
There Are No Insurmountable Barriers: Passage of the Helicobacter pylori VacA Toxin from Bacterial Cytoplasm to Eukaryotic Cell Organelle

Abstract
:1. Introduction
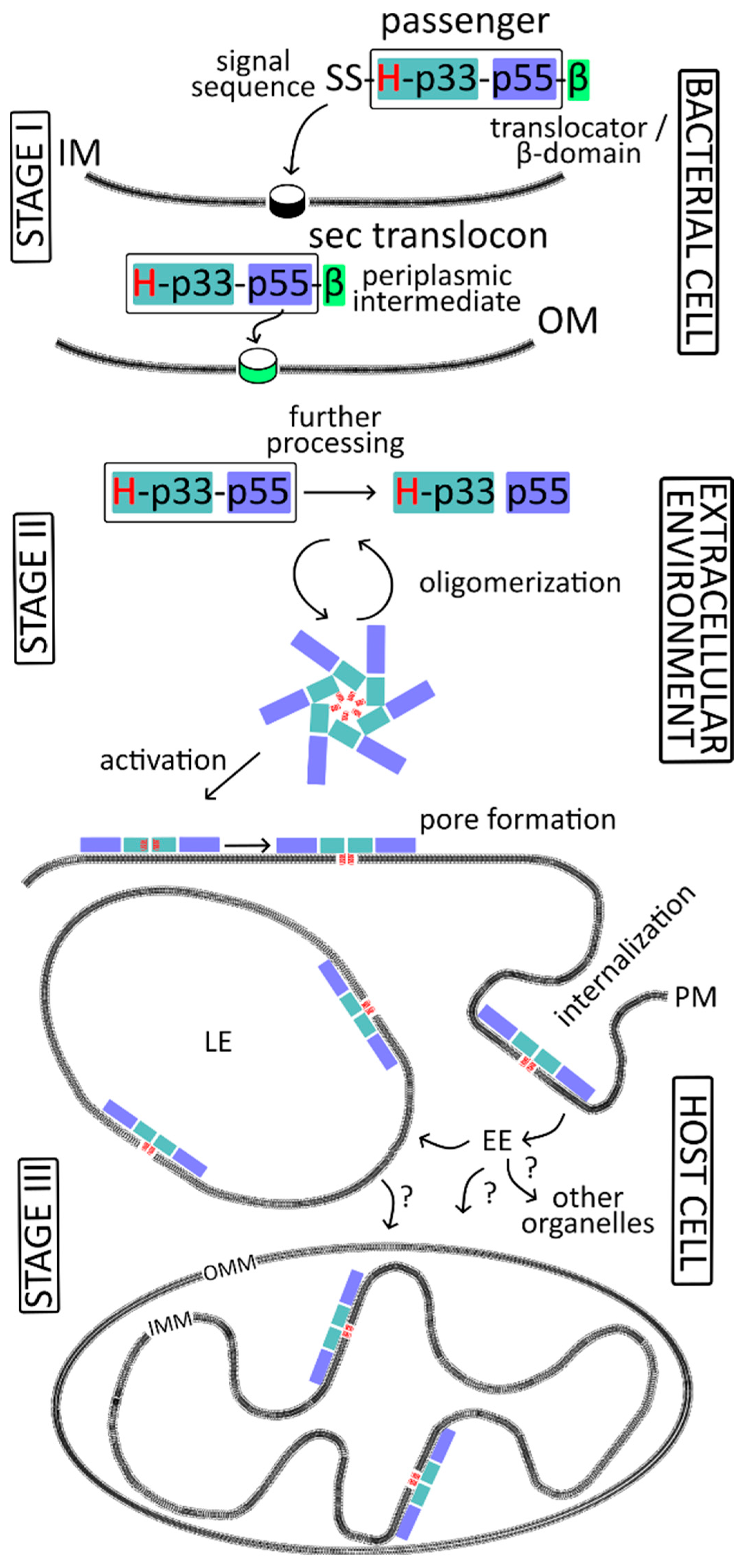
2. Stage I—Events in the Helicobacter pylori Cell
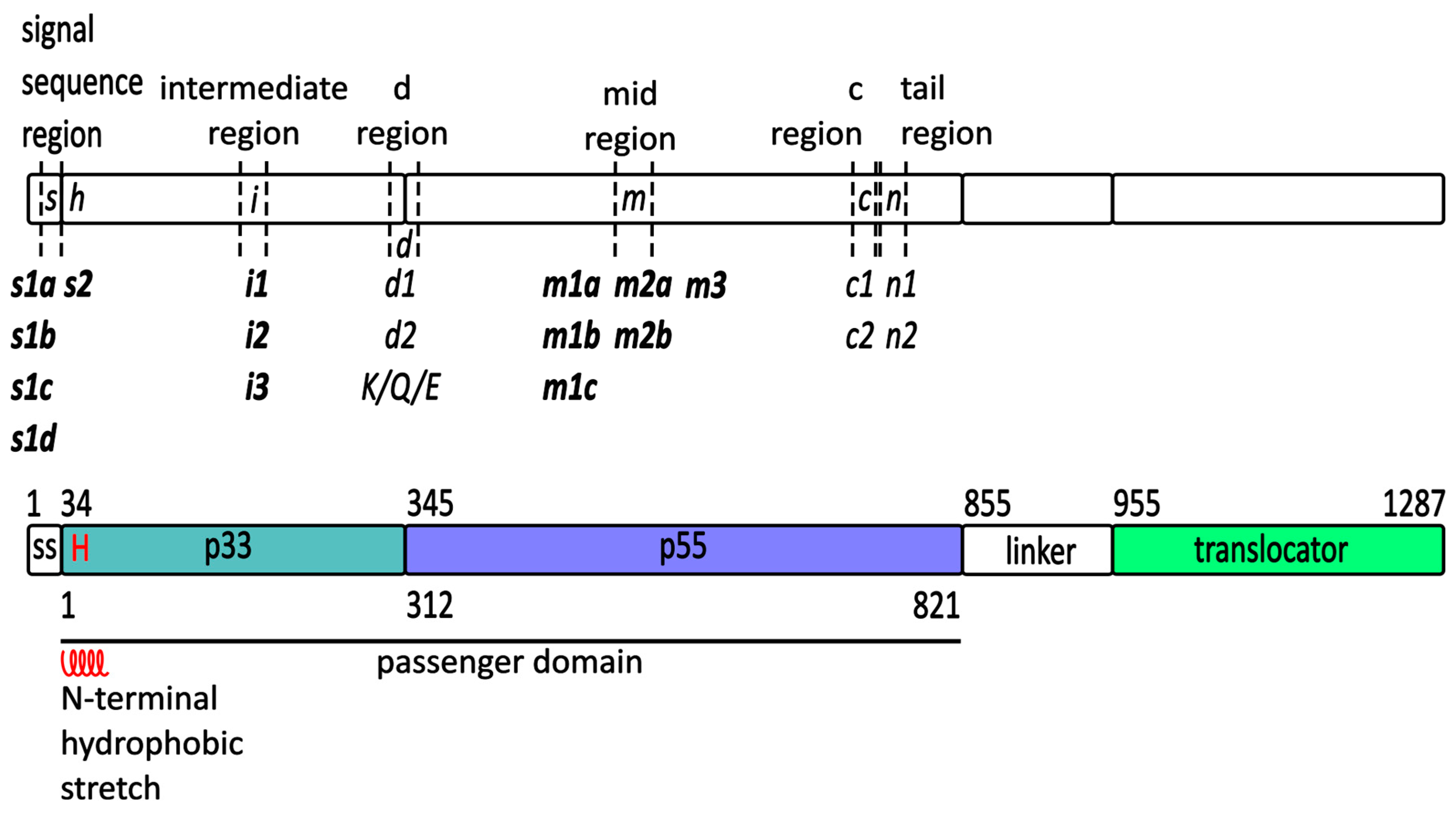
2.1. The vacA Gene
2.2. VacA Protein in the H. pylori Cell
2.2.1. Translocation of VacA across the Inner Membrane
