
Autoinflammatory Features in Gouty Arthritis




Abstract
:1. Introduction
2. Clinical Challenges and Differential Diagnosis
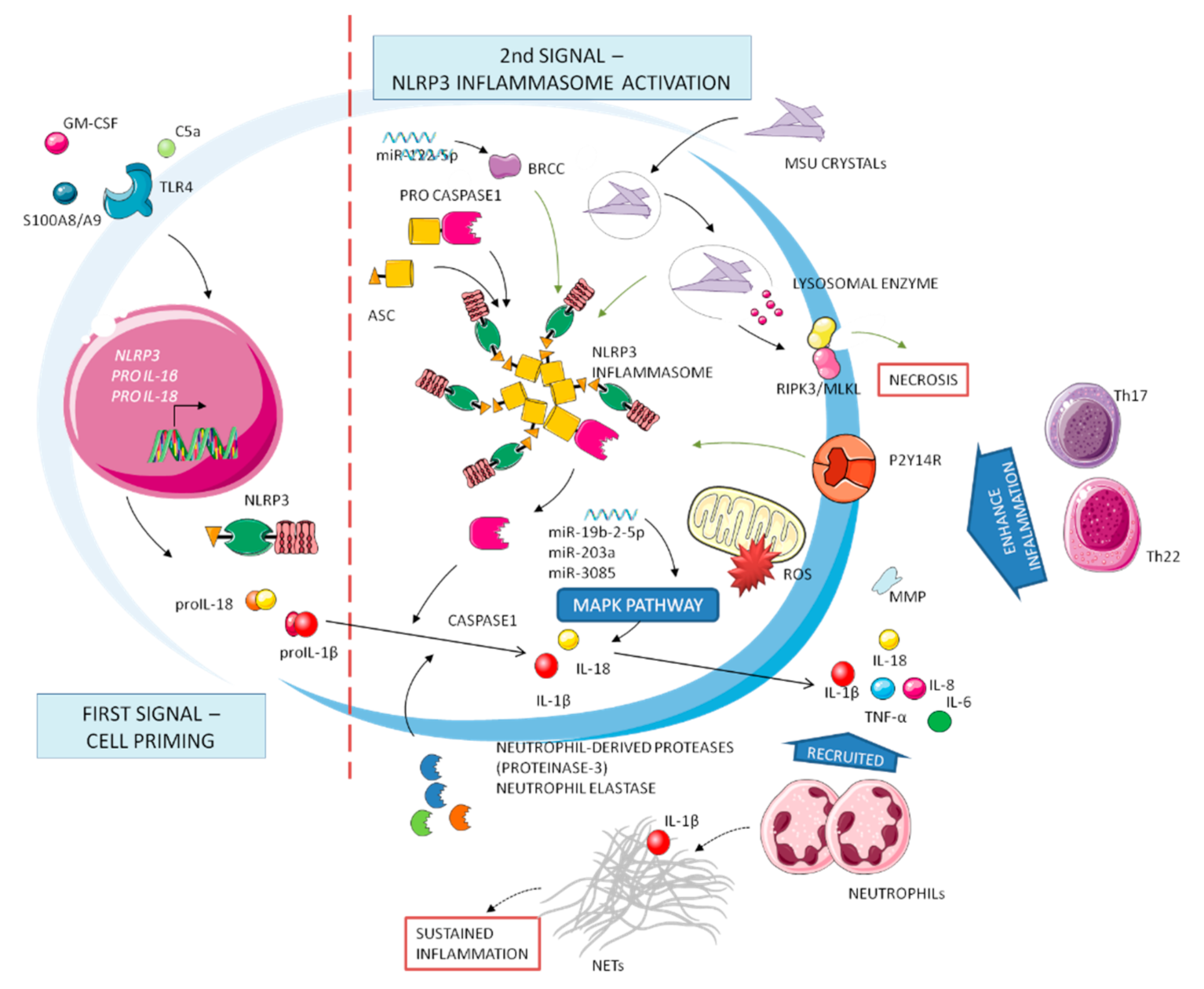
3. Molecular Mechanisms of Gouty Inflammation
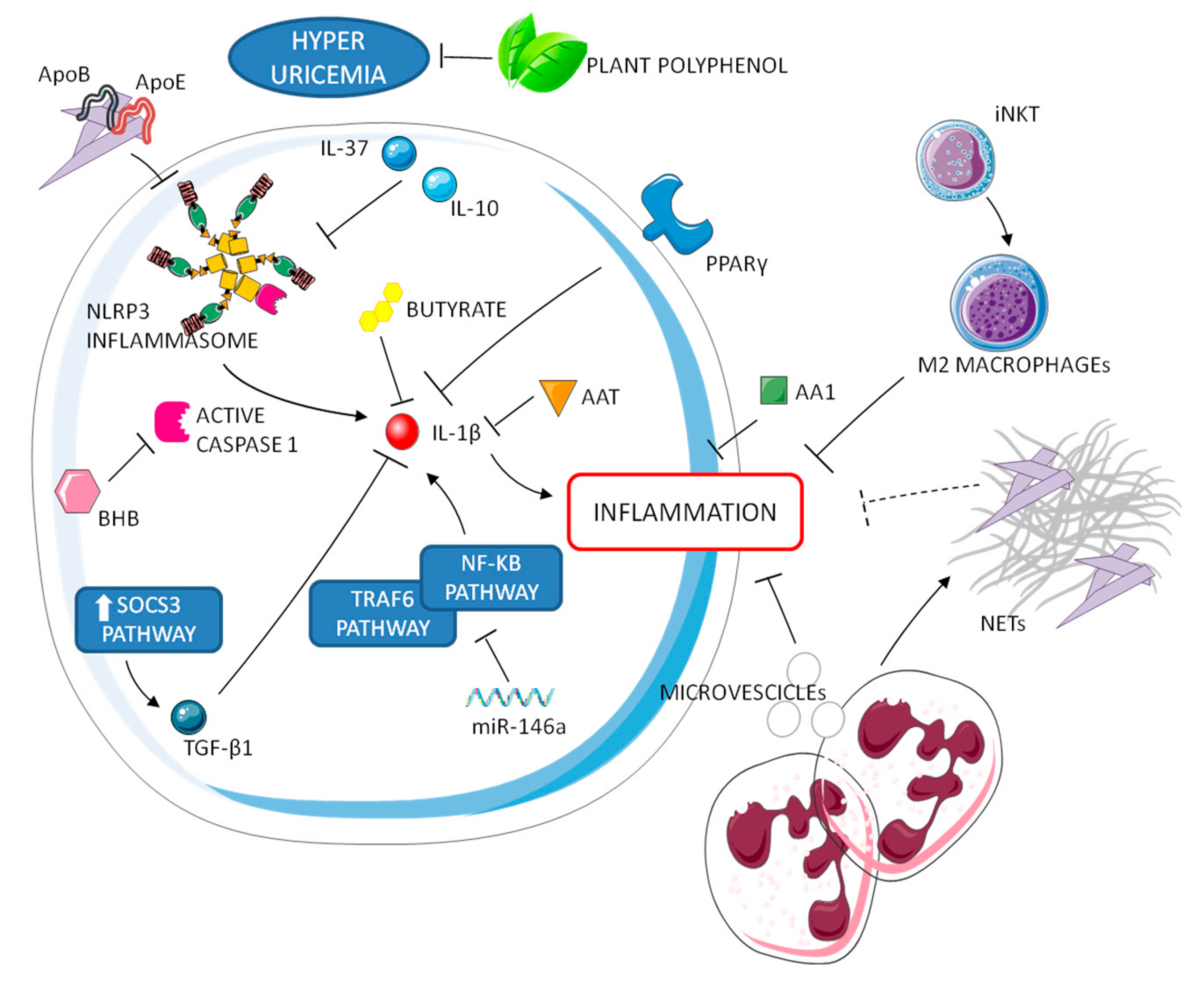
4. Resolution of Gouty Inflammation
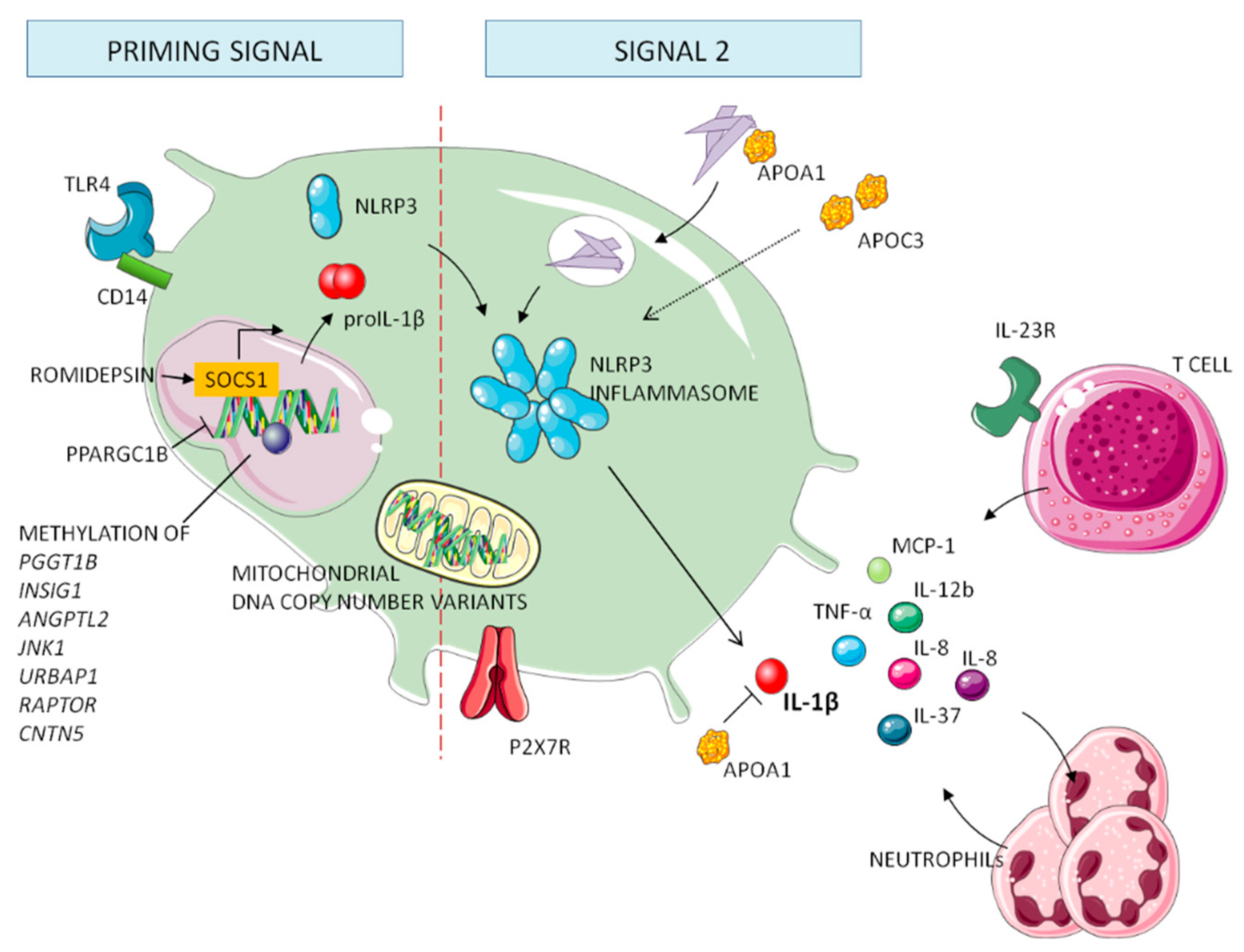
5. Genetics of Gout
5.1. Genes Involved in Processing NLRP3 Inflammasome
5.2. Genes Involved in the Downstream Cascade of NLRP3 Inflammasome
5.3. Mitochondrial and Epigenetic Factors in Gout
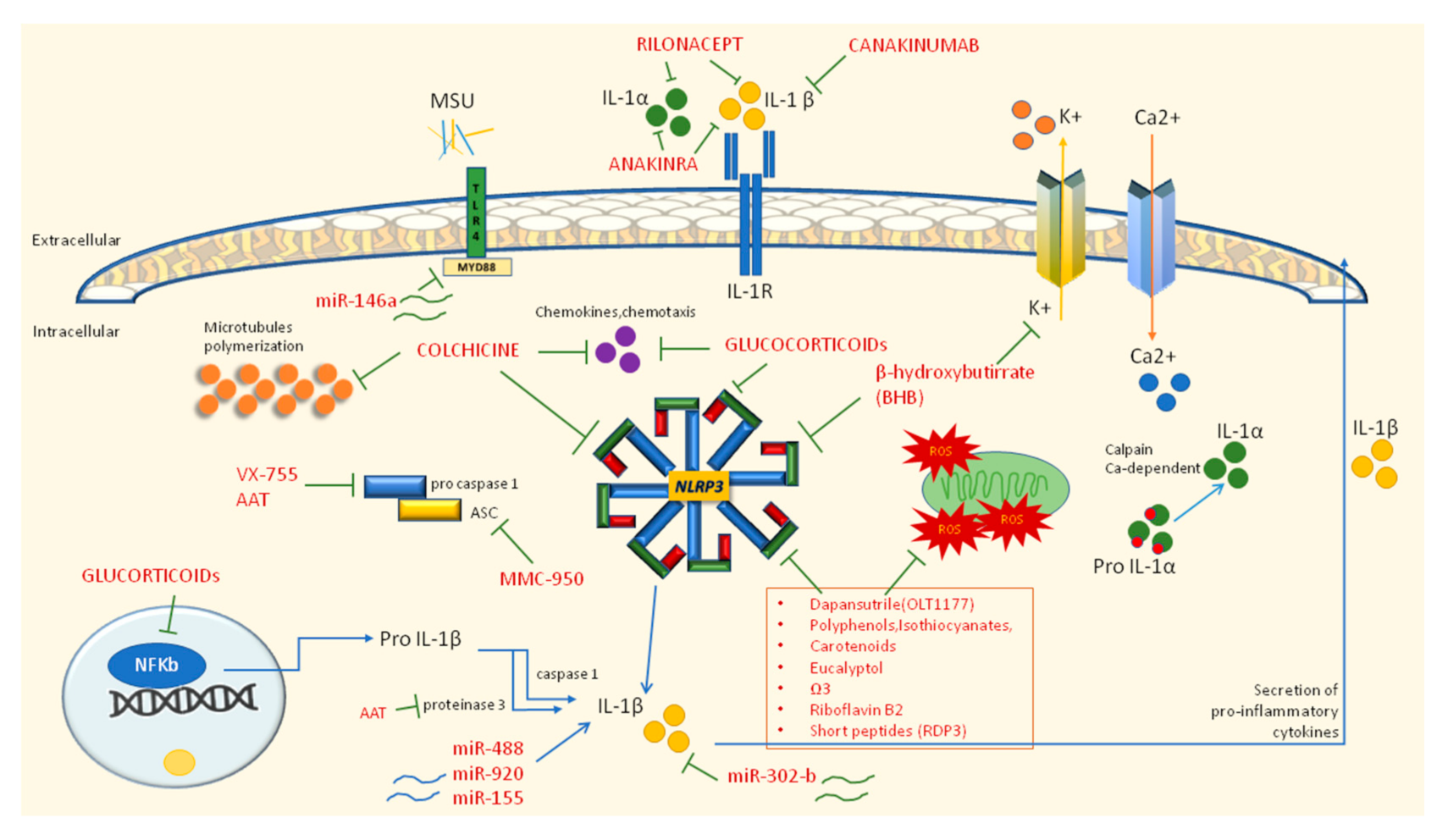
6. Therapeutic Approaches
6.1. First Line Therapy: NSAIDs, Colchicine, and Glucocorticoids
6.2. Second Line Therapy: IL-1 Inhibitors
6.3. Novel Therapies Modulating Inflammatory Pathways
7. Future Perspectives
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kastner, D.L. Autoinflammation: Past, Present, and Future. In Textbook of Autoinflammation; Hashkes, P.J., Laxer, R.M., Simon, A., Eds.; Springer International Publishing: New York City, NY, USA, 2019; Volume 1, pp. 3–15. [Google Scholar]
- McGonagle, D.; McDermott, M.F. A proposed classification of the immunological diseases. PLoS Med. 2006, 3, e297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punzi, L.; Scanu, A.; Galozzi, P.; Luisetto, R.; Spinella, P.; Scirè, C.A.; Oliviero, F. One year in review 2020: Gout. Clin. Exp. Rheumatol. 2020, 38, 807–821. [Google Scholar] [PubMed]
- Dalbeth, N.; Phipps-Green, A.; Frampton, C.; Neogi, T.; Taylor, W.J.; Merriman, T.R. Relationship between serum urate concentration and clinically evident incident gout: An individual participant data analysis. Ann. Rheum. Dis. 2018, 77, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Steiger, S.; Harper, J.L. Mechanisms of spontaneous resolution of acute gouty inflammation. Curr. Rheumatol. Rep. 2014, 16, 392. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Merriman, T.R.; Stamp, L.K. Gout. Lancet 2016, 388, 2039–2052. [Google Scholar] [CrossRef]
- Oliviero, F.; Bindoli, S.; Scanu, A.; Feist, E.; Doria, A.; Galozzi, P.; Sfriso, P. Autoinflammatory Mechanisms in Crystal-Induced Arthritis. Front. Med. 2020, 7. [Google Scholar] [CrossRef]
- Bruun, T.; Rath, E.; Oppegaard, O.; Skrede, S. Beta-Hemolytic Streptococci and Necrotizing Soft Tissue Infections. Adv. Exp. Med. Biol. 2020, 1294, 73–86. [Google Scholar] [CrossRef]
- Shah, D.; Mohan, G.; Flueckiger, P.; Corrigan, F.; Conn, D. Polyarticular Gout Flare Masquerading as Sepsis. Am. J. Med. 2015, 128, e11–e12. [Google Scholar] [CrossRef] [Green Version]
- Goh, C.L.; Lai, F.Y.X.; Chee, A.; Junckerstorff, R. Back pain and fever: When the diagnosis becomes crystal clear. Intern. Med. J. 2018, 48, 480–481. [Google Scholar] [CrossRef]
- Busso, N.; So, A. Mechanisms of inflammation in gout. Arthritis Res. Ther. 2010, 12, 206. [Google Scholar] [CrossRef] [Green Version]
- So, A.K.; Martinon, F. Inflammation in gout: Mechanisms and therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 639–647. [Google Scholar] [CrossRef]
