
Pathophysiology of Early Brain Injury and Its Association with Delayed Cerebral Ischemia in Aneurysmal Subarachnoid Hemorrhage: A Review of Current Literature
 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Risk Factors Associated with aSAH and DCI
3. Pathophysiologic and Hemodynamic Factors Associated with aSAH
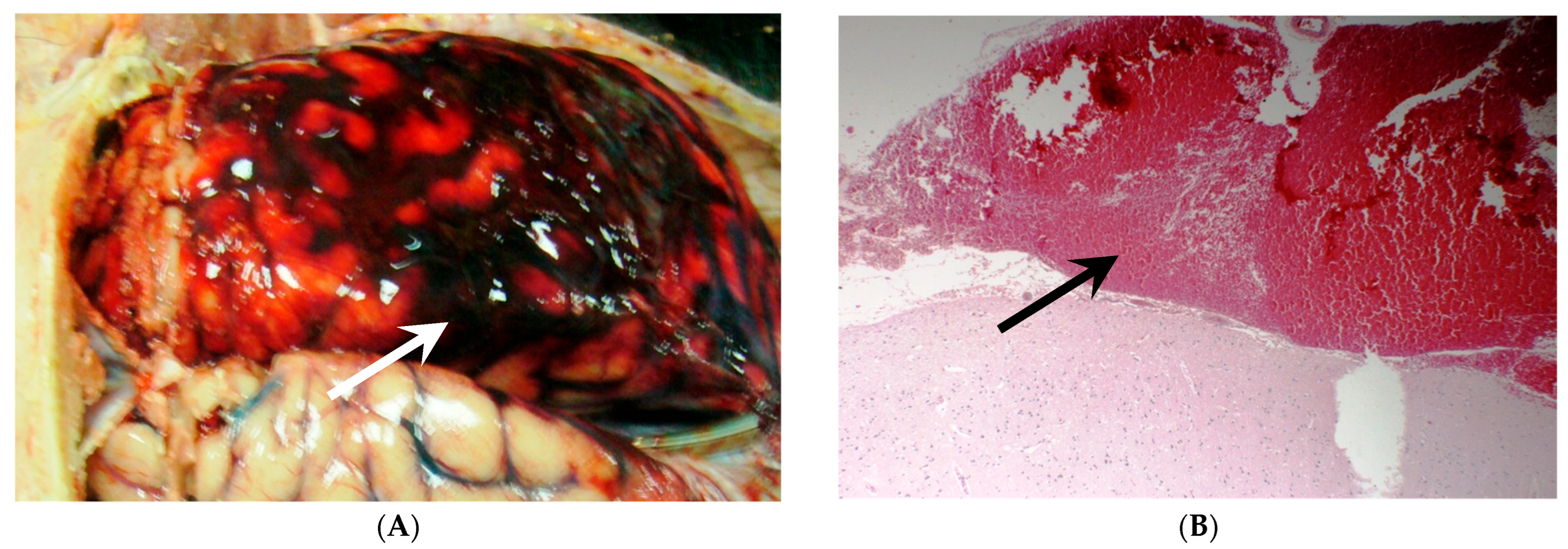
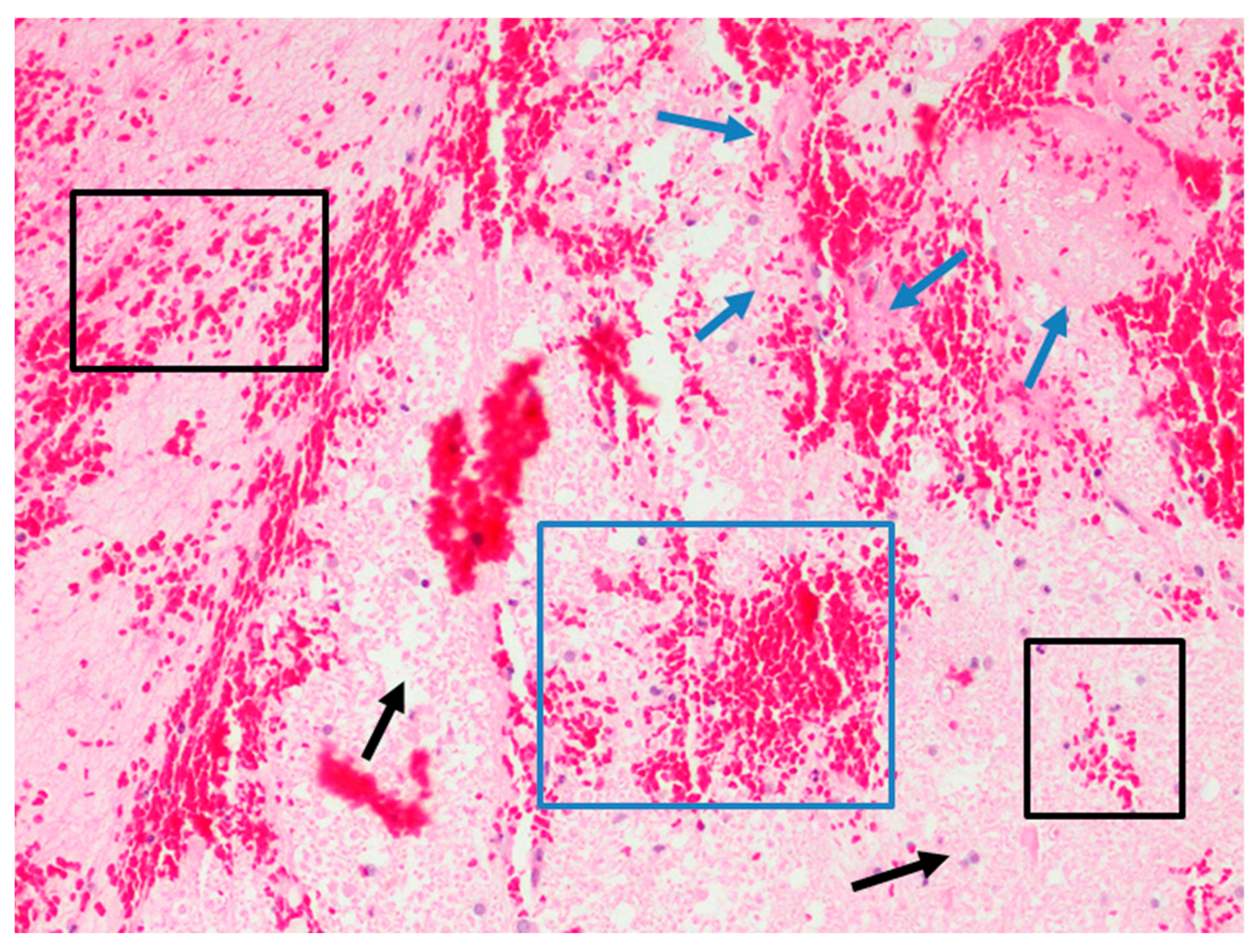
Early Brain Injury
4. Recent Developments
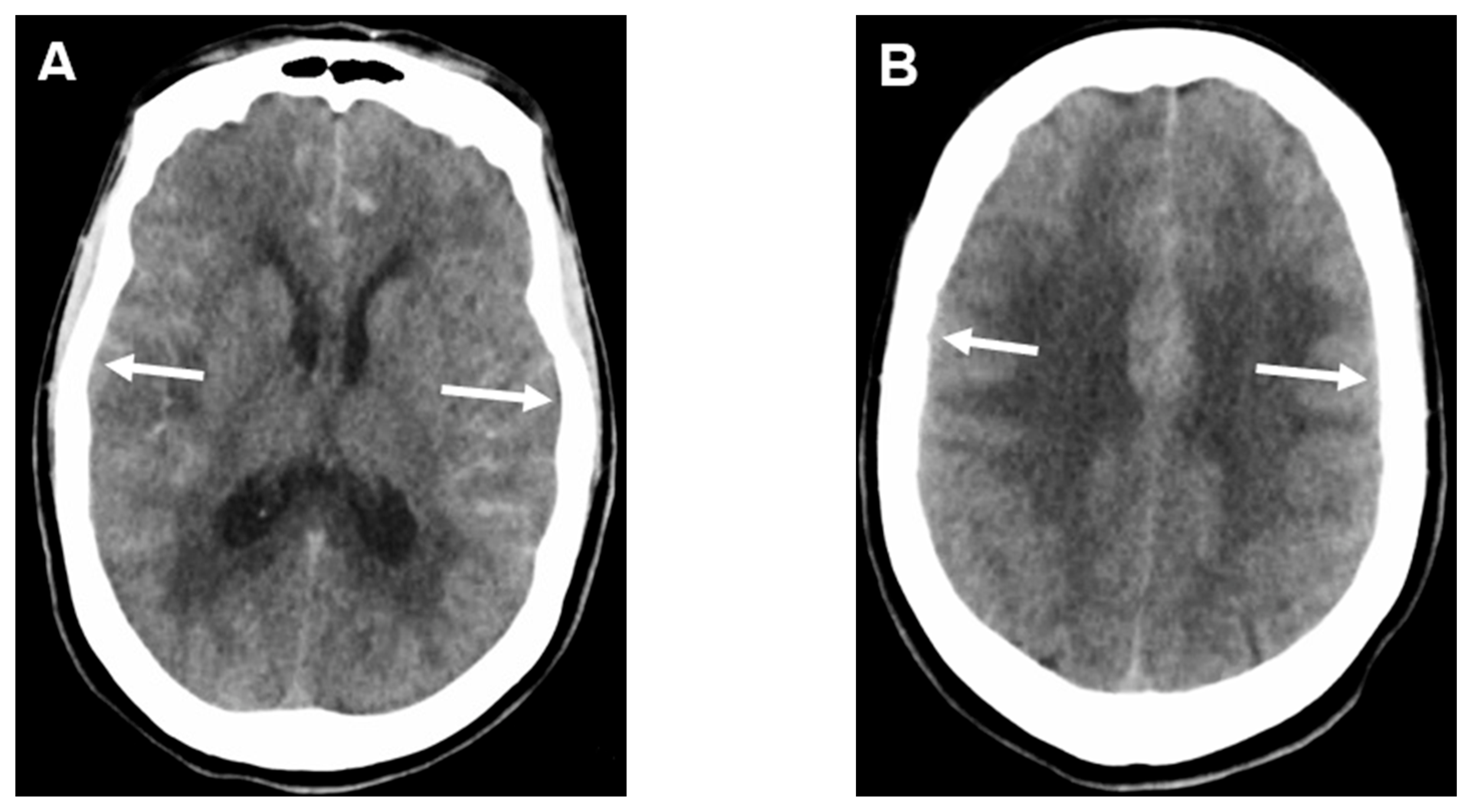
4.1. Subarachnoid Hemorrhage Early Brain Edema Score (SEBES)
4.2. Volumetric Analysis of Edema
4.3. Multimodal Monitoring, Predictive Models, and Diagnosis of Dci
5. Future Directions
Clinical and Risk Factor Assessment
6. Necessity for Ongoing Research
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aSAH | Aneurysmal subarachnoid hemorrhage |
| ADC | Apparent diffusion coefficient |
| APOE | Apolipoprotein E |
| CBF | Cerebral blood flow |
| CPP | Cerebral perfusion pressure |
| CT | Computed tomography |
| DCI | Delayed cerebral ischemia |
| EBI | Early brain injury |
| EEG | Electroencephalogram |
| eNOS | Endothelial nitric oxide synthase |
| EWAS | Epigenome wide association studies |
| FiO2 | Fraction of inspired oxygen |
| GCS | Glasgow Coma Scale |
| GWAS | genome wide association studies |
| Hp | Haptoglobin |
| ICP | Intracranial pressure |
| IL-6 | Interleukin 6 |
| MAP | Mean arterial pressure |
| MIF | Macrophage migratory inhibitory factor |
| MRI | Magnetic resonance imaging |
| NIHSS | National Institute of Health Stroke Scale |
| NIRS | Near-infrared spectroscopy |
| NOS | Nitric oxide synthase |
| Npi | Neurological Pupil index |
| NRG1 | Neuregulin 1 |
| OR | Odds ratio |
| PbtO2 | Brain tissue oxygenation |
| PRS | Polygenic risk scores |
| RYY-1 | Ryanodine-1 |
| SEBES | Subarachnoid hemorrhage early brain edema score |
| wPRS | Weighted PRS |
References
- Etminan, N.; Chang, H.S.; Hackenberg, K.; de Rooij, N.K.; Vergouwen, M.D.I.; Rinkel, G.J.E.; Algra, A. Worldwide Incidence of Aneurysmal Subarachnoid Hemorrhage According to Region, Time Period, Blood Pressure, and Smoking Prevalence in the Population: A Systematic Review and Meta-analysis. JAMA Neurol. 2019, 76, 588–597. [Google Scholar] [CrossRef]
- De Oliveira Manoel, A.L.; Mansur, A.; Silva, G.S.; Germans, M.R.; Jaja, B.N.; Kouzmina, E.; Marotta, T.R.; Abrahamson, S.; Schweizer, T.A.; Spears, J.; et al. Functional Outcome After Poor-Grade Subarachnoid Hemorrhage: A Single-Center Study and Systematic Literature Review. Neurocritical Care 2016, 25, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Nieuwkamp, D.J.; Setz, L.E.; Algra, A.; Linn, F.H.; de Rooij, N.K.; Rinkel, G.J. Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: A meta-analysis. Lancet Neurol. 2009, 8, 635–642. [Google Scholar] [CrossRef]
- Springer, M.V.; Schmidt, J.M.; Wartenberg, K.E.; Frontera, J.A.; Badjatia, N.; Mayer, S.A. Predictors of global cognitive impairment 1 year after subarachnoid hemorrhage. Neurosurgery 2009, 65, 1043–1050. [Google Scholar] [CrossRef]
- Geraghty, J.R.; Testai, F.D. Delayed Cerebral Ischemia after Subarachnoid Hemorrhage: Beyond Vasospasm and Towards a Multifactorial Pathophysiology. Curr. Atheroscler. Rep. 2017, 19, 50. [Google Scholar] [CrossRef]
- Adams, H.P., Jr.; Kassell, N.F.; Torner, J.C.; Haley, E.C., Jr. Predicting cerebral ischemia after aneurysmal subarachnoid hemorrhage: Influences of clinical condition, CT results, and antifibrinolytic therapy. A report of the Cooperative Aneurysm Study. Neurology 1987, 37, 1586–1591. [Google Scholar] [CrossRef]
- Frontera, J.A.; Claassen, J.; Schmidt, J.M.; Wartenberg, K.E.; Temes, R.; Connolly, E.S., Jr.; MacDonald, R.L.; Mayer, S.A. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: The modified fisher scale. Neurosurgery 2006, 59, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Francoeur, C.L.; Mayer, S.A. Management of delayed cerebral ischemia after subarachnoid hemorrhage. Crit. Care 2016, 20, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saripalli, M.; Tan, D.; Chandra, R.V.; Lai, L.T. Predictive Relevance of Early Temperature Elevation on the Risk of Delayed Cerebral Ischemia Development Following Aneurysmal Subarachnoid Hemorrhage. World Neurosurg. 2021, 150, e474–e481. [Google Scholar] [CrossRef] [PubMed]