2.2.2. Periplasmic Transit
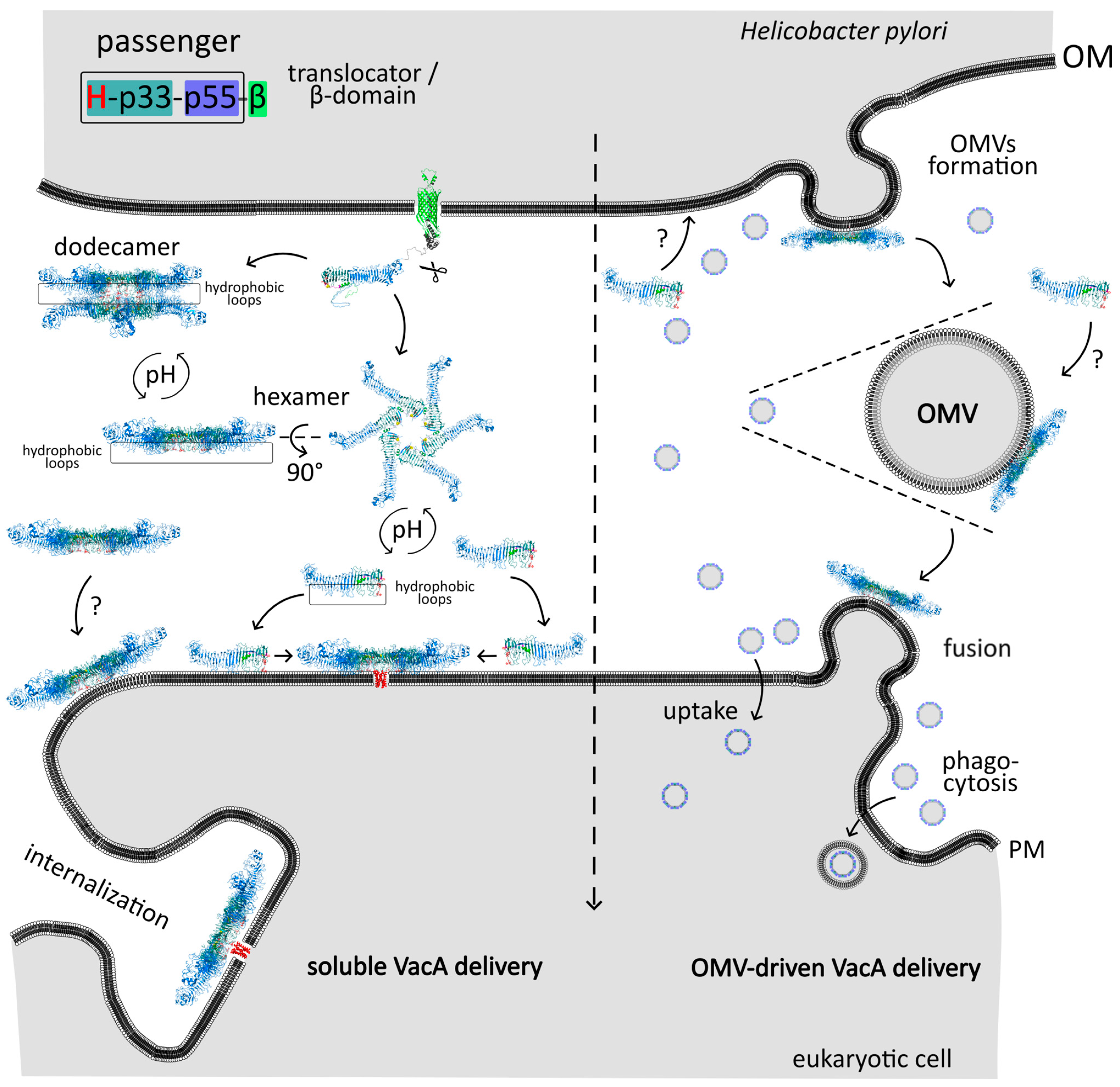
2.2.3. Translocator Domain and Translocation across the Outer Membrane
2.2.4. Release of the Passenger Domain
3. Stage II—Events in the Extracellular Space
3.1. Processing of the Passenger Domain
3.2. Structure of the Mature VacA Toxin
4. Stage III—Events in the Host Cell
4.1. Interaction with the Host Cell Membranes