- Terkeltaub, R. What makes gouty inflammation so variable? BMC Med. 2017, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Joosten, L.A.; Netea, M.G.; Mylona, E.; Koenders, M.I.; Malireddi, R.K.; Oosting, M.; Stienstra, R.; van de Veerdonk, F.L.; Stalenhoef, A.F.; Giamarellos-Bourboulis, E.J.; et al. Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1β production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum. 2010, 62, 3237–3248. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Dalbeth, N.; Haskard, D.O. Mechanisms of inflammation in gout. Rheumatology 2005, 44, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Jiang, W.; Ye, S.; Zhou, M.; Liu, C.; Yang, X.; Hao, K.; Hu, Q. P2Y (14) receptor has a critical role in acute gouty arthritis by regulating pyroptosis of macrophages. Cell Death Dis. 2020, 11, 394. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Szamosi, S.; Kovács, G.E.; Kocsis, E.; Benkő, S. The NLRP3 inflammasome—Interleukin 1 pathway as a therapeutic target in gout. Arch. Biochem. Biophys. 2019, 670, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Netea, M.G.; Fantuzzi, G.; Koenders, M.I.; Helsen, M.M.; Sparrer, H.; Pham, C.T.; van der Meer, J.W.; Dinarello, C.A.; van den Berg, W.B. Inflammatory arthritis in caspase 1 gene-deficient mice: Contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 2009, 60, 3651–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulay, S.R.; Desai, J.; Kumar, S.V.; Eberhard, J.N.; Thomasova, D.; Romoli, S.; Grigorescu, M.; Kulkarni, O.P.; Popper, B.; Vielhauer, V.; et al. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat. Commun. 2016, 7, 10274. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wu, H.; Wang, D.; Yang, Z.; Dong, J. LncRNA ANRIL promotes NLRP3 inflammasome activation in uric acid nephropathy through miR-122-5p/BRCC3 axis. Biochimie 2019, 157, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.T.; Leng, Y.R.; Liu, M.M.; Dong, R.F.; Bian, J.; Yuan, L.L.; Zhang, J.G.; Xia, Y.Z.; Kong, L.Y. MicroRNA and long noncoding RNA involvement in gout and prospects for treatment. Int. Immunopharmacol. 2020, 87, 106842. [Google Scholar] [CrossRef]
- Wang, B.; Chen, S.; Qian, H.; Zheng, Q.; Chen, R.; Liu, Y.; Shi, G. Role of T cells in the pathogenesis and treatment of gout. Int. Immunopharmacol. 2020, 88, 106877. [Google Scholar] [CrossRef] [PubMed]
- Klück, V.; Liu, R.; Joosten, L.A.B. The role of interleukin-1 family members in hyperuricemia and gout. Jt. Bone Spine 2020, 88, 105092. [Google Scholar] [CrossRef]
- Wan, W.; Shi, Y.; Ji, L.; Li, X.; Xu, X.; Zhao, D. Interleukin-37 contributes to the pathogenesis of gout by affecting PDZ domain-containing 1 protein through the nuclear factor-kappa B pathway. J. Int. Med. Res. 2020, 48, 300060520948717. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.; Zeng, C. Update on the epidemiology, genetics, and therapeutic options of hyperuricemia. Am. J. Transl. Res. 2020, 12, 3167–3181. [Google Scholar] [PubMed]
