210–212. [Google Scholar] [CrossRef] [Green Version]
- Mburu, P.; Mustapha, M.; Varela, A.; Weil, D.; El-Amraoui, A.; Holme, R.H.; Rump, A.; Hardisty, R.E.; Blanchard, S.; Coimbra, R.S.; et al. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB. Nat. Genet. 2003, 34, 421–428. [Google Scholar] [CrossRef]
- Bonnet, C.; El-Amraoui, A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): Pathogenesis, molecular diagnosis and therapeutic approaches. Curr. Opin. Neurol. 2012, 25, 42–49. [Google Scholar] [CrossRef]
- Rossor, A.M.; Reilly, M.M.; Sleigh, J.N. Antisense oligonucleotides and other genetic therapies made simple. Pract. Neurol. 2018, 18, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.A. RNAi and double-strand RNA. Genes Dev. 1999, 13, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Lentz, J.J.; Jodelka, F.M.; Hinrich, A.J.; McCaffrey, K.E.; Farris, H.E.; Spalitta, M.J.; Bazan, N.G.; Duelli, D.M.; Rigo, F.; Hastings, M.L. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 2013, 19, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Sheffield, A.M.; Smith, R.J.H. Therapeutic regulation of gene expression in the inner ear using RNA interference. Adv. Otorhinolaryngol. 2009, 66, 13–36. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.B.; Ranum, P.T.; Moteki, H.; Pan, B.; Goodwin, A.T.; Goodman, S.S.; Abbas, P.J.; Holt, J.R.; Smith, R.J.H. RNA Interference Prevents Autosomal-Dominant Hearing Loss. Am. J. Hum. Genet. 2016, 98, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Moteki, H.; Smith, R.J.H. Targeted Allele Suppression Prevents Progressive Hearing Loss in the Mature Murine Model of Human TMC1 Deafness. Mol. Ther. 2019, 27, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [Green Version]
- Zou, B.; Mittal, R.; Grati, M.; Lu, Z.; Shu, Y.; Tao, Y.; Feng, Y.; Xie, D.; Kong, W.; Yang, S.; et al. The application of genome editing in studying hearing loss. Hear. Res. 2015, 327, 102–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorgy, B.; Nist-Lund, C.; Pan, B.; Asai, Y.; Karavitaki, K.D.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Solanes, P.; Spataro, S.; et al. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat. Med. 2019, 25, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Devare, J.; Gubbels, S.; Raphael, Y. Outlook and future of inner ear therapy. Hear. Res. 2018, 368, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, K.; Ishimoto, S.; Minoda, R.; Brough, D.E.; Raphael, Y. Math1 gene transfer generates new cochlear hair cells in mature guinea pigs in vivo. J. Neurosci. 2003, 23, 4395–4400. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Corfas, G.; Liberman, M.C. Round-window delivery of neurotrophin 3 regenerates cochlear synapses after acoustic overexposure. Sci. Rep. 2016, 6, 24907. [Google Scholar] [CrossRef] [Green Version]
- Pfingst, B.E.; Colesa, D.J.; Swiderski, D.L.; Hughes, A.P.; Strahl, S.B.; Sinan, M.; Raphael, Y. Neurotrophin Gene Therapy in Deafened Ears with Cochlear Implants: Long-term Effects on Nerve Survival and Functional Measures. J. Assoc. Res. Otolaryngol. 2017, 18, 731–750. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name; Deafness Locus | AAV Vector | Animal Model | Route of Delivery; Age at Delivery | Target Cells/Outcomes | References |
|---|---|---|---|---|---|
| Vglut3; DFNA25 | AAV1-mVglut3 | Vglut3−/− mice | RWM; P1-P3, P10-P12 | Transduction of almost 100% IHCs, normal ABR thresholds and waveforms at both low and high frequencies, up to 28 weeks post injection. Earlier delivery increases hearing recovery longevity. | [20] |