4.1.1. VacA Receptors
4.1.2. Pore Formation
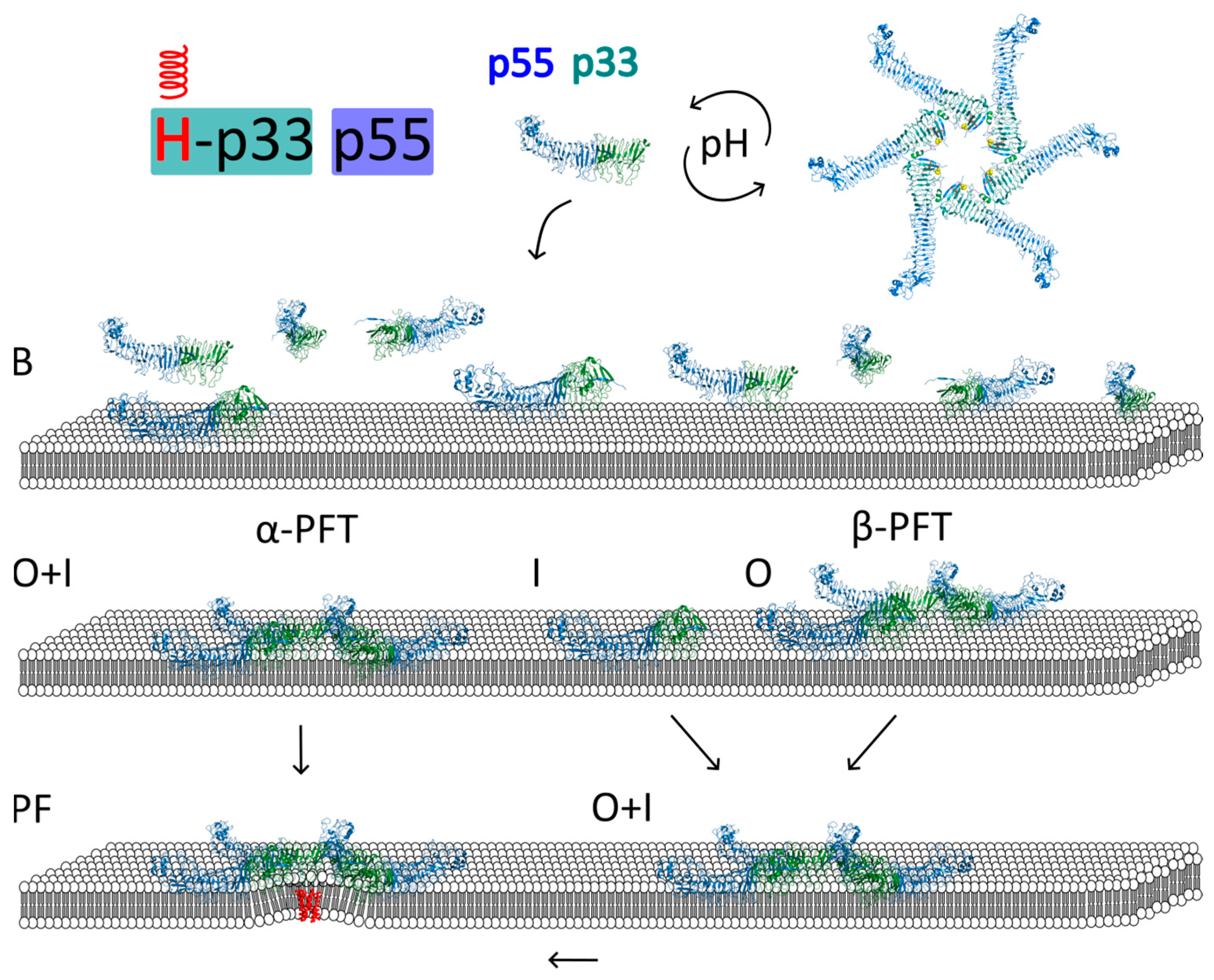
Activation of the Toxin
Models of VacA Interaction with Membrane and Pore Formation
- Regions of p33 insert into the lipid bilayer simultaneously with p88 oligomerization into a hexamer
- Regions of p33 insert into the lipid bilayer before p88 oligomerization into a hexamer
- Regions of p33 insert into the lipid bilayer after p88 oligomerization into a hexamer