- Stewart, S.; Tallon, A.; Taylor, W.J.; Gaffo, A.; Dalbeth, N. How flare prevention outcomes are reported in gout studies: A systematic review and content analysis of randomized controlled trials. Semin. Arthritis Rheum. 2020, 50, 303–313. [Google Scholar] [CrossRef]
- Desai, J.; Steiger, S.; Anders, H.J. Molecular Pathophysiology of Gout. Trends Mol. Med. 2017, 23, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.M.; Ruscetti, F.W.; Palaszynski, E.W.; Falk, L.A.; Oppenheim, J.J.; Keller, J.R. Transforming growth factor beta is a potent inhibitor of interleukin 1 (IL-1) receptor expression: Proposed mechanism of inhibition of IL-1 action. J. Exp. Med. 1990, 172, 737–744. [Google Scholar] [CrossRef]
- Chen, Y.H.; Hsieh, S.C.; Chen, W.Y.; Li, K.J.; Wu, C.H.; Wu, P.C.; Tsai, C.Y.; Yu, C.L. Spontaneous resolution of acute gouty arthritis is associated with rapid induction of the anti-inflammatory factors TGFβ1, IL-10 and soluble TNF receptors and the intracellular cytokine negative regulators CIS and SOCS3. Ann. Rheum. Dis. 2011, 70, 1655–1663. [Google Scholar] [CrossRef]
- Scanu, A.; Oliviero, F.; Gruaz, L.; Sfriso, P.; Pozzuoli, A.; Frezzato, F.; Agostini, C.; Burger, D.; Punzi, L. High-density lipoproteins downregulate CCL2 production in human fibroblast-like synoviocytes stimulated by urate crystals. Arthritis Res. Ther. 2010, 12, R23. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Bravo, E.; Sieck, M.S.; Schumacher, H.R., Jr. Changes in the proteins coating monosodium urate crystals during active and subsiding inflammation. Immunogold studies of synovial fluid from patients with gout and of fluid obtained using the rat subcutaneous air pouch model. Arthritis Rheum. 1993, 36, 1274–1285. [Google Scholar] [CrossRef]
- Akahoshi, T.; Namai, R.; Murakami, Y.; Watanabe, M.; Matsui, T.; Nishimura, A.; Kitasato, H.; Kameya, T.; Kondo, H. Rapid induction of peroxisome proliferator-activated receptor gamma expression in human monocytes by monosodium urate monohydrate crystals. Arthritis Rheum. 2003, 48, 231–239. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourcet, B.; Duez, H. Circadian Control of Inflammasome Pathways: Implications for Circadian Medicine. Front. Immunol. 2020, 11, 1630. [Google Scholar] [CrossRef] [PubMed]
- Galvão, I.; Vago, J.P.; Barroso, L.C.; Tavares, L.P.; Queiroz-Junior, C.M.; Costa, V.V.; Carneiro, F.S.; Ferreira, T.P.; Silva, P.M.; Amaral, F.A.; et al. Annexin A1 promotes timely resolution of inflammation in murine gout. Eur. J. Immunol. 2017, 47, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Pool, B.; Shaw, O.M.; Harper, J.L.; Tan, P.; Franklin, C.; House, M.E.; Cornish, J.; Naot, D. Role of miR-146a in regulation of the acute inflammatory response to monosodium urate crystals. Ann. Rheum. Dis. 2015, 74, 786–790. [Google Scholar] [CrossRef]
- Oliviero, F.; Scanu, A. How Factors Involved in the Resolution of Crystal-Induced Inflammation Target IL-1β. Front. Pharmacol. 2017, 8, 164. [Google Scholar] [CrossRef] [Green Version]
- Oliviero, F.; Scanu, A.; Zamudio-Cuevas, Y.; Punzi, L.; Spinella, P. Anti-inflammatory effects of polyphenols in arthritis. J. Sci. Food Agric. 2018, 98, 1653–1659. [Google Scholar] [CrossRef]
