
Homeostatic Signaling by Cell–Cell Junctions and Its Dysregulation during Cancer Progression

Abstract
:1. Breast Cancer
2. Epithelial-to-Mesenchymal Transition (EMT), Tumor Invasion, and Metastasis
2.1. Physiological Significance
2.2. EMT Confers Invasiveness
2.3. EMT Confers Resistance to Anoikis
2.4. EMT Confers Stemness
2.5. EMT Confers Immunoresistance
2.6. Induction of EMT by Soluble Factors
2.7. Structural Cues Suppress EMT
3. Epithelial Cell–Cell Junctions Suppress EMT
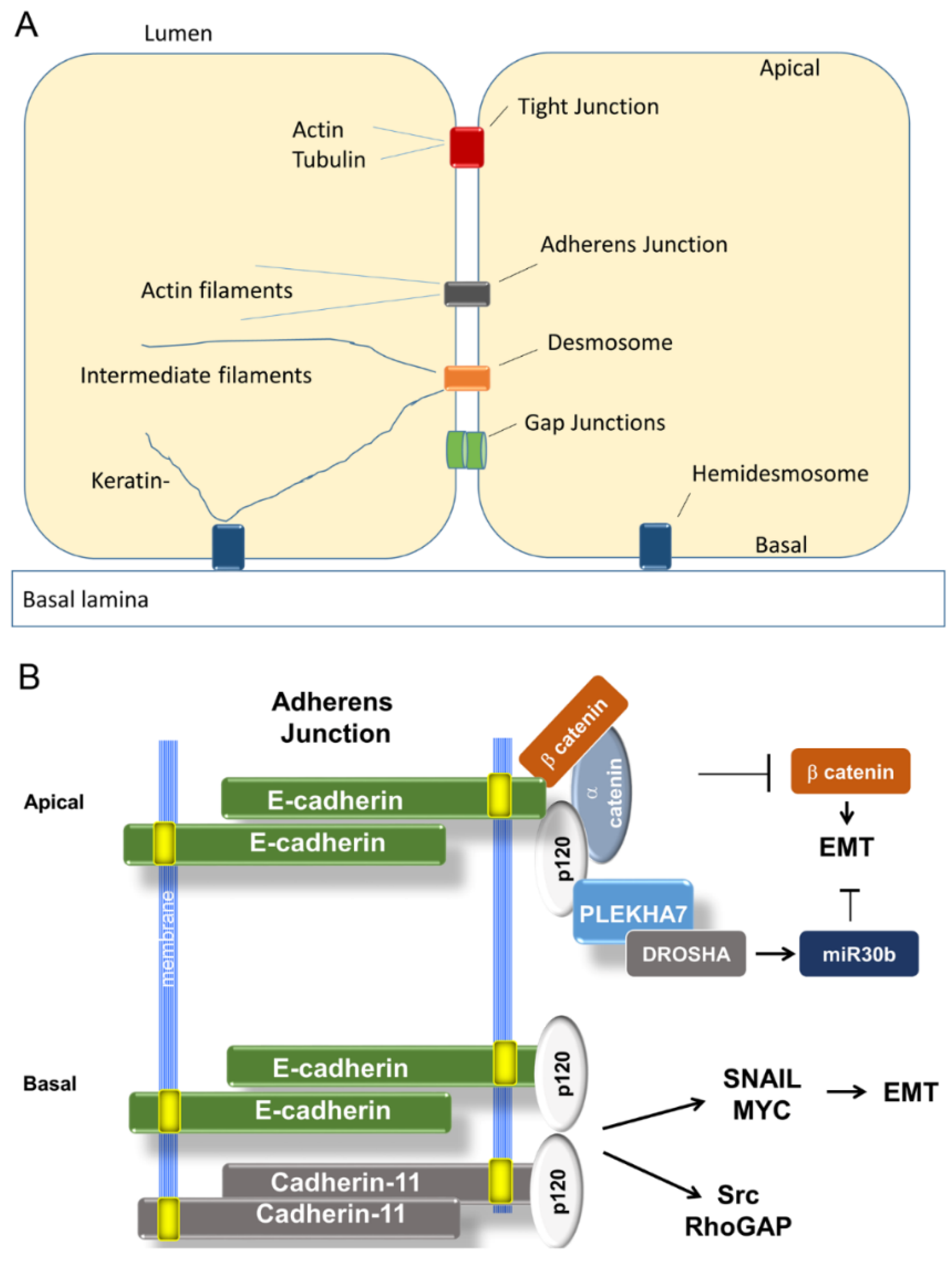
3.1. Adherens Junctions Suppress EMT
3.1.1. Organization of Adherens Junctions (AJ)
3.1.2. Loss of E-Cadherin in Cancer and EMT
3.1.3. Could E-Cadherin Abet Dissemination?
3.1.4. A Tale of Two E-cadherins
3.1.5. R-Cadherin
3.2. Tight Junctions Block EMT
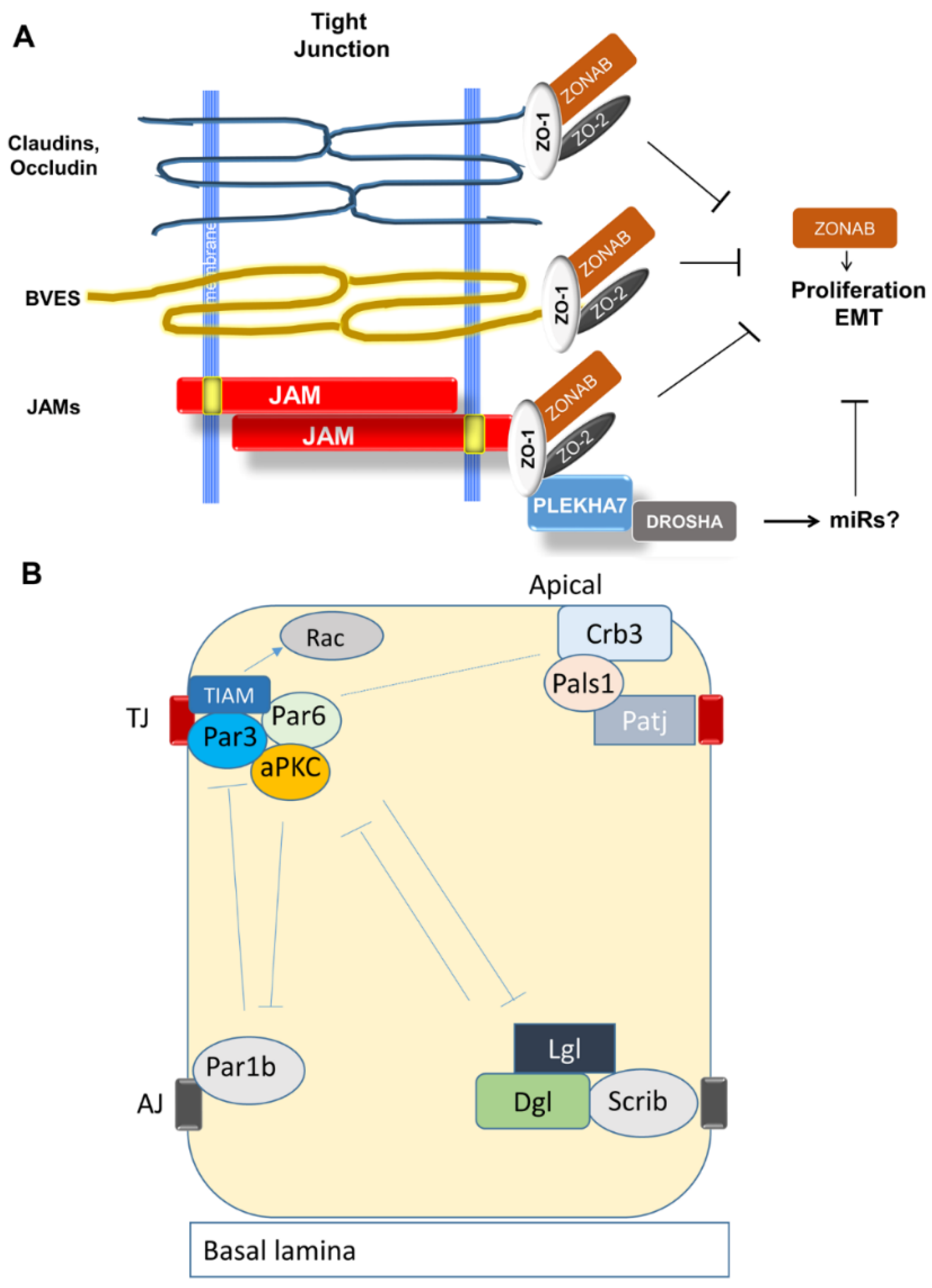
3.2.1. Organization and Function of Tight Junctions (TJ)
3.2.2. Establishment of Apical-Basal Polarity and Formation of TJ
3.2.3. Claudins
3.2.4. Occludin
3.2.5. Blood Vessel Epicardial Substance (BVES)
3.2.6. Junctional Adhesion Molecules (JAMs)
3.2.7. TJ Adaptor and Regulatory Molecules
4. Targeting EMT Therapeutically
5. Conclusions
Acknowledgments
Conflicts of interest
References
- Fidler, I.J. The biology of cancer metastasis. Semin. Cancer Biol. 2011, 21, 71. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev. Dyn. 2005, 233, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Bitting, R.L.; Schaeffer, D.; Somarelli, J.A.; Garcia-Blanco, M.A.; Armstrong, A.J. The role of epithelial plasticity in prostate cancer dissemination and treatment resistance. Cancer Metastasis Rev. 2014, 33, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.N.; Chikwem, A.J.; Solanki, N.R.; Golemis, E.A. Bioinformatic approaches to augment study of epithelial-to-mesenchymal transition in lung cancer. Phys. Genom. 2014, 46, 699–724. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Xie, D.; Cui, J.; Li, Q.; Gao, Y.; Xie, K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin. Cancer Res. 2014, 20, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Shorning, B.Y.; Griffiths, D.; Clarke, A.R. Lkb1 and Pten synergise to suppress mTOR-mediated tumorigenesis and epithelial-mesenchymal transition in the mouse bladder. PLoS ONE 2011, 6, e16209. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [PubMed]
- Gilles, C.; Newgreen, D.F.; Sato, H.; Thompson, E.W. Matrix Metalloproteases and Epithelial-to-Mesenchymal Transition: Implications for Carcinoma Metastasis. In Raise and Fall of Epithelial Phenotype; Landes Bioscience: Austin, TX, USA, 2000. [Google Scholar]
- Derksen, P.W.; Liu, X.; Saridin, F.; van der Gulden, H.; Zevenhoven, J.; Evers, B.; van Beijnum, J.R.; Griffioen, A.W.; Vink, J.; Krimpenfort, P.; et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 2006, 10, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.J.; Zhang, C.C.; Zhou, B.; Zimonjic, D.B.; Mani, S.A.; Kaba, M.; Gifford, A.; Reinhardt, F.; Popescu, N.C.; Guo, W.; et al. Enrichment of a population of mammary gland cells that form mammospheres and have in vivo repopulating activity. Cancer Res. 2007, 67, 8131–8138. [Google Scholar] [CrossRef] [PubMed]