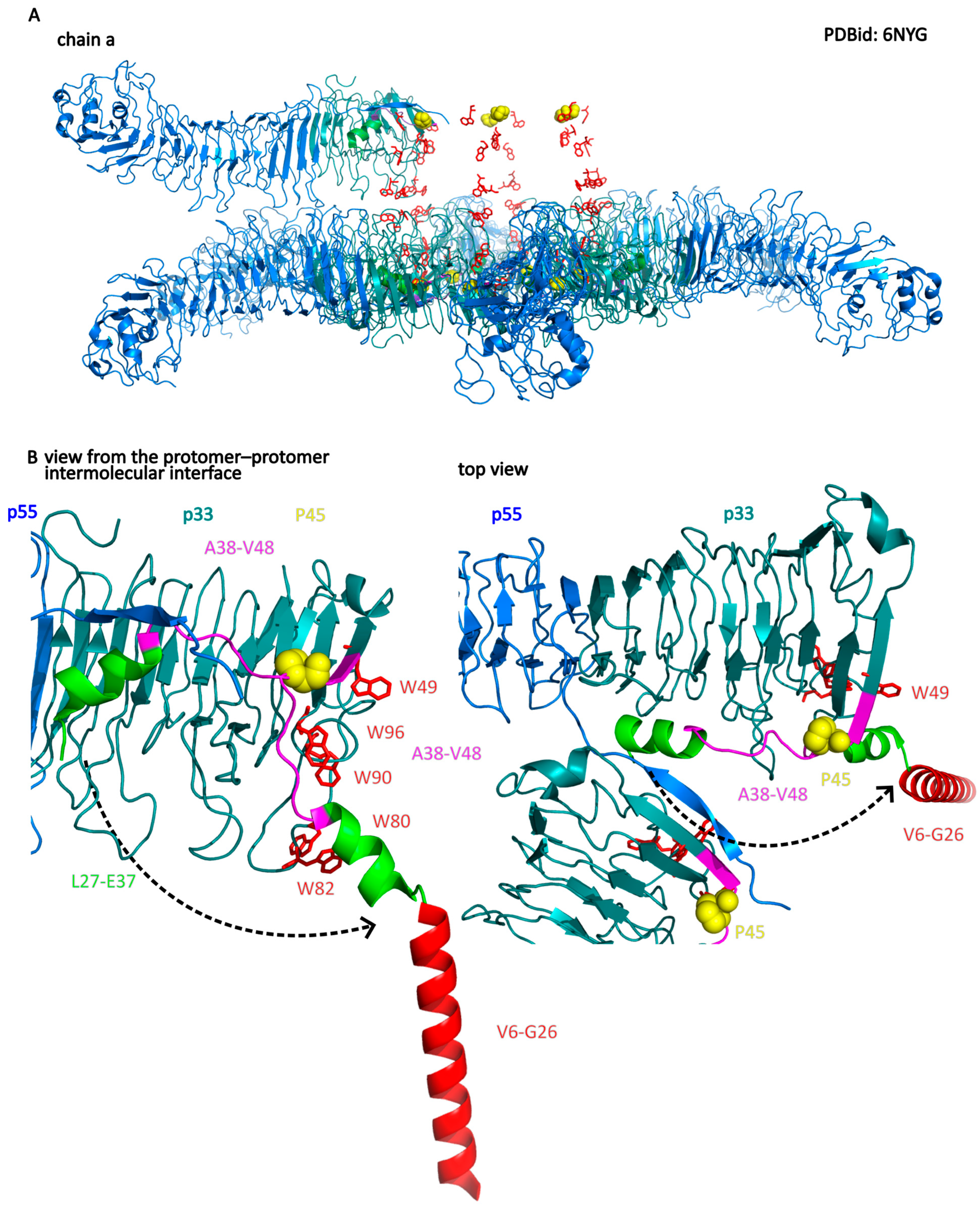
- Regions important for insertion into the membrane and channel formation are located at the N-terminal region of the p33 subdomain. These include helices V6–G26 and W30–E37, with the P40 residue at the loop connected to the W30–E37 helix. The W30–E37 helix is hidden in a protomer–protomer interface in the water-soluble hexamer.