- Rass, V.; Helbok, R. How to diagnose delayed cerebral ischaemia and symptomatic vasospasm and prevent cerebral infarction in patients with subarachnoid haemorrhage. Curr. Opin. Crit. Care 2021, 27, 103–114. [Google Scholar] [CrossRef]
- Ikram, A.; Javaid, M.A.; Ortega-Gutierrez, S.; Selim, M.; Kelangi, S.; Anwar, S.M.H.; Torbey, M.T.; Divani, A.A. Delayed Cerebral Ischemia after Subarachnoid Hemorrhage. J. Stroke Cerebrovasc. Dis. 2021, 30, 106064. [Google Scholar] [CrossRef]
- Schmidt, T.P.; Weiss, M.; Hoellig, A.; Nikoubashman, O.; Schulze-Steinen, H.; Albanna, W.; Clusmann, H.; Schubert, G.A.; Veldeman, M. Revisiting the Timeline of Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage: Toward a Temporal Risk Profile. Neurocritical Care 2022, 37, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Rosengart, A.J.; Schultheiss, K.E.; Tolentino, J.; Macdonald, R.L. Prognostic factors for outcome in patients with aneurysmal subarachnoid hemorrhage. Stroke J. Cereb. Circ. 2007, 38, 2315–2321. [Google Scholar] [CrossRef] [Green Version]
- Vergouwen, M.D.; Vermeulen, M.; van Gijn, J.; Rinkel, G.J.; Wijdicks, E.F.; Muizelaar, J.P.; Mendelow, A.D.; Juvela, S.; Yonas, H.; Terbrugge, K.G.; et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: Proposal of a multidisciplinary research group. Stroke J. Cereb. Circ. 2010, 41, 2391–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raatikainen, E.; Vahtera, A.; Kuitunen, A.; Junttila, E.; Huhtala, H.; Ronkainen, A.; Pyysalo, L.; Kiiski, H. Prognostic value of the 2010 consensus definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J. Neurol. Sci. 2021, 420, 117261. [Google Scholar] [CrossRef]
- Etminan, N.; Vergouwen, M.D.; Macdonald, R.L. Angiographic vasospasm versus cerebral infarction as outcome measures after aneurysmal subarachnoid hemorrhage. Acta Neurochir. Suppl. 2013, 115, 33–40. [Google Scholar] [CrossRef]
- Foreman, B. The Pathophysiology of Delayed Cerebral Ischemia. J. Clin. Neurophysiol. 2016, 33, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Higashida, R.T.; Keller, E.; Mayer, S.A.; Molyneux, A.; Raabe, A.; Vajkoczy, P.; Wanke, I.; Bach, D.; Frey, A.; et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: A randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 2011, 10, 618–625. [Google Scholar] [CrossRef]
- Ya, X.; Zhang, C.; Zhang, S.; Zhang, Q.; Cao, Y.; Wang, S.; Zhao, J. The Relationship Between Smoking and Delayed Cerebral Ischemia After Intracranial Aneurysm Rupture: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 625087. [Google Scholar] [CrossRef]
- Duan, W.; Pan, Y.; Wang, C.; Wang, Y.; Zhao, X.; Wang, Y.; Liu, L.; CNSR Investigators. Risk Factors and Clinical Impact of Delayed Cerebral Ischemia after Aneurysmal Subarachnoid Hemorrhage: Analysis from the China National Stroke Registry. Neuroepidemiology 2018, 50, 128–136. [Google Scholar] [CrossRef]
- Crobeddu, E.; Mittal, M.K.; Dupont, S.; Wijdicks, E.F.; Lanzino, G.; Rabinstein, A.A. Predicting the lack of development of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Stroke J. Cereb. Circ. 2012, 43, 697–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, M.K.; Ruigrok, Y.M. Genetics of Intracranial Aneurysms. Stroke J. Cereb. Circ. 2021, 52, 3004–3012. [Google Scholar] [CrossRef]
- Gaastra, B.; Alexander, S.; Bakker, M.K.; Bhagat, H.; Bijlenga, P.; Blackburn, S.; Collins, M.K.; Dore, S.; Griessenauer, C.; Hendrix, P.; et al. Genome-Wide Association Study of Clinical Outcome After Aneurysmal Subarachnoid Haemorrhage: Protocol. Transl. Stroke Res. 2022, 13, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Rinkel, G.J. Intracranial aneurysm screening: Indications and advice for practice. Lancet Neurol. 2005, 4, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Korja, M.; Silventoinen, K.; McCarron, P.; Zdravkovic, S.; Skytthe, A.; Haapanen, A.; de Faire, U.; Pedersen, N.L.; Christensen, K.; Koskenvuo, M.; et al. Genetic epidemiology of spontaneous subarachnoid hemorrhage: Nordic Twin Study. Stroke J. Cereb. Circ. 2010, 41, 2458–2462. [Google Scholar] [CrossRef] [Green Version]