- Murohashi, M.; Hinohara, K.; Kuroda, M.; Isagawa, T.; Tsuji, S.; Kobayashi, S.; Umezawa, K.; Tojo, A.; Aburatani, H.; Gotoh, N.; et al. Gene set enrichment analysis provides insight into novel signalling pathways in breast cancer stem cells. Br. J. Cancer 2010, 102, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Andre, F.; De C.remoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [PubMed]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Thiery, J.P.; Mami-Chouaib, F.; et al. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy 2013, 9, 1104–1106. [Google Scholar] [CrossRef] [PubMed]
- Alsuliman, A.; Colak, D.; Al-Harazi, O.; Fitwi, H.; Tulbah, A.; Al-Tweigeri, T.; Chouaib, S. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: Significance in claudin-low breast cancer cells. Mol. Cancer 2015, 14, 149. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Oft, M.; Heider, K.H.; Beug, H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr. Biol. 1998, 8, 1243–1252. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Awad, P.; Campisi, J.; Desprez, P.Y. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2012, 5, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Santisteban, M.; Reiman, J.M.; Asiedu, M.K.; Behrens, M.D.; Nassar, A.; Kalli, K.R.; Haluska, P.; Ingle, J.N.; Hartmann, L.C.; Manjili, M.H.; et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009, 69, 2887–2895. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.K.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. TGFβ/TNFα-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011, 71, 4707–4719. [Google Scholar] [CrossRef] [PubMed]
- Bednarz-Knoll, N.; Alix-Panabières, C.; Pantel, K. Plasticity of disseminating cancer cells in patients with epithelial malignancies. Cancer Metastasis Rev. 2012, 31, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Wu, Q.; Acquafondata, M.; Dhir, R.; Wells, A. Partial mesenchymal to epithelial reverting transition in breast and prostate cancer metastases. Cancer Microenviron. 2012, 5, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Walia, V.; Elble, R.C. Enrichment for breast cancer cells with stem/progenitor properties by differential adhesion. Stem. Cells Dev. 2010, 19, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Walia, V.; Yu, Y.; Cao, D.; Sun, M.; McLean, J.R.; Hollier, B.G.; Cheng, J.; Mani, S.A.; Rao, K.; Premkumar, L.; et al. Loss of breast epithelial marker hCLCA2 promotes epithelial-to-mesenchymal transition and indicates higher risk of metastasis. Oncogene 2012, 31, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H; Berk, A; Kaiser, C.A; Krieger, M; Scott, M.P.; Bretscher, A.; Ploegh, H.; Matsudaira, P. Integrating cells into tissues. In Molecular Cell Biology, 6th ed.; Ahr, K., Ed.; W.H. Freeman and Co.: New York, NY, USA, 2008; pp. 801–841. [Google Scholar]
- Ivanov, D.B.; Philippova, M.P.; Tkachuk, V.A. Structure and functions of classical cadherins. Biochemistry 2001, 66, 1174–1186. [Google Scholar] [PubMed]
- Albergaria, A.; Ribeiro, A.S.; Vieira, A.F.; Sousa, B.; Nobre, A.R.; Seruca, R.; Schmitt, F.; Paredes, J. P-cadherin role in normal breast development and cancer. Int. J. Dev. Biol. 2011, 55, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Theveneau, E.; Mayor, R. Cadherins in collective cell migration of mesenchymal cells. Curr. Opin. Cell Biol. 2012, 24, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Brieher, W.M.; Yap, A.S. Cadherin junctions and their cytoskeleton(s). Curr. Opin. Cell Biol. 2013, 25, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Lehembre, F.; Yilmaz, M.; Wicki, A.; Schomber, T.; Strittmatter, K.; Ziegler, D.; Kren, A.; Went, P.; Derksen, P.W.; Berns, A.; et al. NCAM-induced focal adhesion assembly: A functional switch upon loss of E-cadherin. EMBO J. 2008, 27, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Shih, W.; Yamada, S. N-cadherin-mediated cell–cell adhesion promotes cell migration in a three-dimensional matrix. J. Cell Sci. 2012, 125, 3661–3670. [Google Scholar] [CrossRef] [PubMed]
- Kotb, A.M.; Hierholzer, A.; Kemler, R. Replacement of E-cadherin by N-cadherin in the mammary gland leads to fibrocystic changes and tumor formation. Breast Cancer Res. 2011, 13, R104. [Google Scholar] [CrossRef] [PubMed]
- Oka, H.; Shiozaki, H.; Kobayashi, K.; Inoue, M.; Tahara, H.; Kobayashi, T.; Takatsuka, Y.; Matsuyoshi, N.; Hirano, S.; Takeichi, M.; et al. Expression of E-cadherin cell adhesion molecules in human breast cancer tissues and its relationship to metastasis. Cancer Res. 1993, 53, 1696–1701. [Google Scholar] [PubMed]
- Umbas, R.; Isaacs, W.B.; Bringuier, P.P.; Schaafsma, H.E.; Karthaus, H.F.; Oosterhof, G.O.; Debruyne, F.M.; Schalken, J.A. Decreased E-cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res. 1994, 54, 3929–3933. [Google Scholar] [PubMed]
- Schipper, J.H.; Frixen, U.H.; Behrens, J.; Unger, A.; Jahnke, K.; Birchmeier, W. E-cadherin expression in squamous cell carcinomas of head and neck: Inverse correlation with tumor dedifferentiation and lymph node metastasis. Cancer Res. 1991, 51, 6328–6237. [Google Scholar] [PubMed]
- Frixen, U.H.; Behrens, J.; Sachs, M.; Eberle, G.; Voss, B.; Warda, A.; Löchner, D.; Birchmeier, W. E-cadherin-mediated cell–cell adhesion prevents invasiveness of human carcinoma cells. J. Cell Biol. 1991, 113, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Herman, J.G.; Lapidus, R.G.; Chopra, H.; Xu, R.; Jarrard, D.F.; Isaacs, W.B.; Pitha, P.M.; Davidson, N.E.; Baylin, S.B. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995, 55, 5195–5199. [Google Scholar] [PubMed]
- Strathdee, G. Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin. Cancer Biol. 2002, 12, 373–379. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P.; Yamada, K.M.; Thiery, J.P. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J. Cell Biol. 1997, 137, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.J.; Moreno-Bueno, G.; Sarrio, D.; Locascio, A.; Cano, A.; Palacios, J.; Nieto, M.A. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 2002, 21, 3241–3246. [Google Scholar] [CrossRef] [PubMed]
- Fendrich, V.; Waldmann, J.; Feldmann, G.; Schlosser, K.; König, A.; Ramaswamy, A.; Bartsch, D.K.; Karakas, E. Unique expression pattern of the EMT markers Snail, Twist and E-cadherin in benign and malignant parathyroid neoplasia. Eur. J. Endocrinol. 2009, 160, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Satake, S.; Nakayama, F.; Matsumoto, M.; Ohnuma, K.; Komori, T.; Semba, S.; Ito, A.; Yokozaki, H. Snail-associated epithelial-mesenchymal transition promotes oesophageal squamous cell carcinoma motility and progression. J. Pathol. 2008, 215, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, H.J.; Jang, M.H.; Gwak, J.M.; Lee, K.S.; Kim, E.J.; Kim, H.J.; Lee, H.E.; Park, S.Y. Epithelial-mesenchymal transition increases during the progression of in situ to invasive basal-like breast cancer. Hum. Pathol. 