- The interaction of the VacA hexamer with the membrane initially occurs via a cluster of tryptophan residues (W49, W80, W82, W90, and W96) located near the inner rim of the hexamer and the helix W30–E37. Association with the membrane can also be mediated by electrostatic interactions between positively charged amino acid residues of the bottom side of the hexamer (facing another hexamer in dodecamer) and anionic phospholipids.
- Interaction with membrane lipids induces a change in the position of the W30–E37 helix, while P40 acts as a hinge for this movement. This leads to the exposure of the hydrophobic N-terminus (p33 domain), which forms a helix bundle within the membrane. Importantly, the structure of most of the hexamer’s elements remains unchanged.

Roles of Specific VacA Regions in Membrane Binding, Oligomerization, and Membrane Insertion
- The “s” region (signal region), localized at the N-terminus of the VacA preprotein, is identical to the signal sequence in the s1 subtype (and is completely removed during export from the cytoplasm), while in the s2 subtype, the cleavage site of the signal sequence is different. As a result, the VacA protein is longer by 12 amino acid residues at the N-terminus. Its processing during VacA export affects the ability of membrane insertion and pore formation by the secreted toxin. Consequently, the N-terminal sequence of the mature toxin in the s1 type is different than that of the s2 type. This is associated with the different properties of both forms. The s1 type fully exhibits vacuolating activity, while the s2 type lacks detectable cytotoxic activity [158].
- The “m” region (middle region) located within the p55 domain with the most common types (m1 or m2) is associated with differences in the ability of VacA to bind to distinct cell types [111,160,161] and exhibit cytotoxin activity [16,111,160,161,162]. The m1 and m2 forms seem to have different cell-binding specificity. The m2 form induces vacuolization in the primary gastric cells of the RK-13 cell line, but, contrary to the m1 form, it is not able to cause vacuolization in HeLa cells [160]. Several different chimeric variants in the m1/m2 mid-region (R460-G793) of VacA were tested for vacuolating activity and confirmed differences between the m1 and m2 forms of VacA in inducing vacuole formation in RK-13 and HeLa cells [163,164,165]. The experiments allowed for the specification of amino acids 460–569 within the p55 domain to be responsible for cell binding [164] and showed no significant role of the m2 variant 21-amino acid insert on vacuolization activity of VacA [163]. Different posttranslational modifications of RPTPα from HeLa may be responsible for the reduced susceptibility to m2 VacA [111]. These data favor a protein receptor-mediated interaction of VacA with plasma membrane.
VacA Pore Structure and Function
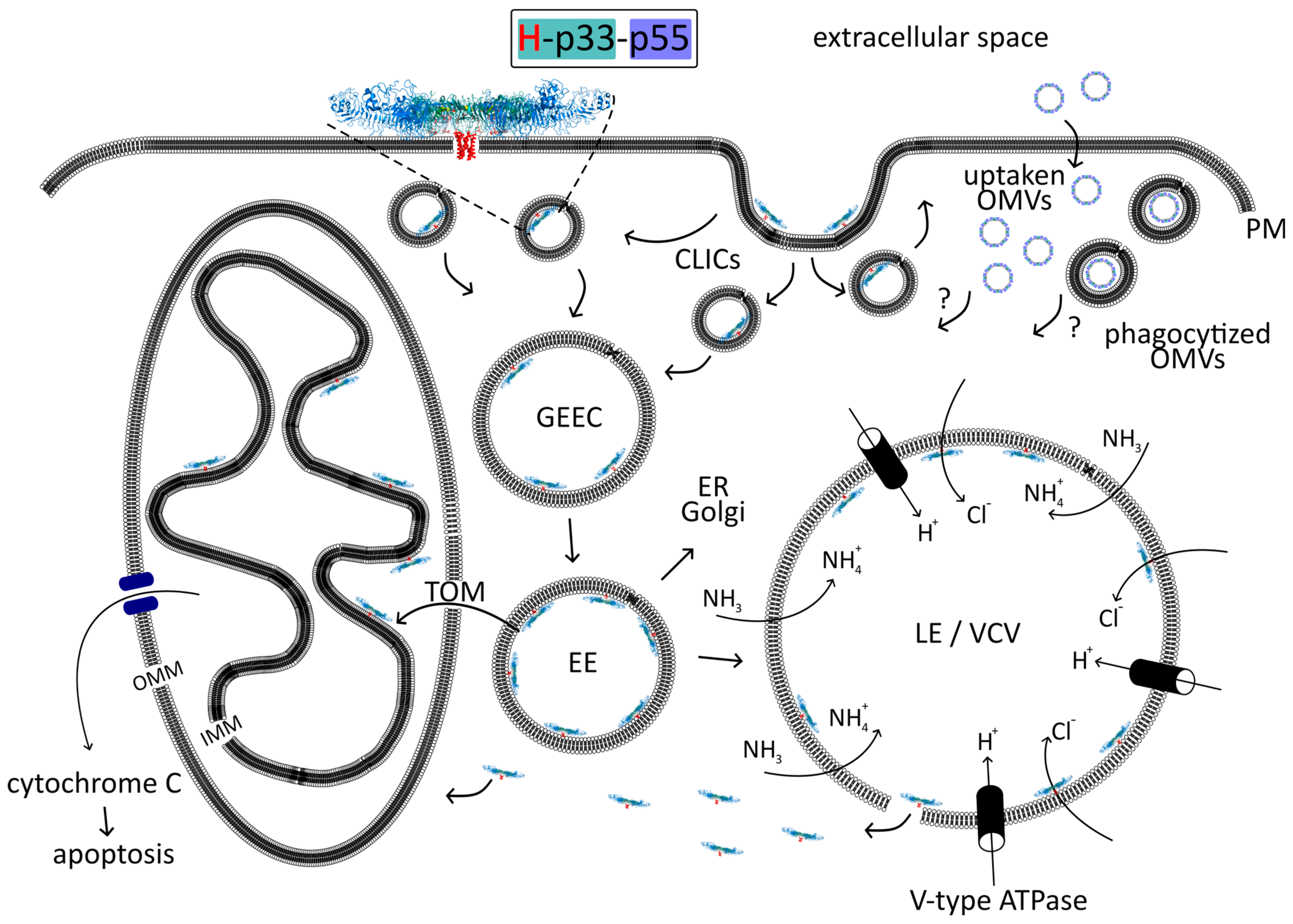
4.1.3. Internalization
4.2. Spread of VacA in the Host Cell
4.2.1. Endosomes
CLIC/GEEC (GPI-AP) Endocytic Pathway of VacA
Early Endosomes
Late Endosomes/Lysosomes
4.2.2. Mitochondria/Apoptosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murray, P.R.; Rosenthal, K.S.; Pfaller, M.A. Medical Microbiology; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Foegeding, N.J.; Caston, R.R.; McClain, M.S.; Ohi, M.D.; Cover, T.L. An Overview of Helicobacter pylori VacA Toxin Biology. Toxins 2016, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Blanke, S.R. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 2005, 3, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Leunk, R.D.; Johnson, P.T.; David, B.C.; Kraft, W.G.; Morgan, D.R. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J. Med. Microbiol. 1988, 26, 93–99. [Google Scholar] [CrossRef]