- Bakker, M.K.; van der Spek, R.A.A.; van Rheenen, W.; Morel, S.; Bourcier, R.; Hostettler, I.C.; Alg, V.S.; van Eijk, K.R.; Koido, M.; Akiyama, M.; et al. Genome-wide association study of intracranial aneurysms identifies 17 risk loci and genetic overlap with clinical risk factors. Nat. Genet. 2020, 52, 1303–1313. [Google Scholar] [CrossRef]
- Theodotou, C.B.; Snelling, B.M.; Sur, S.; Haussen, D.C.; Peterson, E.C.; Elhammady, M.S. Genetic associations of intracranial aneurysm formation and sub-arachnoid hemorrhage. Asian J. Neurosurg. 2017, 12, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Solodovnikova, Y.; Ivaniuk, A.; Marusich, T.; Son, A. Meta-analysis of associations of genetic polymorphisms with cerebral vasospasm and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Acta Neurol. Belg. 2021, 122, 1547–1556. [Google Scholar] [CrossRef]
- Gaastra, B.; Ren, D.; Alexander, S.; Bennett, E.R.; Bielawski, D.M.; Blackburn, S.L.; Borsody, M.K.; Dore, S.; Galea, J.; Garland, P.; et al. Haptoglobin genotype and aneurysmal subarachnoid hemorrhage: Individual patient data analysis. Neurology 2019, 92, e2150–e2164. [Google Scholar] [CrossRef] [Green Version]
- Gaastra, B.; Glazier, J.; Bulters, D.; Galea, I. Haptoglobin Genotype and Outcome after Subarachnoid Haemorrhage: New Insights from a Meta-Analysis. Oxid. Med. Cell. Longev. 2017, 2017, 6747940. [Google Scholar] [CrossRef] [Green Version]
- McInnes, M.D.; Bossuyt, P.M. Pitfalls of Systematic Reviews and Meta-Analyses in Imaging Research. Radiology 2015, 277, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Devaux, Y.; Audibert, G.; Zhang, L.; Bracard, S.; Colnat-Coulbois, S.; Klein, O.; Zannad, F.; Charpentier, C.; Longrois, D.; et al. Gene expression profile of blood cells for the prediction of delayed cerebral ischemia after intracranial aneurysm rupture: A pilot study in humans. Cerebrovasc. Dis. 2013, 36, 236–242. [Google Scholar] [CrossRef]
- Kim, B.J.; Kim, Y.; Youn, D.H.; Park, J.J.; Rhim, J.K.; Kim, H.C.; Kang, K.; Jeon, J.P. Genome-wide blood DNA methylation analysis in patients with delayed cerebral ischemia after subarachnoid hemorrhage. Sci. Rep. 2020, 10, 11419. [Google Scholar] [CrossRef] [PubMed]
- Heinsberg, L.W.; Weeks, D.E.; Alexander, S.A.; Minster, R.L.; Sherwood, P.R.; Poloyac, S.M.; Deslouches, S.; Crago, E.A.; Conley, Y.P. Iron homeostasis pathway DNA methylation trajectories reveal a role for STEAP3 metalloreductase in patient outcomes after aneurysmal subarachnoid hemorrhage. Epigenetics Commun. 2021, 1, 4. [Google Scholar] [CrossRef]
- Guttmacher, A.E.; Porteous, M.E.; McInerney, J.D. Educating health-care professionals about genetics and genomics. Nat. Rev. Genet. 2007, 8, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar] [CrossRef]
- Slunecka, J.L.; van der Zee, M.D.; Beck, J.J.; Johnson, B.N.; Finnicum, C.T.; Pool, R.; Hottenga, J.J.; de Geus, E.J.C.; Ehli, E.A. Implementation and implications for polygenic risk scores in healthcare. Hum. Genom. 2021, 15, 46. [Google Scholar] [CrossRef]
- Hong, E.P.; Youn, D.H.; Kim, B.J.; Lee, J.J.; Na, D.; Ahn, J.H.; Park, J.J.; Rhim, J.K.; Kim, H.C.; Jeon, H.J.; et al. Genome-wide polygenic risk impact on intracranial aneurysms and acute ischemic stroke. PLoS ONE 2022, 17, e0265581. [Google Scholar] [CrossRef]
- Sehba, F.A.; Pluta, R.M.; Macdonald, R.L. Brain injury after transient global cerebral ischemia and subarachnoid hemorrhage. Stroke Res. Treat. 2013, 2013, 827154. [Google Scholar] [CrossRef] [Green Version]
- Weimer, J.M.; Jones, S.E.; Frontera, J.A. Acute Cytotoxic and Vasogenic Edema after Subarachnoid Hemorrhage: A Quantitative MRI Study. AJNR Am. J. Neuroradiol. 2017, 38, 928–934. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, Y.; Uchikawa, H.; Kajiwara, S.; Morioka, M. Central sympathetic nerve activation in subarachnoid hemorrhage. J. Neurochem. 2022, 160, 34–50. [Google Scholar] [CrossRef]
- Crompton, M.R. Hypothalamic lesions following the rupture of cerebral berry aneurysms. Brain 1963, 86, 301–314. [Google Scholar] [CrossRef]
- Lee, S.J.; Jang, S.H. Hypothalamic injury in spontaneous subarachnoid hemorrhage: A diffusion tensor imaging study. Clin. Auton. Res. 2021, 31, 321–322. [Google Scholar] [CrossRef]