2013, 44, 2581–2589. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.C.; Leite, C.F.; Cardoso, S.V.; Loyola, A.M.; Faria, P.R.; Souza, P.E.; Horta, M.C. Expression of epithelial-mesenchymal transition markers at the invasive front of oral squamous cell carcinoma. J. Appl. Oral. Sci. 2015, 23, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Wildenberg, G.A.; Dohn, M.R.; Carnahan, R.H.; Davis, M.A.; Lobdell, N.A.; Settleman, J.; Reynolds, A.B. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell 2006, 127, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.A.; Ireton, R.C.; Reynolds, A.B. A core function for p120-catenin in cadherin turnover. J. Cell Biol. 2003, 163, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Ngok, S.P.; Pulimeno, P.; Feathers, R.W.; Carpio, L.R.; Baker, T.R.; Carr, J.M.; Yan, I.K.; Borges, S.; Perez, E.A.; et al. Distinct E-cadherin-based complexes regulate cell behavior through miRNA processing or Src and p120 catenin activity. Nat. Cell Biol. 2015, 17, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.J.; Nusse, R. Convergence of Wnt, β-catenin, and cadherin pathways. Science 2004, 303, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lu, Z.; Hay, E.D. Direct evidence for a role of β-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol. Int. 2002, 26, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Mateus, A.R.; Seruca, R.; Machado, J.C.; Keller, G.; Oliveira, M.J.; Suriano, G.; Luber, B. EGFR regulates RhoA-GTP dependent cell motility in E-cadherin mutant cells. Hum. Mol. Genet. 2007, 16, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Laprise, P.; Langlois, M.J.; Boucher, M.J.; Jobin, C.; Rivard, N. Down-regulation of MEK/ERK signaling by E-cadherin-dependent PI3K/Akt pathway in differentiating intestinal epithelial cells. J. Cell Physiol. 2004, 199, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Shamir, E.R.; Pappalardo, E.; Jorgens, D.M.; Coutinho, K.; Tsai, W.T.; Aziz, K.; Auer, M.; Tran, P.T.; Bader, J.S.; Ewald, A.J. Twist1-induced dissemination preserves epithelial identity and requires E-cadherin. J. Cell Biol. 2014, 204, 839–856. [Google Scholar] [CrossRef] [PubMed]
- Soto, E.; Yanagisawa, M.; Marlow, L.A.; Copland, J.A.; Perez, E.A.; Anastasiadis, P.Z. p120 catenin induces opposing effects on tumor cell growth depending on E-cadherin expression. J. Cell Biol. 2008, 183, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Agiostratidou, G.; Li, M.; Suyama, K.; Badano, I.; Keren, R.; Chung, S.; Anzovino, A.; Hulit, J.; Qian, B.; Bouzahzah, B.; et al. Loss of retinal cadherin facilitates mammary tumor progression and metastasis. Cancer Res. 2009, 69, 5030–5038. [Google Scholar] [CrossRef] [PubMed]
- Matter, K.; Aijaz, S.; Tsapara, A.; Balda, M.S. Mammalian tight junctions in the regulation of epithelial differentiation and proliferation. Curr. Opin. Cell Biol. 2005, 17, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.J.; McCaffrey, L. Emerging role of cell polarity proteins in breast cancer progression and metastasis. Breast Cancer 2014, 6, 15–27. [Google Scholar] [PubMed]