- Cover, T.L.; Halter, S.A.; Blaser, M.J. Characterization of HeLa cell vacuoles induced by Helicobacter pylori broth culture supernatant. Hum. Pathol. 1992, 23, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Czajkowsky, D.M.; Iwamoto, H.; Szabo, G.; Cover, T.L.; Shao, Z. Mimicry of a host anion channel by a Helicobacter pylori pore-forming toxin. Biophys. J. 2005, 89, 3093–3101. [Google Scholar] [CrossRef] [PubMed]
- Wayne, R. Chapter 2—Plasma Membrane. In Plant Cell Biology, 2nd ed.; Wayne, R., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 17–53. [Google Scholar]
- Ma, Y.; Poole, K.; Goyette, J.; Gaus, K. Introducing Membrane Charge and Membrane Potential to T Cell Signaling. Front. Immunol. 2017, 8, 1513. [Google Scholar] [CrossRef] [PubMed]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Schmitt, W.; Haas, R. Genetic analysis of the Helicobacter pylori vacuolating cytotoxin: Structural similarities with the IgA protease type of exported protein. Mol. Microbiol. 1994, 12, 307–319. [Google Scholar] [CrossRef]
- Kern, B.; Jain, U.; Utsch, C.; Otto, A.; Busch, B.; Jiménez-Soto, L.; Becher, D.; Haas, R. Characterization of Helicobacter pylori VacA-containing vacuoles (VCVs), VacA intracellular trafficking and interference with calcium signalling in T lymphocytes. Cell Microbiol. 2015, 17, 1811–1832. [Google Scholar] [CrossRef]
- Rassow, J. Helicobacter pylori vacuolating toxin A and apoptosis. Cell Commun. Signal 2011, 9, 26. [Google Scholar] [CrossRef]
- Raghunathan, K.; Foegeding, N.J.; Campbell, A.M.; Cover, T.L.; Ohi, M.D.; Kenworthy, A.K. Determinants of Raft Partitioning of the Helicobacter pylori Pore-Forming Toxin VacA. Infect. Immun. 2018, 86, e00872-17. [Google Scholar] [CrossRef] [PubMed]
- Telford, J.L.; Ghiara, P.; Dell’Orco, M.; Comanducci, M.; Burroni, D.; Bugnoli, M.; Tecce, M.F.; Censini, S.; Covacci, A.; Xiang, Z.; et al. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med. 1994, 179, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Tummuru, M.K.; Cao, P.; Thompson, S.A.; Blaser, M.J. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J. Biol. Chem. 1994, 269, 10566–10573. [Google Scholar] [CrossRef]
- Atherton, J.C.; Cao, P.; Peek, R.M., Jr.; Tummuru, M.K.; Blaser, M.J.; Cover, T.L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem. 1995, 270, 17771–17777. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Peek, R.M., Jr. Diet, microbial virulence, and Helicobacter pylori-induced gastric cancer. Gut Microbes 2013, 4, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, M.H.; Cover, T.L. Mutational analysis of the vacA promoter provides insight into gene transcription in Helicobacter pylori. J. Bacteriol. 1999, 181, 2261–2266. [Google Scholar] [CrossRef] [PubMed]
- Amilon, K.R.; Letley, D.P.; Winter, J.A.; Robinson, K.; Atherton, J.C. Expression of the Helicobacter pylori virulence factor vacuolating cytotoxin A (vacA) is influenced by a potential stem-loop structure in the 5’ untranslated region of the transcript. Mol. Microbiol. 2015, 98, 831–846. [Google Scholar] [CrossRef]
- Barnard, F.M.; Loughlin, M.F.; Fainberg, H.P.; Messenger, M.P.; Ussery, D.W.; Williams, P.; Jenks, P.J. Global regulation of virulence and the stress response by CsrA in the highly adapted human gastric pathogen Helicobacter pylori. Mol. Microbiol. 2004, 51, 15–32. [Google Scholar] [CrossRef]