| Gjb2 (Cx26); DFNB1 | AAv2/1-CB7-Gjb2 AAV2/1-CB7-Gjb2-GFP | Cx26fl/flFoxg1-Cre+/− mice | Cochleostomy; P0-P1 | High transduction efficiency of supporting cells, especially outer sulcus cells. Very low transduction of sensory hair cells, but improved hair cells and spiral ganglion neurons survival. No restoration of hearing sensitivity. | [35] |
| AAV2/5-CMV-Gjb2 | Cx26fl/flP0-Cre mice | RWM; P0, P42 | Transduction of supporting cells, the spiral ligament fibrocytes and the spiral limbus. Preserved structure of OHCs, IHCs, and supporting cells, and improved ABR thresholds at neonatal stage, but not in adult mice. | [86] | |
| Tmc1; DFNB7/11 Tmc1Bth/+; DFNA36 | AAV2/1-CBA-Tmc1 AAV2/1-CBA-Tmc2 | Tmc1−/− and Tmc1Bth mice | RWM; P0-P2 | Restoration of IHCs function. Partial recovery of ABR thresholds in Tmc1−/− mice injected either with either AAV2/1-CBA-Tmc1 or AAV2/1-CBA-Tmc2. No recovery of OHC function, due to low viral transduction rates in OHCs. | [22] |
| AAV2/Anc80L65-CMV-Tmc1-WPRE, AAV2/Anc80L65-CMV-Tmc2-WPRE, AAV2/Anc80L65-CMV-Tmc1EX1-WPRE, AAV2/Anc80L65-CMV-EGFP-WPRE | Wild-type and Tmc1−/− mice | RWM; P0-P2, P4, P7, P14, and P30 | Cochlea: High transduction of both IHCs and OHCs at neonatal stage. Rescued sensory function in mature hair cells, and enhanced hair cell survival. Partial recovery of ABR thresholds, especially at the low frequencies. Vestibular end-organs: High transduction of vestibular hair cells, and enhanced viability of hair cells. Restoration of vestibular behavior and balance function even at mature stages. | [75] | |
| Whrn; DFNB31; USH2D | AAV2/8-CMV-Whrn-GFP | Whirler (Whrnwi/wi) mice | RWM; P1-P5 | Transduction of ~15% of IHCs, and any of OHCs. No improvement in hearing sensitivity. Restoration of stereocilia length and hair bundle morphology and increase in IHCs survival. | [87] |
| AAV2/8-CMV-Whrn-GFP | Whirler (Whrnwi/wi) mice | PSCC; P4 | Cochlea: Normal stereocilia bundles morphology. Successful transduction of IHCs, and partial restoration of hearing for at least 4 months. Vestibular end-organs: Efficient transduction of vestibular hair cell. Restoration of utricular hair cells morphology. Normal vestibular behavior and balance function for at least 4 months, and improved vestibular evoked potentials (VsEPs). | [88] | |
| Pjvk; DFNB59 | AAV2/8-Pjvk-IRES-eGFP AAV8-Pjvk-IRES-eGFP | Pjvk−/− mice | RWM; P3 | Partial restoration of ABR thresholds, normal ABR waveforms and wave amplitudes. | [21] |
| MsrB3; DFNB74 | rAAV2/1-CMV-MsrB3-GFP | MsrB3−/− mice | Otocysts; E12.5 | High transduction efficiency of both IHCs and OHCs in all cochlear turns. Preserved hair cells, rescued morphology of stereociliary bundles, and normal ABR thresholds at both low and high frequencies at P28. | [89] |
| Clrn1; Ush3A | AAV2/2-CAG-Clrn1-UTRs AAV2/8-CAG-Clrn1-UTRs | Clrn1−/− and KO-TgAC1 mice (Transgene Atoh1-enhacer-Clrn-1 UTRs) | RWM; P1-P3 | Preserved hair bundle morphology at P100. Normal click-evoked ABR thresholds and waveforms at P100. | [90] |
| AAV2/8-CAG-Clrn1-IRES-eGFP | Clrn1ex4−/− and Clrn1ex4fl/fl Myo15−Cre+/− mice | RWM; P1-P3 | High transduction of both IHCs and OHCs. An almost complete rescue of hearing for low and high frequencies in Clrn1ex4fl/flMyo15-Cre+/−mice. Prevention of the synaptic defects and durably preservation of the stereocilia hair bundles morphology up to P12. | [26] | |
| AAV2/9.PHP.B-CBA-Clrn1-eGFP | Clrn−/−mice | RWM; P0-P1, P30 | Cochlea: High transduction of both IHCs and OHCs at neonatal stage. Almost all IHCs from apex to base transduced, but no OHC transduction at adult stage. Robust hearing rescue at low frequencies. Vestibular end-organs: Robust transduction of vestibular hair cells | [27] | |