- Ebnet, K.; Suzuki, A.; Horikoshi, Y.; Hirose, T.; Meyer Zu Brickwedde, M.K.; Ohno, S.; Vestweber, D. The cell polarity protein ASIP/PAR-3 directly associates with junctional adhesion molecule (JAM). EMBO J. 2001, 20, 3738–3748. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Anderson, J.M. Claudin interactions in and out of the tight junction. Tissue Barriers 2013, 1, e25247. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Capaldo, C.; Gumbiner, B.M.; Macara, I.G. The mammalian Scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J. Cell Biol. 2005, 171, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Bilder, D.; Perrimon, N. Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature 2000, 403, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Macara, I.G. Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat. Cell Biol. 2005, 7, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Hirata, M.; Kamimura, K.; Maniwa, R.; Yamanaka, T.; Mizuno, K.; Kishikawa, M.; Hirose, H.; Amano, Y.; Izumi, N.; et al. aPKC acts upstream of PAR-1b in both the establishment and maintenance of mammalian epithelial polarity. Curr. Biol. 2004, 14, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, J.; Umeda, K.; Tsukita, S.; Furuse, M.; Tsukita, S. Requirement of ZO-1 for the formation of belt-like adherens junctions during epithelial cell polarization. J. Cell Biol. 2007, 176, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Anderson, J.M. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann. N. Y. Acad. Sci. 2009, 1165, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.R.; Zhang, Y.; Ozdamar, B.; Ogunjimi, A.A.; Alexandrova, E.; Thomsen, G.H.; Wrana, J.L. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 2003, 302, 1775–1779. [Google Scholar] [CrossRef] [PubMed]
- Barrios-Rodiles, M.; Brown, K.R.; Ozdamar, B.; Bose, R.; Liu, Z.; Donovan, R.S.; Shinjo, F.; Liu, Y.; Dembowy, J.; Taylor, I.W.; et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science 2005, 307, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Danforth, D.N., Jr.; Sgagias, M.K. Tumor necrosis factor α enhances secretion of transforming growth factor β2 in MCF-7 breast cancer cells. Clin. Cancer Res. 1996, 2, 827–835. [Google Scholar] [PubMed]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Sasaki, H.; Fujimoto, K.; Tsukita, S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 1998, 143, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M. Overcoming barriers in the study of tight junction functions: From occludin to claudin. Genes Cells 1998, 3, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Anderson, J.M. Claudins and epithelial paracellular transport. Annu. Rev. Physiol. 2006, 68, 403–429. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, J.; Matsuda, M.; Furuse, M.; Tsukita, S. Regulation of tight junctions during the epithelium-mesenchyme transition: Direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci. 2003, 116, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Miwa, N.; Furuse, M.; Tsukita, S.; Niikawa, N.; Nakamura, Y.; Furukawa, Y. Involvement of claudin-1 in the β-catenin/Tcf signaling pathway and its frequent upregulation in human colorectal cancers. Oncol. Res. 2001, 12, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Singh, A.B.; Deane, N.G.; No, Y.; Shiou, S.R.; Schmidt, C.; Neff, J.; Washington, M.K.; Beauchamp, R.D. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J. Clin. Invest. 