- Mitroulis, I.; Kambas, K.; Chrysanthopoulou, A.; Skendros, P.; Apostolidou, E.; Kourtzelis, I.; Drosos, G.I.; Boumpas, D.T.; Ritis, K. Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS ONE 2011, 6, e29318. [Google Scholar] [CrossRef]
- Davidsson, L.; Dahlstrand Rudin, A.; Sanchez Klose, F.P.; Buck, A.; Björkman, L.; Christenson, K.; Bylund, J. In Vivo Transmigrated Human Neutrophils Are Highly Primed for Intracellular Radical Production Induced by Monosodium Urate Crystals. Int. J. Mol. Sci. 2020, 21, 3750. [Google Scholar] [CrossRef]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhöfer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, E.; Gamberucci, A.; Lucherini, O.M.; Alì, A.; Simpatico, A.; Lorenzini, S.; Lazzerini, P.E.; Tripodi, S.; Frediani, B.; Selvi, E. Neutrophil extracellular traps release in gout and pseudogout depends on number of crystals regardless of leukocyte count. Rheumatology 2021, keab087. [Google Scholar] [CrossRef] [PubMed]
- Apostolidou, E.; Skendros, P.; Kambas, K.; Mitroulis, I.; Konstantinidis, T.; Chrysanthopoulou, A.; Nakos, K.; Tsironidou, V.; Koffa, M.; Boumpas, D.T.; et al. Neutrophil extracellular traps regulate IL-1β-mediated inflammation in familial Mediterranean fever. Ann. Rheum. Dis. 2016, 75, 269–277. [Google Scholar] [CrossRef]
- Cumpelik, A.; Ankli, B.; Zecher, D.; Schifferli, J.A. Neutrophil microvesicles resolve gout by inhibiting C5a-mediated priming of the inflammasome. Ann. Rheum. Dis. 2016, 75, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yang, Q.; Zhang, Q.; Yin, C.; Zhou, L.; Zhou, J.; Wang, Y.; Mi, Q.S. Invariant Natural Killer T Cells Ameliorate Monosodium Urate Crystal-Induced Gouty Inflammation in Mice. Front. Immunol. 2017, 8, 1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galozzi, P.; Maschio, L.; Carraro, S.; Scanu, A.; Facco, M.; Oliviero, F. M2 macrophages as resolvers of crystal-induced inflammation. Rheumatology 2021, keab122. [Google Scholar] [CrossRef]
- Baggio, C.; Sfriso, P.; Cignarella, A.; Galozzi, P.; Scanu, A.; Mastrotto, F.; Favero, M.; Ramonda, R.; Oliviero, F. Phagocytosis and inflammation in crystal-induced arthritis: A synovial fluid and in vitro study. Clin. Exp. Rheumatol. 2020, in press. [Google Scholar]
- Tai, V.; Merriman, T.R.; Dalbeth, N. Genetic advances in gout: Potential applications in clinical practice. Curr. Opin. Rheumatol. 2019, 31, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Tseng, C.C.; Yen, J.H.; Chang, J.G.; Chou, W.C.; Chu, H.W.; Chang, S.J.; Liao, W.T. ABCG2 contributes to the development of gout and hyperuricemia in a genome-wide association study. Sci. Rep. 2018, 8, 3137. [Google Scholar] [CrossRef]
- Karaarslan, A.; Kobak, S.; Kaya, I.; Intepe, N.; Orman, M.; Berdelı, A. Prevalence and significance of MEFV gene mutations in patients with gouty arthritis. Rheumatol. Int. 2016, 36, 1585–1589. [Google Scholar] [CrossRef]
- Salehzadeh, F.; Mohammadikebar, Y.; Haghi, R.N.; Asl, S.H.; Enteshary, A. Familial Mediterranean Fever Gene Mutations and Gout as an Auto-Inflammatory Arthropathy. Med. Arch. 2019, 73, 55–57. [Google Scholar] [CrossRef]