- Naredi, S.; Lambert, G.; Eden, E.; Zall, S.; Runnerstam, M.; Rydenhag, B.; Friberg, P. Increased sympathetic nervous activity in patients with nontraumatic subarachnoid hemorrhage. Stroke J. Cereb. Circ. 2000, 31, 901–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, Y.; Hasegawa, Y.; Hayashi, K.; Cao, C.; Hamasaki, T.; Kawano, T.; Mukasa, A.; Kim-Mitsuyama, S. The Stabilization of Central Sympathetic Nerve Activation by Renal Denervation Prevents Cerebral Vasospasm after Subarachnoid Hemorrhage in Rats. Transl. Stroke Res. 2020, 11, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, F.; Kanamaru, H.; Asada, R.; Suzuki, Y.; Nampei, M.; Nakajima, H.; Oinaka, H.; Suzuki, H. Roles of glutamate in brain injuries after subarachnoid hemorrhage. Histol. Histopathol. 2022, 37, 1041–1051. [Google Scholar] [CrossRef]
- Wan, W.H.; Ang, B.T.; Wang, E. The Cushing Response: A case for a review of its role as a physiological reflex. J. Clin. Neurosci. 2008, 15, 223–228. [Google Scholar] [CrossRef]
- Schmidt, E.A.; Czosnyka, Z.; Momjian, S.; Czosnyka, M.; Bech, R.A.; Pickard, J.D. Intracranial baroreflex yielding an early cushing response in human. Acta Neurochir. Suppl. 2005, 95, 253–256. [Google Scholar] [CrossRef]
- Chen, S.; Li, Q.; Wu, H.; Krafft, P.R.; Wang, Z.; Zhang, J.H. The harmful effects of subarachnoid hemorrhage on extracerebral organs. Biomed. Res. Int. 2014, 2014, 858496. [Google Scholar] [CrossRef] [Green Version]
- Meyfroidt, G.; Baguley, I.J.; Menon, D.K. Paroxysmal sympathetic hyperactivity: The storm after acute brain injury. Lancet Neurol. 2017, 16, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Osgood, M.L. Aneurysmal Subarachnoid Hemorrhage: Review of the Pathophysiology and Management Strategies. Curr. Neurol. Neurosci. Rep. 2021, 21, 50. [Google Scholar] [CrossRef] [PubMed]
- Calandra, T.; Bucala, R. Macrophage Migration Inhibitory Factor (MIF): A Glucocorticoid Counter-Regulator within the Immune System. Crit. Rev. Immunol. 2017, 37, 359–370. [Google Scholar] [CrossRef]
- Chen, Y.H.; Cheng, Z.Y.; Shao, L.H.; Shentu, H.S.; Fu, B. Macrophage migration inhibitory factor as a serum prognostic marker in patients with aneurysmal subarachnoid hemorrhage. Clin. Chim. Acta 2017, 473, 60–64. [Google Scholar] [CrossRef]
- Koda, M.; Nishio, Y.; Hashimoto, M.; Kamada, T.; Koshizuka, S.; Yoshinaga, K.; Onodera, S.; Nishihira, J.; Moriya, H.; Yamazaki, M. Up-regulation of macrophage migration-inhibitory factor expression after compression-induced spinal cord injury in rats. Acta Neuropathol. 2004, 108, 31–36. [Google Scholar] [CrossRef]
- Su, Y.; Wang, Y.; Zhou, Y.; Zhu, Z.; Zhang, Q.; Zhang, X.; Wang, W.; Gu, X.; Guo, A.; Wang, Y. Macrophage migration inhibitory factor activates inflammatory responses of astrocytes through interaction with CD74 receptor. Oncotarget 2017, 8, 2719–2730. [Google Scholar] [CrossRef] [Green Version]
- Inacio, A.R.; Ruscher, K.; Leng, L.; Bucala, R.; Deierborg, T. Macrophage migration inhibitory factor promotes cell death and aggravates neurologic deficits after experimental stroke. J. Cereb. Blood Flow Metab. 2011, 31, 1093–1106. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.S.; Chen, W.; Liu, S.; Zhang, Y.Y.; Li, X.H. Serum macrophage migration inhibitory factor levels are associated with infarct volumes and long-term outcomes in patients with acute ischemic stroke. Int. J. Neurosci. 2017, 127, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.B.; Yu, W.H.; Dong, X.Q.; Zhang, Z.Y.; Du, Q.; Zhu, Q.; Che, Z.H.; Wang, H.; Shen, Y.F.; Jiang, L. Serum macrophage migration inhibitory factor concentrations correlate with prognosis of traumatic brain injury. Clin. Chim. Acta 2017, 469, 99–104. [Google Scholar] [CrossRef]
- Lin, Q.; Cai, J.Y.; Lu, C.; Sun, J.; Ba, H.J.; Chen, M.H.; Chen, X.D.; Dai, J.X.; Lin, J.H. Macrophage migration inhibitory factor levels in serum from patients with acute intracerebral hemorrhage: Potential contribution to prognosis. Clin. Chim. Acta 2017, 472, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Peng, J.; Pang, J.; Wan, W.; Zhong, C.; Peng, T.; Bao, K.; Jiang, Y. The Association Between Serum Macrophage Migration Inhibitory Factor and Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. Neurotox. Res. 2020, 37, 397–405. [Google Scholar] [CrossRef]