2005, 115, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Leotlela, P.D.; Wade, M.S.; Duray, P.H.; Rhode, M.J.; Brown, H.F.; Rosenthal, D.T.; Dissanayake, S.K.; Earley, R.; Indig, F.E.; Nickoloff, B.J.; et al. Claudin-1 overexpression in melanoma is regulated by PKC and contributes to melanoma cell motility. Oncogene 2007, 26, 3846–3856. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.C.; Pan, S.H.; Yang, S.C.; Yu, S.L.; Che, T.F.; Lin, C.W.; Tsai, M.S.; Chang, G.C.; Wu, C.H.; Wu, Y.Y.; et al. Claudin-1 is a metastasis suppressor and correlates with clinical outcome in lung adenocarcinoma. Am. J. Respir. Crit. Care Med. 2009, 179, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Moodie, A.; Blanchard, A.A.; Leygue, E.; Myal, Y. Claudin 1 in Breast Cancer: New Insights. J. Clin. Med. 2015, 4, 1960–1976. [Google Scholar] [CrossRef] [PubMed]
- Osanai, M.; Murata, M.; Chiba, H.; Kojima, T.; Sawada, N. Epigenetic silencing of claudin-6 promotes anchorage-independent growth of breast carcinoma cells. Cancer Sci. 2007, 98, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, S.L.; Argani, P.; Korz, D.; Evron, E.; Raman, V.; Garrett, E.; Rein, A.; Sauter, G.; Kallioniemi, O.P.; Sukumar, S. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 2003, 22, 2021–2033. [Google Scholar] [CrossRef] [PubMed]
- Philip, R.; Heiler, S.; Mu, W.; Buchler, M.W.; Zoller, M.; Thuma, F. Claudin-7 promotes the epithelial-mesenchymal transition in human colorectal cancer. Oncotarget 2015, 6, 2046–2063. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Harrison, G.M.; Watkins, G.; Jiang, W.G. Claudin-16 reduces the aggressive behavior of human breast cancer cells. J. Cell Biochem. 2008, 105, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Kimbung, S.; Kovacs, A.; Bendahl, P.O.; Malmstrom, P.; Ferno, M.; Hatschek, T.; Hedenfalk, I. Claudin-2 is an independent negative prognostic factor in breast cancer and specifically predicts early liver recurrences. Mol. Oncol. 2014, 8, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J. Cell Biol. 2001, 153, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Lanigan, F.; McKiernan, E.; Brennan, D.J.; Hegarty, S.; Millikan, R.C.; McBryan, J.; Jirstrom, K.; Landberg, G.; Martin, F.; Duffy, M.J.; et al. Increased claudin-4 expression is associated with poor prognosis and high tumour grade in breast cancer. Int. J. Cancer 2009, 124, 2088–2097. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, K.J.; Agarwal, R.; Morin, P.J. The claudin gene family: Expression in normal and neoplastic tissues. BMC Cancer 2006, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Soini, Y. Expression of claudins 1, 2, 3, 4, 5 and 7 in various types of tumours. Histopathology 2005, 46, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Escudero-Esparza, A.; Jiang, W.G.; Martin, T.A. Claudin-5 is involved in breast cancer cell motility through the N-WASP and ROCK signalling pathways. J. Exp. Clin. Cancer Res. 2012, 31, 43. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Mansel, R.E.; Jiang, W.G. Loss of occludin leads to the progression of human breast cancer. Int. J. Mol. Med. 2010, 26, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Osanai, M.; Murata, M.; Nishikiori, N.; Chiba, H.; Kojima, T.; Sawada, N. Epigenetic silencing of occludin promotes tumorigenic and metastatic properties of cancer cells via modulations of unique sets of apoptosis-associated genes. Cancer Res. 2006, 66, 9125–9133. [Google Scholar] [CrossRef] [PubMed]