- Qing, Y.F.; Zhou, J.G.; Zhang, Q.B.; Wang, D.S.; Li, M.; Yang, Q.B.; Huang, C.P.; Yin, L.; Pan, S.Y.; Xie, W.G.; et al. Association of TLR4 Gene rs2149356 polymorphism with primary gouty arthritis in a case-control study. PLoS ONE 2013, 8, e64845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, P.; Ma, H.; Viriyakosol, S.; Terkeltaub, R.; Liu-Bryan, R. Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J. Immunol. 2006, 177, 6370–6378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.; Luo, J.; Fu, Q.; Shang, K.; Wei, Y.; Wang, Y.; Li, Y.; Chen, J. Decreased Expression of CD14 in MSU-Mediated Inflammation May Be Associated with Spontaneous Remission of Acute Gout. J. Immunol. Res. 2019, 2019, 7143241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.H.; Cheng, M.; Tang, J.P.; Dai, X.J.; Zhang, Y.; Li, X.P.; Liu, Q.; Wang, Y.L. Single nucleotide polymorphisms associated with P2X7R function regulate the onset of gouty arthritis. PLoS ONE 2017, 12, e0181685. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.C.; Jan Wu, Y.J.; Chung, W.H.; Lee, Y.S.; Chin, S.W.; Chen, T.J.; Chang, Y.S.; Chen, D.Y.; Hung, S.I. Genetic variants of PPAR-gamma coactivator 1B augment NLRP3-mediated inflammation in gouty arthritis. Rheumatology 2017, 56, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, H.; Phipps-Green, A.J.; Topless, R.; Smith, M.D.; Hill, C.; Lester, S.; Rischmueller, M.; Janssen, M.; Jansen, T.L.; Joosten, L.A.; et al. Replication of association of the apolipoprotein A1-C3-A4 gene cluster with the risk of gout. Rheumatology 2016, 55, 1421–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zewinger, S.; Reiser, J.; Jankowski, V.; Alansary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 2020, 21, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Hu, C.; Luo, L.; Fang, F.; Chen, Y.; Li, J.; Peng, Z.; Pan, H. Clinical features and short-term outcomes of 221 patients with COVID-19 in Wuhan, China. J. Clin. Virol. 2020, 127, 104364. [Google Scholar] [CrossRef]
- Chen, Y.; Ren, X.; Li, C.; Xing, S.; Fu, Z.; Yuan, Y.; Wang, R.; Wang, Y.; Lv, W. CARD8 rs2043211 polymorphism is associated with gout in a Chinese male population. Cell. Physiol. Biochem. 2015, 35, 1394–1400. [Google Scholar] [CrossRef]
- Chang, S.J.; Tsai, P.C.; Chen, C.J.; Lai, H.M.; Ko, Y.C. The polymorphism -863C/A in tumour necrosis factor-alpha gene contributes an independent association to gout. Rheumatology 2007, 46, 1662–1666. [Google Scholar] [CrossRef] [Green Version]
- McKinney, C.; Stamp, L.K.; Dalbeth, N.; Topless, R.K.; Day, R.O.; Kannangara, D.R.; Williams, K.M.; Janssen, M.; Jansen, T.L.; Joosten, L.A.; et al. Multiplicative interaction of functional inflammasome genetic variants in determining the risk of gout. Arthritis Res. Ther. 2015, 17, 288. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yin, C.; Chu, N.; Han, L.; Li, C. IL-8 -251T/A and IL-12B 1188A/C polymorphisms are associated with gout in a Chinese male population. Scand. J. Rheumatol. 2013, 42, 150–158. [Google Scholar] [CrossRef]