- Hayman, E.G.; Wessell, A.; Gerzanich, V.; Sheth, K.N.; Simard, J.M. Mechanisms of Global Cerebral Edema Formation in Aneurysmal Subarachnoid Hemorrhage. Neurocritical Care 2017, 26, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Dreier, J.P.; Lemale, C.L.; Kola, V.; Friedman, A.; Schoknecht, K. Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology 2018, 134, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Wartenberg, K.E.; Sheth, S.J.; Michael Schmidt, J.; Frontera, J.A.; Rincon, F.; Ostapkovich, N.; Fernandez, L.; Badjatia, N.; Sander Connolly, E.; Khandji, A.; et al. Acute ischemic injury on diffusion-weighted magnetic resonance imaging after poor grade subarachnoid hemorrhage. Neurocritical Care 2011, 14, 407–415. [Google Scholar] [CrossRef]
- Ahn, S.H.; Savarraj, J.P.; Pervez, M.; Jones, W.; Park, J.; Jeon, S.B.; Kwon, S.U.; Chang, T.R.; Lee, K.; Kim, D.H.; et al. The Subarachnoid Hemorrhage Early Brain Edema Score Predicts Delayed Cerebral Ischemia and Clinical Outcomes. Neurosurgery 2018, 83, 137–145. [Google Scholar] [CrossRef]
- Sehba, F.A.; Hou, J.; Pluta, R.M.; Zhang, J.H. The importance of early brain injury after subarachnoid hemorrhage. Prog. Neurobiol. 2012, 97, 14–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R.M.; Hansen-Schwartz, J.; Dreier, J.; Vajkoczy, P.; Macdonald, R.L.; Nishizawa, S.; Kasuya, H.; Wellman, G.; Keller, E.; Zauner, A.; et al. Cerebral vasospasm following subarachnoid hemorrhage: Time for a new world of thought. Neurol. Res. 2009, 31, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Frontera, J.A.; Provencio, J.J.; Sehba, F.A.; McIntyre, T.M.; Nowacki, A.S.; Gordon, E.; Weimer, J.M.; Aledort, L. The Role of Platelet Activation and Inflammation in Early Brain Injury Following Subarachnoid Hemorrhage. Neurocritical Care 2017, 26, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Winkler, M.K.L.; Major, S.; Horst, V.; Lublinsky, S.; Kola, V.; Lemale, C.L.; Kang, E.J.; Maslarova, A.; Salur, I.; et al. Spreading depolarizations in ischaemia after subarachnoid haemorrhage, a diagnostic phase III study. Brain 2022, 145, 1264–1284. [Google Scholar] [CrossRef]
- Owen, B.; Vangala, A.; Fritch, C.; Alsarah, A.A.; Jones, T.; Davis, H.; Shuttleworth, C.W.; Carlson, A.P. Cerebral Autoregulation Correlation with Outcomes and Spreading Depolarization in Aneurysmal Subarachnoid Hemorrhage. Stroke J. Cereb. Circ. 2022, 53, 1975–1983. [Google Scholar] [CrossRef]
- Carlson, A.P.; Abbas, M.; Alunday, R.L.; Qeadan, F.; Shuttleworth, C.W. Spreading depolarization in acute brain injury inhibited by ketamine: A prospective, randomized, multiple crossover trial. J. Neurosurg. 2018, 130, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Reinhart, K.M.; Humphrey, A.; Brennan, K.C.; Carlson, A.P.; Shuttleworth, C.W. Memantine Improves Recovery After Spreading Depolarization in Brain Slices and can be Considered for Future Clinical Trials. Neurocritical Care 2021, 35, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Barber, P.A.; Demchuk, A.M.; Zhang, J.; Buchan, A.M. Validity and reliability of a quantitative computed tomography score in predicting outcome of hyperacute stroke before thrombolytic therapy. ASPECTS Study Group. Alberta Stroke Programme Early CT Score. Lancet 2000, 355, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Said, M.; Gumus, M.; Herten, A.; Dinger, T.F.; Chihi, M.; Darkwah Oppong, M.; Deuschl, C.; Wrede, K.H.; Kleinschnitz, C.; Sure, U.; et al. Subarachnoid Hemorrhage Early Brain Edema Score (SEBES) as a radiographic marker of clinically relevant intracranial hypertension and unfavorable outcome after subarachnoid hemorrhage. Eur. J. Neurol. 2021, 28, 4051–4059. [Google Scholar] [CrossRef] [PubMed]
- Rass, V.; Ianosi, B.A.; Wegmann, A.; Gaasch, M.; Schiefecker, A.J.; Kofler, M.; Lindner, A.; Addis, A.; Almashad, S.S.; Rhomberg, P.; et al. Delayed Resolution of Cerebral Edema Is Associated with Poor Outcome After Nontraumatic Subarachnoid Hemorrhage. Stroke J. Cereb. Circ. 2019, 50, 828–836. [Google Scholar] [CrossRef]