- Beeman, N.; Webb, P.G.; Baumgartner, H.K. Occludin is required for apoptosis when claudin-claudin interactions are disrupted. Cell Death Dis. 2012, 3, e273. [Google Scholar] [CrossRef] [PubMed]
- Beeman, N.E.; Baumgartner, H.K.; Webb, P.G.; Schaack, J.B.; Neville, M.C. Disruption of occludin function in polarized epithelial cells activates the extrinsic pathway of apoptosis leading to cell extrusion without loss of transepithelial resistance. BMC Cell Biol. 2009, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Ninan, V.T.; Koshi, K.T.; Niyamthullah, M.M.; Jacob, C.K.; Gopalakrishnan, G.; Pandey, A.P.; Shastry, J.C. A comparative study of methods of estimating renal size in normal adults. Nephrol. Dial. Transpl. 1990, 5, 851–854. [Google Scholar] [CrossRef]
- Williams, C.S.; Zhang, B.; Smith, J.J.; Jayagopal, A.; Barrett, C.W.; Pino, C.; Russ, P.; Presley, S.H.; Peng, D.; Rosenblatt, D.O.; et al. BVES regulates EMT in human corneal and colon cancer cells and is silenced via promoter methylation in human colorectal carcinoma. J. Clin. Invest. 2011, 121, 4056–4069. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Nie, M.; Matter, K.; Balda, M.S. Rho signaling and tight junction functions. Physiology 2010, 25, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Tian, Y.; Zhang, W.; Wei, F.; Yang, J.; Luo, X.; Zhou, T.; Hou, B.; Qian, S.; Deng, X.; et al. Junctional adhesion molecule-A, an epithelial-mesenchymal transition inducer, correlates with metastasis and poor prognosis in human nasopharyngeal cancer. Carcinogenesis 2015, 36, 41–48. [Google Scholar] [CrossRef] [PubMed]
- McSherry, E.A.; McGee, S.F.; Jirstrom, K.; Doyle, E.M.; Brennan, D.J.; Landberg, G.; Dervan, P.A.; Hopkins, A.M.; Gallagher, W.M. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int. J. Cancer 2009, 125, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Gotte, M.; Mohr, C.; Koo, C.Y.; Stock, C.; Vaske, A.K.; Viola, M.; Ibrahim, S.A.; Peddibhotla, S.; Teng, Y.H.; Low, J.Y.; et al. miR-145-dependent targeting of junctional adhesion molecule A and modulation of fascin expression are associated with reduced breast cancer cell motility and invasiveness. Oncogene 2010, 29, 6569–6580. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.U.; Naik, T.U.; Suckow, A.T.; Duncan, M.K.; Naik, U.P. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res. 2008, 68, 2194–2203. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Garrett, M.D.; Matter, K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 2003, 160, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Huerta, M.; Munoz, R.; Tapia, R.; Soto-Reyes, E.; Ramirez, L.; Recillas-Targa, F.; González-Mariscal, L.; López-Bayghen, E. Cyclin D1 is transcriptionally down-regulated by ZO-2 via an E box and the transcription factor c-Myc. Mol. Biol. Cell 2007, 18, 4826–4836. [Google Scholar] [CrossRef] [PubMed]
- Tapia, R.; Huerta, M.; Islas, S.; Avila-Flores, A.; Lopez-Bayghen, E.; Weiske, J.; Huber, O.; González-Mariscal, L. Zona occludens-2 inhibits cyclin D1 expression and cell proliferation and exhibits changes in localization along the cell cycle. Mol. Biol. Cell 2009, 20, 1102–1117. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, A.; Tschernutter, M.; Bainbridge, J.W.; Balaggan, K.S.; Mowat, F.; West, E.L.; Munro, P.M.; Thrasher, A.J.; Matter, K.; Balda, M.S.; et al. The tight junction associated signalling proteins ZO-1 and ZONAB regulate retinal pigment epithelium homeostasis in mice. PLoS ONE 2010, 5, e15730. [Google Scholar] [CrossRef] [PubMed]