- Merrell, D.S.; Thompson, L.J.; Kim, C.C.; Mitchell, H.; Tompkins, L.S.; Lee, A.; Falkow, S. Growth phase-dependent response of Helicobacter pylori to iron starvation. Infect. Immun. 2003, 71, 6510–6525. [Google Scholar] [CrossRef]
- van Amsterdam, K.; van Vliet, A.H.; Kusters, J.G.; Feller, M.; Dankert, J.; van der Ende, A. Induced Helicobacter pylori vacuolating cytotoxin VacA expression after initial colonisation of human gastric epithelial cells. FEMS Immunol. Med. Microbiol. 2003, 39, 251–256. [Google Scholar] [CrossRef]
- Gancz, H.; Jones, K.R.; Merrell, D.S. Sodium chloride affects Helicobacter pylori growth and gene expression. J. Bacteriol. 2008, 190, 4100–4105. [Google Scholar] [CrossRef] [PubMed]
- Caston, R.R.; Loh, J.T.; Voss, B.J.; McDonald, W.H.; Scholz, M.B.; McClain, M.S.; Cover, T.L. Effect of environmental salt concentration on the Helicobacter pylori exoproteome. J. Proteom. 2019, 202, 103374. [Google Scholar] [CrossRef] [PubMed]
- van Doorn, L.J.; Figueiredo, C.; Sanna, R.; Pena, S.; Midolo, P.; Ng, E.K.; Atherton, J.C.; Blaser, M.J.; Quint, W.G. Expanding allelic diversity of Helicobacter pylori vacA. J. Clin. Microbiol. 1998, 36, 2597–2603. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Sapién, G.; Cerritos, R.; González-Valencia, G.; Méndez, J.L.; Cravioto, A.; Morales-Espinosa, R. Novel VacA S1d Signal Sequence Found in Helicobacter Pylori from Mexican Children with Recurrent Abdominal Pain. SOJ Microbiol. Infect. Dis. 2017, 5, 1–7. [Google Scholar] [CrossRef]
- Rhead, J.L.; Letley, D.P.; Mohammadi, M.; Hussein, N.; Mohagheghi, M.A.; Eshagh Hosseini, M.; Atherton, J.C. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 2007, 133, 926–936. [Google Scholar] [CrossRef]
- Chung, C.; Olivares, A.; Torres, E.; Yilmaz, O.; Cohen, H.; Perez-Perez, G. Diversity of VacA intermediate region among Helicobacter pylori strains from several regions of the world. J. Clin. Microbiol. 2010, 48, 690–696. [Google Scholar] [CrossRef]
- Han, S.R.; Schreiber, H.J.; Bhakdi, S.; Loos, M.; Maeurer, M.J. vacA genotypes and genetic diversity in clinical isolates of Helicobacter pylori. Clin. Diagn. Lab. Immunol. 1998, 5, 139–145. [Google Scholar] [CrossRef]
- Strobel, S.; Bereswill, S.; Balig, P.; Allgaier, P.; Sonntag, H.G.; Kist, M. Identification and analysis of a new vacA genotype variant of Helicobacter pylori in different patient groups in Germany. J. Clin. Microbiol. 1998, 36, 1285–1289. [Google Scholar] [CrossRef]
- Pan, Z.J.; Berg, D.E.; van der Hulst, R.W.; Su, W.W.; Raudonikiene, A.; Xiao, S.D.; Dankert, J.; Tytgat, G.N.; van der Ende, A. Prevalence of vacuolating cytotoxin production and distribution of distinct vacA alleles in Helicobacter pylori from China. J. Infect. Dis. 1998, 178, 220–226. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.K.; Kersulyte, D.; Jeong, J.Y.; Datta, S.; Ito, Y.; Chowdhury, A.; Chowdhury, S.; Santra, A.; Bhattacharya, S.K.; Azuma, T.; et al. Distinctiveness of genotypes of Helicobacter pylori in Calcutta, India. J. Bacteriol. 2000, 182, 3219–3227. [Google Scholar] [CrossRef]
- Ogiwara, H.; Sugimoto, M.; Ohno, T.; Vilaichone, R.K.; Mahachai, V.; Graham, D.Y.; Yamaoka, Y. Role of deletion located between the intermediate and middle regions of the Helicobacter pylori vacA gene in cases of gastroduodenal diseases. J. Clin. Microbiol. 2009, 47, 3493–3500. [Google Scholar] [CrossRef] [PubMed]