- Yuan, J.Y.; Chen, Y.; Kumar, A.; Zlepper, Z.; Jayaraman, K.; Aung, W.Y.; Clarke, J.V.; Allen, M.; Athiraman, U.; Osbun, J.; et al. Automated Quantification of Reduced Sulcal Volume Identifies Early Brain Injury After Aneurysmal Subarachnoid Hemorrhage. Stroke J. Cereb. Circ. 2021, 52, 1380–1389. [Google Scholar] [CrossRef]
- Veldeman, M.; Albanna, W.; Weiss, M.; Conzen, C.; Schmidt, T.P.; Schulze-Steinen, H.; Wiesmann, M.; Clusmann, H.; Schubert, G.A. Invasive neuromonitoring with an extended definition of delayed cerebral ischemia is associated with improved outcome after poor-grade subarachnoid hemorrhage. J. Neurosurg. 2020, 134, 1527–1534. [Google Scholar] [CrossRef]
- Veldeman, M.; Albanna, W.; Weiss, M.; Park, S.; Hoellig, A.; Clusmann, H.; Helbok, R.; Temel, Y.; Alexander Schubert, G. Invasive Multimodal Neuromonitoring in Aneurysmal Subarachnoid Hemorrhage: A Systematic Review. Stroke J. Cereb. Circ. 2021, 52, 3624–3632. [Google Scholar] [CrossRef]
- Lazaridis, C. Brain Shock-Toward Pathophysiologic Phenotyping in Traumatic Brain Injury. Crit. Care Explor. 2022, 4, e0724. [Google Scholar] [CrossRef]
- Zahra, K.; Gopal, N.; Freeman, W.D.; Turnbull, M.T. Using Cerebral Metabolites to Guide Precision Medicine for Subarachnoid Hemorrhage: Lactate and Pyruvate. Metabolites 2019, 9, 245. [Google Scholar] [CrossRef] [Green Version]
- Hosmann, A.; Schnackenburg, P.; Rauscher, S.; Hopf, A.; Bohl, I.; Engel, A.; Brugger, J.; Graf, A.; Plochl, W.; Reinprecht, A.; et al. Brain Tissue Oxygen Response as Indicator for Cerebral Lactate Levels in Aneurysmal Subarachnoid Hemorrhage Patients. J. Neurosurg. Anesthesiol. 2022, 34, 193–200. [Google Scholar] [CrossRef]
- Megjhani, M.; Weiss, M.; Ford, J.; Terilli, K.; Kastenholz, N.; Nametz, D.; Kwon, S.B.; Velazquez, A.; Agarwal, S.; Roh, D.J.; et al. Optimal Cerebral Perfusion Pressure and Brain Tissue Oxygen in Aneurysmal Subarachnoid Hemorrhage. Stroke J. Cereb. Circ. 2023, 54, 189–197. [Google Scholar] [CrossRef]
- De Courson, H.; Proust-Lima, C.; Tuaz, E.; Georges, D.; Verchere, E.; Biais, M. Relationship Between Brain Tissue Oxygen and Near-Infrared Spectroscopy in Patients with Nontraumatic Subarachnoid Hemorrhage. Neurocritical Care 2022, 37, 620–628. [Google Scholar] [CrossRef]
- Aoun, S.G.; Stutzman, S.E.; Vo, P.N.; El Ahmadieh, T.Y.; Osman, M.; Neeley, O.; Plitt, A.; Caruso, J.P.; Aiyagari, V.; Atem, F.; et al. Detection of delayed cerebral ischemia using objective pupillometry in patients with aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2019, 132, 27–32. [Google Scholar] [CrossRef]
- Yu, Z.; Wen, D.; Zheng, J.; Guo, R.; Li, H.; You, C.; Ma, L. Predictive Accuracy of Alpha-Delta Ratio on Quantitative Electroencephalography for Delayed Cerebral Ischemia in Patients with Aneurysmal Subarachnoid Hemorrhage: Meta-Analysis. World Neurosurg. 2019, 126, e510–e516. [Google Scholar] [CrossRef]
- Baang, H.Y.; Chen, H.Y.; Herman, A.L.; Gilmore, E.J.; Hirsch, L.J.; Sheth, K.N.; Petersen, N.H.; Zafar, S.F.; Rosenthal, E.S.; Westover, M.B.; et al. The Utility of Quantitative EEG in Detecting Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. J. Clin. Neurophysiol. 2022, 39, 207–215. [Google Scholar] [CrossRef]
- Rosenthal, E.S.; Biswal, S.; Zafar, S.F.; O’Connor, K.L.; Bechek, S.; Shenoy, A.V.; Boyle, E.J.; Shafi, M.M.; Gilmore, E.J.; Foreman, B.P.; et al. Continuous electroencephalography predicts delayed cerebral ischemia after subarachnoid hemorrhage: A prospective study of diagnostic accuracy. Ann. Neurol. 2018, 83, 958–969. [Google Scholar] [CrossRef]
- Cremers, C.H.; van der Schaaf, I.C.; Wensink, E.; Greving, J.P.; Rinkel, G.J.; Velthuis, B.K.; Vergouwen, M.D. CT perfusion and delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage: A systematic review and meta-analysis. J. Cereb. Blood Flow Metab. 2014, 34, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Mir, D.I.; Gupta, A.; Dunning, A.; Puchi, L.; Robinson, C.L.; Epstein, H.A.; Sanelli, P.C. CT perfusion for detection of delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage: A systematic review and meta-analysis. AJNR Am. J. Neuroradiol. 2014, 35, 866–871. [Google Scholar] [CrossRef] [Green Version]