- Paschoud, S.; Jond, L.; Guerrera, D.; Citi, S. PLEKHA7 modulates epithelial tight junction barrier function. Tissue Barriers 2014, 2, e28755. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Bottinger, E.P. TGF-β and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. A specific inhibitor of TGF-β receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia 2005, 7, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Lv, J.; Ye, X.L.; Sun, M.Y.; Xu, Q.; Liu, C.H.; Min, L.H.; Li, H.P.; Liu, P.; Ding, X. Sorafenib inhibits transforming growth factor β1-mediated epithelial-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology 2011, 53, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kono, E.; Tran, C.P.; Miyazaki, H.; Yamashiro, J.; Shimomura, T.; Fazli, L.; Wada, R.; Huang, J.; Vessella, R.L.; et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 2010, 16, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Vyas, P.; Enver, T. Molecular targeting of cancer stem cells. Cell Stem. Cell 2009, 5, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ramena, G.; Elble, R.C. The role of cancer stem cells in relapse of solid tumors. Front. Biosci. 2012, 4, 1528–1541. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Struhl, K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc. Natl. Acad. Sci. USA 2013, 110, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhang, J.; Qian, X.; Han, L.; Zhang, K.; Chen, L.; Liu, J.; Ren, Y.; Yang, M.; Zhang, A.; et al. AC1MMYR2, an inhibitor of dicer-mediated biogenesis of Oncomir miR-21, reverses epithelial-mesenchymal transition and suppresses tumor growth and progression. Cancer Res. 2013, 73, 5519–5531. [Google Scholar] [CrossRef] [PubMed]
- Ramena, G.; Yin, Y.; Yu, Y.; Walia, V.; Elble, R.C. CLCA2 interactor EVA1 is required for mammary epithelial cell differentiation. PLoS ONE 2016, in press. [Google Scholar]



{kind=link}
{kind=link}
| Type of Claudin | Tumor Suppressor | Oncogene |
|---|---|---|
| Claudin-1 | Lung cancer [93] | Colon cancer [94,95] |
| - | Melanoma [96] | |
| Breast cancer [97] | Breast cancer [97] | |
| Claudin-2 | - | Breast cancer [98,99] |
| Claudin-3 | - | Breast cancer [100,101] |
| Claudin-4 | - | Breast cancer [100,101,102] |
| Claudin-5 | - | Breast cancer [102,103] |
| Claudin-6 | Breast cancer [104] | - |
| Claudin-7 | Breast cancer [105] | Colorectal cancer [106] |
| Claudin-16 | Breast cancer [107] | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Elble, R.C. Homeostatic Signaling by Cell–Cell Junctions and Its Dysregulation during Cancer Progression. J. Clin. Med. 2016, 5, 26. https://doi.org/10.3390/jcm5020026
Yu Y, Elble RC. Homeostatic Signaling by Cell–Cell Junctions and Its Dysregulation during Cancer Progression. Journal of Clinical Medicine. 2016; 5(2):26. https://doi.org/10.3390/jcm5020026
Chicago/Turabian StyleYu, Yang, and Randolph C. Elble. 2016. "Homeostatic Signaling by Cell–Cell Junctions and Its Dysregulation during Cancer Progression" Journal of Clinical Medicine 5, no. 2: 26. https://doi.org/10.3390/jcm5020026