- Liu, S.; He, H.; Yu, R.; Han, L.; Wang, C.; Cui, Y.; Li, C. The rs7517847 polymorphism in the IL-23R gene is associated with gout in a Chinese Han male population. Mod. Rheumatol. 2015, 25, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Klück, V.; van Deuren, R.C.; Cavalli, G.; Shaukat, A.; Arts, P.; Cleophas, M.C.; Crișan, T.O.; Tausche, A.K.; Riches, P.; Dalbeth, N.; et al. Rare genetic variants in interleukin-37 link this anti-inflammatory cytokine to the pathogenesis and treatment of gout. Ann. Rheum. Dis. 2020, 79, 536–544. [Google Scholar] [CrossRef]
- Gosling, A.L.; Boocock, J.; Dalbeth, N.; Harré Hindmarsh, J.; Stamp, L.K.; Stahl, E.A.; Choi, H.K.; Matisoo-Smith, E.A.; Merriman, T.R. Mitochondrial genetic variation and gout in Māori and Pacific people living in Aotearoa New Zealand. Ann. Rheum. Dis. 2018, 77, 571–578. [Google Scholar] [CrossRef]
- Tseng, C.C.; Wong, M.C.; Liao, W.T.; Chen, C.J.; Lee, S.C.; Yen, J.H.; Chang, S.J. Systemic Investigation of Promoter-wide Methylome and Genome Variations in Gout. Int. J. Mol. Sci. 2020, 21, 4702. [Google Scholar] [CrossRef] [PubMed]
- Cleophas, M.C.P.; Crişan, T.O.; Klück, V.; Hoogerbrugge, N.; Netea-Maier, R.T.; Dinarello, C.A.; Netea, M.G.; Joosten, L.A.B. Romidepsin suppresses monosodium urate crystal-induced cytokine production through upregulation of suppressor of cytokine signaling 1 expression. Arthritis Res. Ther. 2019, 21, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FitzGerald, J.D.; Dalbeth, N.; Mikuls, T.; Brignardello-Petersen, R.; Guyatt, G.; Abeles, A.M.; Gelber, A.C.; Harrold, L.R.; Khanna, D.; King, C.; et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis Care Res. 2020, 72, 744–760. [Google Scholar] [CrossRef]
- Dalbeth, N.; Lauterio, T.J.; Wolfe, H.R. Mechanism of action of colchicine in the treatment of gout. Clin. Ther. 2014, 36, 1465–1479. [Google Scholar] [CrossRef] [Green Version]
- So, A.; De Smedt, T.; Revaz, S.; Tschopp, J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 2007, 9, R28. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Fields, T.; Mancuso, C.A.; Bass, A.R.; Vasanth, L. Anakinra’s efficacy is variable in refractory gout: Report of ten cases. Semin. Arthritis Rheum. 2010, 40, 210–214. [Google Scholar] [CrossRef]
- Terkeltaub, R.A.; Schumacher, H.R.; Carter, J.D.; Baraf, H.S.; Evans, R.R.; Wang, J.; King-Davis, S.; Weinstein, S.P. Rilonacept in the treatment of acute gouty arthritis: A randomized, controlled clinical trial using indomethacin as the active comparator. Arthritis Res. Ther. 2013, 15, R25. [Google Scholar] [CrossRef] [Green Version]
- So, A.; De Meulemeester, M.; Pikhlak, A.; Yücel, A.E.; Richard, D.; Murphy, V.; Arulmani, U.; Sallstig, P.; Schlesinger, N. Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: Results of a multicenter, phase II, dose-ranging study. Arthritis Rheum. 2010, 62, 3064–3076. [Google Scholar] [CrossRef]