- Cremers, C.H.; Vos, P.C.; van der Schaaf, I.C.; Velthuis, B.K.; Vergouwen, M.D.; Rinkel, G.J.; Dankbaar, J.W. CT perfusion during delayed cerebral ischemia after subarachnoid hemorrhage: Distinction between reversible ischemia and ischemia progressing to infarction. Neuroradiology 2015, 57, 897–902. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, E.D.; Gobin, Y.P.; Riina, H.; Johnson, C.E.; Tsiouris, A.J.; Comunale, J.; Sanelli, P.C. Role of CT perfusion imaging in the diagnosis and treatment of vasospasm. Imaging Med. 2011, 3, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Taran, S.; Mandell, D.M.; McCredie, V.A. CT Perfusion for the Detection of Delayed Cerebral Ischemia in the Presence of Neurologic Confounders. Neurocritical Care 2020, 33, 317–322. [Google Scholar] [CrossRef]
- Shi, D.; Jin, D.; Cai, W.; Zhu, Q.; Dou, X.; Fan, G.; Shen, J.; Xu, L. Serial low-dose quantitative CT perfusion for the evaluation of delayed cerebral ischaemia following aneurysmal subarachnoid haemorrhage. Clin. Radiol. 2020, 75, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Zhou, Y.; Wang, M.; Yang, C.; Yuan, Q.; Fang, X. Whole-brain CT perfusion on admission predicts delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage. Eur. J. Radiol. 2019, 116, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.W.; Prater, A.; Kallas, O.; Abidi, S.A.; Howard, B.M.; Tong, F.; Agarwal, S.; Yaghi, S.; Dehkharghani, S. Diagnostic Performance of Computed Tomography Angiography and Computed Tomography Perfusion Tissue Time-to-Maximum in Vasospasm Following Aneurysmal Subarachnoid Hemorrhage. J. Am. Heart. Assoc. 2022, 11, e023828. [Google Scholar] [CrossRef] [PubMed]
- Rajajee, V. Grading scales in subarachnoid hemorrhage—Many options, but do we have a winner? Eur. J. Neurol. 2018, 25, 207–208. [Google Scholar] [CrossRef]
- De Oliveira Manoel, A.L.; Jaja, B.N.; Germans, M.R.; Yan, H.; Qian, W.; Kouzmina, E.; Marotta, T.R.; Turkel-Parrella, D.; Schweizer, T.A.; Macdonald, R.L.; et al. The VASOGRADE: A Simple Grading Scale for Prediction of Delayed Cerebral Ischemia After Subarachnoid Hemorrhage. Stroke J. Cereb. Circ. 2015, 46, 1826–1831. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.H.; Ouyang, B.; John, S.; Conners, J.J.; Garg, R.; Bleck, T.P.; Temes, R.E.; Cutting, S.; Prabhakaran, S. Risk stratification for the in-hospital mortality in subarachnoid hemorrhage: The HAIR score. Neurocritical Care 2014, 21, 14–19. [Google Scholar] [CrossRef]
- Cahill, J.; Calvert, J.W.; Zhang, J.H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2006, 26, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Xu, Q.; Li, A. Nomogram for predicting delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage in the Chinese population. J. Stroke Cerebrovasc. Dis. 2020, 29, 105005. [Google Scholar] [CrossRef]
- Megjhani, M.; Terilli, K.; Weiss, M.; Savarraj, J.; Chen, L.H.; Alkhachroum, A.; Roh, D.J.; Agarwal, S.; Connolly, E.S., Jr.; Velazquez, A.; et al. Dynamic Detection of Delayed Cerebral Ischemia: A Study in 3 Centers. Stroke J. Cereb. Circ. 2021, 52, 1370–1379. [Google Scholar] [CrossRef]
- Ray, B.; Pandav, V.M.; Mathews, E.A.; Thompson, D.M.; Ford, L.; Yearout, L.K.; Bohnstedt, B.N.; Chaudhary, S.; Dale, G.L.; Prodan, C.I. Coated-Platelet Trends Predict Short-Term Clinical OutcomeAfter Subarachnoid Hemorrhage. Transl. Stroke Res. 2018, 9, 459–470. [Google Scholar] [CrossRef]
- Ramos, L.A.; van der Steen, W.E.; Sales Barros, R.; Majoie, C.; van den Berg, R.; Verbaan, D.; Vandertop, W.P.; Zijlstra, I.; Zwinderman, A.H.; Strijkers, G.J.; et al. Machine learning improves prediction of delayed cerebral ischemia in patients with subarachnoid hemorrhage. J. Neurointerventional Surg. 2019, 11, 497–502. [Google Scholar] [CrossRef]
- Savarraj, J.P.J.; Hergenroeder, G.W.; Zhu, L.; Chang, T.; Park, S.; Megjhani, M.; Vahidy, F.S.; Zhao, Z.; Kitagawa, R.S.; Choi, H.A. Machine Learning to Predict Delayed Cerebral Ischemia and Outcomes in Subarachnoid Hemorrhage. Neurology 2021, 96, e553–e562. [Google Scholar] [CrossRef]