| Ush1c; DFNB18 | AAV2/Anc80L65-CMV-Harma1 AAV2/Anc80L65-CMV-Harmb1 | Ush1c c.216G>A knockin mice (Acadian mutation) | RWM; P0-P1, P10-P12 | Cochlea: High transduction efficiency of both IHCs and OHCs. Normal ABR thresholds in mice injected with harmonin-b1 alone or harmonin-a1/b1 together, particularly at low frequencies. Normal hair bundle morphology along the entire organ of Corti at 6 weeks of age. Vestibular end-organs: Restoration of balance behaviors | [25] |
| Sans; USH1G | AAV2/1-CAG-Sans-eGFP AAV2/2-CAG-Sans-eGFP AAV2/5-CAG-Sans-eGFP AAV2/8-CAG-Sans-eGFP | Ush1g−/− mice | RWM; P2.5 | Cochlea: AAV2/1 and AAV2/2 and AAV2/5 injections mostly transduced supporting cells of the organ of Corti. AAV2/8 injection transduced IHCs with greater efficiency at the apex of the cochlea than at the base, whereas OHCs were transduced with roughly the same efficiency at the base and at the apex. Restoration of hair bundle morphology, and improved hearing thresholds. Vestibular end-organs: AAV2/8 transduced the vast majority of vestibular hair cells, restored morphology of stereociliary bundles, and durably rescued balance defects. | [23] |
| Lhfpl5; DFNB66/67 | Exo-AAV1-CBA-GFP Exo-AAV1-CBA-HA-Lhfpl5 | Wild-type and Lhfpl5−/− mice | RWM; Cochleostomy; P0-P1 | Cochlea: Both RWM and cochleostomy injection transduced with high efficiently both IHCs and OHCs. Cochleostomy also transduced spiral ganglion neurons and supporting cells. Improved hearing thresholds at frequencies from 4 to 22 kHz. Vestibular end-organs: Robust transduction of vestibular hair cells via both RWM and cochleostomy injections. Restoration of balance behaviors. | [36] |
| Otof; DFNB9 | Dual vector: AAv2/quadY-F-smCBA-Otof N term-AA1-816-SD and AAv2/quadY-F-smCBA-Otof C term-AA817-1992-SA-pA. ALK bridge 3′ to SD and 5′ to SA, respectively. | Otof−/− mice | RWM; P10, P17, and P30 | High transduction efficiency of IHCs. Durable restoration of otoferlin expression in transduced inner hair cells. Normal ABR thresholds for both click and tone-burst stimuli in treated mice. Restoration of ABR wave I latency, and partial recovery of ABR wave I amplitude. | [73] |
| Dual 5′-AAv2/6-TS and 5′-AAV2/6-hybrid: hbA-CMVe-eGFP-P2A, 5′-Otof CDS-exon 1-21-SD. Dual-3′AAV2/6-TS and 3′-AAV2/6-hybrid: SA-3′Otof CDS-exon 22-46-WPRE-pA. ALK bridge 3′ to SD and 5′ to SA, respectively | Otof−/− mice | RWM; P6-P7 | Highly efficient transduction of IHCs, supporting cells, and spiral ganglion neurons. Full recovery of fast exocytosis in Otof−/− IHCs. Normal click-evoked ABR thresholds and waveforms (particularly, waves II-V), and increased ABR wave amplitudes. | [74] | |
| Slc26a4; DFNB4 | rAAV2/1-CMV-Slc26a4-tGFP | Slc26a4−/− and Slc26a4tm1Dontuh/tm1Dontuh mice | Otocysts; endolymphatic sac; E12.5 | Cochlea: Restoration of hearing function, but variable hearing phenotype between injected mice. Preservation of both OHCs and IHCs at 5 weeks of age. Vestibular end-organs: Transient pendrin expression prevented enlargement of the membranous labyrinth but failed to restore otoconia formation and the acquisition vestibular function. | [91] |
| Non-Viral Vector | Transgene | Animal Model | Route of Delivery | Targeted Cells/ Outcomes | References |
|---|---|---|---|---|---|
| Cationic Liposomes | |||||
| Liposomes | β−gal plasmid | Guinea pig | RWM or cochleostomy and osmotic minipump infusion | Spiral limbus, spiral ligament, Reissner’s membrane, and spiral ganglion neurons | [95] |
| Liposomes | eGFP plasmid | Mouse | Gelfoam on RWM | Auditory hair cells, spiral ganglion neurons, spiral ligament, and stria vascularis | [92,96,97] |
| Lipofectamine 2000 | Math1 plasmid (pcDNA6.2/C-EGFP-Math1) | Rat | Organ of Corti-derived cell line | Transfection of fibrocytes, spiral ganglion neuron and hair cell-like cells. Very low transfection efficiency (2.9%) | [94] |