- Schlesinger, N.; Alten, R.E.; Bardin, T.; Schumacher, H.R.; Bloch, M.; Gimona, A.; Krammer, G.; Murphy, V.; Richard, D.; So, A.K. Canakinumab for acute gouty arthritis in patients with limited treatment options: Results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann. Rheum. Dis. 2012, 71, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wang, Y.; Wu, R.; He, Y.; Su, Q.; Shi, G. MicroRNA-488 and-920 regulate the production of proinflammatory cytokines in acute gouty arthritis. Arthritis Res. Ther. 2017, 19, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Li, X.; Liu, Y.; Xia, Y.; Chang, R.; Zhang, C. Inflammasome inhibitors: Promising therapeutic approaches against cancer. J. Hematol. Oncol. 2019, 12, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, T.; Rust, M.; Kaiser, S.N.; Slowik, A.; Beyer, C.; Koczulla, A.R.; Schulz, J.B.; Habib, P.; Bach, J.P. α1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid β (1–42)-stimulated murine astrocytes. J. Neuroinflamm. 2018, 15, 282. [Google Scholar] [CrossRef]
- Klück, V.; Jansen, T.; Janssen, M.; Comarniceanu, A.; Efdé, M.; Tengesdal, I.W.; Schraa, K.; Cleophas, M.C.P.; Scribner, C.L.; Skouras, D.B.; et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: An open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020, 2, e270–e280. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Asher, J.L.; Molony, R.D.; Shaw, A.C.; Zeiss, C.J.; Wang, C.; Morozova-Roche, L.A.; Herzog, R.I.; Iwasaki, A.; Dixit, V.D. β-Hydroxybutyrate Deactivates Neutrophil NLRP3 Inflammasome to Relieve Gout Flares. Cell Rep. 2017, 18, 2077–2087. [Google Scholar] [CrossRef]
- Qiao, C.Y.; Li, Y.; Shang, Y.; Jiang, M.; Liu, J.; Zhan, Z.Y.; Ye, H.; Lin, Y.C.; Jiao, J.Y.; Sun, R.H.; et al. Management of Gout-associated MSU crystals-induced NLRP3 inflammasome activation by procyanidin B2: Targeting IL-1β and Cathepsin B in macrophages. Inflammopharmacology 2020, 28, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Li, H.; Ou, G.; Ren, L.; Yang, X.; Zeng, M. Curcumin attenuates MSU crystal-induced inflammation by inhibiting the degradation of IκBα and blocking mitochondrial damage. Arthritis Res. Ther. 2019, 21, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Liu, B.; Wang, P.; Li, X.; Li, Y.; Zheng, X.; Tai, Y.; Wang, C.; Liu, B. Eucalyptol alleviates inflammation and pain responses in a mouse model of gout arthritis. Br. J. Pharmacol. 2020, 177, 2042–2057. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.; Lee, G.S. Riboflavin, vitamin B2, attenuates NLRP3, NLRC4, AIM2, and non-canonical inflammasomes by the inhibition of caspase-1 activity. Sci. Rep. 2020, 10, 19091. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Spinetti, T.; Tardivel, A.; Castillo, R.; Bourquin, C.; Guarda, G.; Tian, Z.; Tschopp, J.; Zhou, R. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 2013, 38, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Meng, B.; Zeng, L.; Yin, S.; Hu, Y.; Li, S.; Fu, Y.; Zhang, X.; Xie, C.; Shu, L.; et al. Discovery of a novel rice-derived peptide with significant anti-gout potency. Food Funct. 2020, 11, 10542–10553. [Google Scholar] [CrossRef]