- Goursaud, S.; de Lizarrondo, S.M.; Grolleau, F.; Chagnot, A.; Agin, V.; Maubert, E.; Gauberti, M.; Vivien, D.; Ali, C.; Gakuba, C. Delayed Cerebral Ischemia After Subarachnoid Hemorrhage: Is There a Relevant Experimental Model? A Systematic Review of Preclinical Literature. Front. Cardiovasc. Med. 2021, 8, 752769. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clinical Deterioration Due to DCI | Occurrence of focal neurological impairment (such as hemiparesis, aphasia, apraxia, hemianopia, or neglect), or a decrease of at least 2 points on the GCS (either on the total score or on one of its individual components [eye, motor on either side, verbal]). This should last for at least 1 h, is not apparent immediately after aneurysm occlusion, and cannot be attributed to other causes by means of clinical assessment, CT or MRI scanning of the brain, and appropriate laboratory studies. |
| Cerebral Infarction Due to DCI | Presence of cerebral infarction on CT or MR scan of the brain within 6 weeks after SAH, or on the latest CT or MR scan made before death within 6 weeks, or proven at autopsy, not present on the CT or MR scan between 24 and 48 h after early aneurysm occlusion, and not attributable to other causes such as surgical clipping or endovascular treatment. Hypodensities on CT imaging resulting from ventricular catheter or intraparenchymal hematoma should not be regarded as cerebral infarctions from DCI. |
| Neurophysiological Parameters | Subtypes and Notes |
|---|---|
| Cerebral Autoregulation |
|
| Cerebral Blood Flow |
|
| Cerebral Oxygenation |
|
| Intracranial Pressure |
|
| Electroencephalography (EEG) |
|
| Pupillometry |
|
| Cerebral Microdialysis |
|
| Pathophysiologic Type | Neuromonitoring Result | Underlying Pathophysiology and Management |
|---|---|---|
| Flow Dependent | ↓: PbtO2, glucose, pyruvate ↑: lactate, LPR | Suboptimal CBF → optimize hemodynamic parameters and CPP |
|
Flow Independent Oxygen Diffusion Limitation | ↓: PbtO2 ↑: lactate, LPR | Intracellular/interstitial edema → appropriately manage cerebral edema |
|
Flow Independent Energy Production (Mitochondrial) Failure | ↓: lactate, LPR, possibly pyruvate ↑: glucose | Management is unclear |
|
Flow Independent Microvascular Shunting | ↓: PbtO2 (from ↑CBF) ↑: glucose, lactate | Microvascular shunting → appropriately manage ICP |
| Low Extraction | ↓: PbtO2, pyruvate ↑: lactate, LPR | Hypoxemic, anemic or high-affinity hypoxia → treat appropriate underlying cause to improve oxygenation |
| Hypermetabolic | ↓: PbtO2, glucose, pyruvate ↑: lactate, LPR Similar profile to flow dependent | Increase in metabolic demand → Avoid hyperthermia, seizures, CSD; consider sedation if appropriate |
| Score | Purpose | Score components | Findings |
|---|---|---|---|
| VASOGRADE (N = 746) | DCI risk stratification | Green: mF 1–2 AND WFNS 1–2 Yellow: mF 3–4 AND WFNS 1–3 Red: WFNS 4–5 regardless of mF grade | Yellow: tendency to DCI compared to Green Red: 3-fold increased risk of DCI compared to Green |
| HAIR (N = 400–score development) (N = 302 –score validation) | In-hospital mortality risk stratification | Score 0–8 total HH: 1–3 = 0 pts; 4 = 1 pt; 5 = 4 pts Age: <60 = 0 pts; 60–80 = 1 pt; ≥80 = 2 pts IVH: No = 0 pts; Yes = 1 pt Re-bleed (within 24hrs): No = 0 pts; Yes = 1 pt | Increase in HAIR score increase in rate of in-hospital mortality |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsbrook, D.L.; Di Napoli, M.; Bhatia, K.; Desai, M.; Hinduja, A.; Rubinos, C.A.; Mansueto, G.; Singh, P.; Domeniconi, G.G.; Ikram, A.; et al. Pathophysiology of Early Brain Injury and Its Association with Delayed Cerebral Ischemia in Aneurysmal Subarachnoid Hemorrhage: A Review of Current Literature. J. Clin. Med. 2023, 12, 1015. https://doi.org/10.3390/jcm12031015
Alsbrook DL, Di Napoli M, Bhatia K, Desai M, Hinduja A, Rubinos CA, Mansueto G, Singh P, Domeniconi GG, Ikram A, et al. Pathophysiology of Early Brain Injury and Its Association with Delayed Cerebral Ischemia in Aneurysmal Subarachnoid Hemorrhage: A Review of Current Literature. Journal of Clinical Medicine. 2023; 12(3):1015. https://doi.org/10.3390/jcm12031015
Chicago/Turabian StyleAlsbrook, Diana L., Mario Di Napoli, Kunal Bhatia, Masoom Desai, Archana Hinduja, Clio A. Rubinos, Gelsomina Mansueto, Puneetpal Singh, Gustavo G. Domeniconi, Asad Ikram, and et al. 2023. "Pathophysiology of Early Brain Injury and Its Association with Delayed Cerebral Ischemia in Aneurysmal Subarachnoid Hemorrhage: A Review of Current Literature" Journal of Clinical Medicine 12, no. 3: 1015. https://doi.org/10.3390/jcm12031015