| Lipofectamine 2000, Lipofectamine RNAiMax | Cas9:sgRNA complexes fused to (−30) GFP-Cre | Atoh1-GFP mice | Cochleostomy | Up to 20% Cas9-mediated genome modification in outer hair cells (loss of GFP expression near the injection site after 10 days) | [98] |
| Lipofectamine 2000 | Cas9:sgRNA complexes targeting the Tmc1Bth allele | Tmc1Bth/+ mice | Cochleostomy | Higher hair cell survival rates, improvement in ABR thresholds for the frequencies between 8–23 kHz, and greater ABR waves amplitudes with almost normal waveform pattern. | [99] |
| Polymeric nanoparticles | |||||
| Poly (lactic-co-glycolic acid) nanoparticles (PLGA) | Rhodamine | Guinea pig | Gelfoam on RWM | Scala tympani | [101] |
| Polybrene | Integrin subunits antisense oligonucleotides | Rat | Organ of Corti-derived cell line | Efficient inhibition of integrin subunits expression. | [102] |
| Polyethylenimine | eGFP plasmid | Guinea pig | Cochleostomy and osmotic minipump infusion | GFP expression in fibrocytes lining the scala vestibuli and scala tympani, mesenchymal and epithelial cells of Reissner’ membrane, and fibrocytes of the spiral ligament. No transfection in the organ of Corti or stria vascularis. | [103] |
| Dendritic polymer (hyperbranched poly-L-Lysine nanoparticle; HPNP) | eGFP plasmid | Rat | Gelatin sponge on RWM | Efficient GFP expression in hair cells, supporting cells, the stria vascularis marginal cells, the spiral ligament fibrocytes, and spiral ganglion neurons | [104] |
| Biolistic (Gene Gun) | |||||
| Gold particles | pEGFP-MyoXVa | Mouse | Organ of Corti explants | MyoXVa-GFP expression at the tips of stereocilia | [105] |
| Gold particles | pEGFP-MyoXVa or pEGFP-Whrn | Myo15ash2 or Whrnwi mice | Organ of Corti explants | Restoration of hair bundle staircase shape in both cochlear and vestibular GFP-positive hair cells | [106] |
| Electroporation | |||||
| Electroporation | Math1-eGFP plasmids | Mouse | Organ of Corti explants | GFP expression in greater epithelial ridge and stereociliary bundles in the hair cells | [107,108] |
| Electroporation | Atoh1 (Math1)-GFP plasmid | Mouse | In utero (microinjected into the E11.5 otic vesicle) | Efficient GFP expression in hair cells and supporting cells. | [109] |
| Electroporation | pCMV-Cx30-eGFP plasmid | Cx30−/− mice | In utero (microinjected into the E11.5 otic vesicle) | Efficient CX30 expression in spiral limbus, organ of Corti, stria vascularis, spiral ligament, and spiral ganglion neurons at P30. Normal ABR thresholds and endocochlear potential at P30 | [110] |
| Electroporation | Transcription factors: Sox2, Neurog1, and Neurod1 | Mouse | Organ of Corti explants | Ectopic expression of these transcription factors in nonsensory regions of cochlear explant cultures induce the formation of neuronal cells | [111] |
| “Close-field” electroporation through cochlear implant electrodes | BDNF-GFP | Guinea pig deafened by kanamycin-furosemide treatment | RWM cochlear implant | Stimulated survival and regeneration of spiral ganglion neurons. | [112] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delmaghani, S.; El-Amraoui, A. Inner Ear Gene Therapies Take Off: Current Promises and Future Challenges. J. Clin. Med. 2020, 9, 2309. https://doi.org/10.3390/jcm9072309
Delmaghani S, El-Amraoui A. Inner Ear Gene Therapies Take Off: Current Promises and Future Challenges. Journal of Clinical Medicine. 2020; 9(7):2309. https://doi.org/10.3390/jcm9072309
Chicago/Turabian StyleDelmaghani, Sedigheh, and Aziz El-Amraoui. 2020. "Inner Ear Gene Therapies Take Off: Current Promises and Future Challenges" Journal of Clinical Medicine 9, no. 7: 2309. https://doi.org/10.3390/jcm9072309