- Pellegrini, C.; Fornai, M.; Antonioli, L.; Blandizzi, C.; Calderone, V. Phytochemicals as Novel Therapeutic Strategies for NLRP3 Inflammasome-Related Neurological, Metabolic, and Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 2876. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Lee, H.E.; Moon, S.J.; Ko, K.M.; Koh, J.H.; Seok, J.K.; Min, J.K.; Heo, T.H.; Kang, H.C.; Cho, Y.Y.; et al. Direct Binding to NLRP3 Pyrin Domain as a Novel Strategy to Prevent NLRP3-Driven Inflammation and Gouty Arthritis. Arthritis Rheumatol. 2020, 72, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Oliviero, F.; Zamudio-Cuevas, Y.; Belluzzi, E.; Andretto, L.; Scanu, A.; Favero, M.; Ramonda, R.; Ravagnan, G.; López-Reyes, A.; Spinella, P.; et al. Polydatin and Resveratrol Inhibit the Inflammatory Process Induced by Urate and Pyrophosphate Crystals in THP-1 Cells. Foods 2019, 8, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Pan, Y.; Li, W.; Guan, P.; You, C. The Role of Noncoding RNAs in Gout. Endocrinology 2020, 161. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti IL-1 | |||
| Dosage | Target | Reference | |
| Anakinra | 100 mg daily | IL-1 receptor | [72] |
| Canakinumab | 150 mg at baseline | IL-1β | [76] |
| Rilonacept (trap protein) | 320 mg at baseline | Trap-fusion protein blocking both IL-α and Il-1β | [74] |
| lncRNA (miRNA-488, miRNA-920) | NA | IL-β | [77] |
| IL-1β processing inhibitors | |||
| Dosage | Target | Reference | |
| VX-765 (belnacasan) | NA | Caspase I | [78] |
| A1AT | NA | Caspase I | [79] |
| MMC-950 (CRID3) | NA | ASC complex | [37] |
| NLRP3 inhibitors | |||
| Dosage | Target | Reference | |
| Glucocorticoids | variable | NLRP3 (indirectly) NF-κB pathway | [6] |
| Colchicine | 1 mg/day (followed by 0.5 mg after 30 min on day 1) | Microtubules polymerization, Chemokines, chemotaxis, NLRP3 | [6] |
| Dapansutrile (OLT 1177) | 100 mg/day, 300 mg/day, 1000 mg/day, or 2000 mg/day orally for 8 days | NLRP3 | [80] |
| Beta-hydroxybutyrate (BHB) | NA | NLRP3, K+ channels | [81] |
| Polyphenols present in food (ProcyanidinB2, Curcumin, Epigallocatechingallate) | NA | NLRP3 | [37,82,83] |
| Carotenoids (Beta-carotene) Other compounds: Eucalyptol, Omega3 FAs, small peptides (RDP3), vitamins (riboflavin-B2) | NA | NLRP3 | [82,84,85,86,87,88] |
| Receptor inhibitors | |||
| Dosage | Target | Reference | |
| lncRNA (miRNA-146a) | NA | Myd88/TLR4 | [47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galozzi, P.; Bindoli, S.; Doria, A.; Oliviero, F.; Sfriso, P. Autoinflammatory Features in Gouty Arthritis. J. Clin. Med. 2021, 10, 1880. https://doi.org/10.3390/jcm10091880
Galozzi P, Bindoli S, Doria A, Oliviero F, Sfriso P. Autoinflammatory Features in Gouty Arthritis. Journal of Clinical Medicine. 2021; 10(9):1880. https://doi.org/10.3390/jcm10091880
Chicago/Turabian StyleGalozzi, Paola, Sara Bindoli, Andrea Doria, Francesca Oliviero, and Paolo Sfriso. 2021. "Autoinflammatory Features in Gouty Arthritis" Journal of Clinical Medicine 10, no. 9: 1880. https://doi.org/10.3390/jcm10091880