- Soyfoo, D.M.; Doomah, Y.H.; Xu, D.; Zhang, C.; Sang, H.M.; Liu, Y.Y.; Zhang, G.X.; Jiang, J.X.; Xu, S.F. New genotypes of Helicobacter pylori VacA d-region identified from global strains. BMC Mol. Cell Biol. 2021, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- Bakhti, S.Z.; Latifi-Navid, S.; Mohammadi, S.; Zahri, S.; Bakhti, F.S.; Feizi, F.; Yazdanbod, A.; Siavoshi, F. Relevance of Helicobacter pylori vacA 3’-end Region Polymorphism to Gastric Cancer. Helicobacter 2016, 21, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Thi Huyen Trang, T.; Thanh Binh, T.; Yamaoka, Y. Relationship between vacA Types and Development of Gastroduodenal Diseases. Toxins 2016, 8, 182. [Google Scholar] [CrossRef]
- Junaid, M.; Linn, A.K.; Javadi, M.B.; Al-Gubare, S.; Ali, N.; Katzenmeier, G. Vacuolating cytotoxin A (VacA)—A multi-talented pore-forming toxin from Helicobacter pylori. Toxicon 2016, 118, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.J.; Blanke, S.R. Remodeling the host environment: Modulation of the gastric epithelium by the Helicobacter pylori vacuolating toxin (VacA). Front. Cell Infect. Microbiol. 2012, 2, 37. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Dooley, C.P.; Blaser, M.J. Characterization of and human serologic response to proteins in Helicobacter pylori broth culture supernatants with vacuolizing cytotoxin activity. Infect. Immun. 1990, 58, 603–610. [Google Scholar] [CrossRef]
- Covacci, A.; Censini, S.; Bugnoli, M.; Petracca, R.; Burroni, D.; Macchia, G.; Massone, A.; Papini, E.; Xiang, Z.; Figura, N.; et al. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 1993, 90, 5791–5795. [Google Scholar] [CrossRef]
- Phadnis, S.H.; Ilver, D.; Janzon, L.; Normark, S.; Westblom, T.U. Pathological significance and molecular characterization of the vacuolating toxin gene of Helicobacter pylori. Infect. Immun. 1994, 62, 1557–1565. [Google Scholar] [CrossRef]
- Rapisarda, C.; Fronzes, R. Secretion Systems Used by Bacteria to Subvert Host Functions. Curr. Issues Mol. Biol. 2018, 25, 1–42. [Google Scholar] [CrossRef]
- Fiocca, R.; Necchi, V.; Sommi, P.; Ricci, V.; Telford, J.; Cover, T.L.; Solcia, E. Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J. Pathol. 1999, 188, 220–226. [Google Scholar] [CrossRef]
- Cover, T.L.; Blaser, M.J. Purification and characterization of the vacuolating toxin from Helicobacter pylori. J. Biol. Chem. 1992, 267, 10570–10575. [Google Scholar] [CrossRef] [PubMed]
- Troman, L.; Collinson, I. Pushing the Envelope: The Mysterious Journey Through the Bacterial Secretory Machinery, and Beyond. Front. Microbiol. 2021, 12, 782900. [Google Scholar] [CrossRef] [PubMed]
- Smets, D.; Loos, M.S.; Karamanou, S.; Economou, A. Protein Transport Across the Bacterial Plasma Membrane by the Sec Pathway. Protein J. 2019, 38, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Liechti, G.; Goldberg, J.B. Outer membrane biogenesis in Escherichia coli, Neisseria meningitidis, and Helicobacter pylori: Paradigm deviations in H. pylori. Front. Cell Infect. Microbiol. 2012, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Leyton, D.L.; Rossiter, A.E.; Henderson, I.R. From self sufficiency to dependence: Mechanisms and factors important for autotransporter biogenesis. Nat. Rev. Microbiol. 2012, 10, 213–225. [Google Scholar] [CrossRef]
- Ambroziak, P.; Rzepka, I.; Skorko-Glonek, J. SecA—A multidomain and multitask bacterial export protein. Acta Biochim. Pol. 2021, 68, 427–436. [Google Scholar] [CrossRef]
- Macošek, J.; Mas, G.; Hiller, S. Redefining Molecular Chaperones as Chaotropes. Front. Mol. Biosci. 2021, 8, 683132. [Google Scholar] [CrossRef]
- Troman, L.; Alvira, S.; Daum, B.; Gold, V.A.M.; Collinson, I. Interaction of the periplasmic chaperone SurA with the inner membrane protein secretion (SEC) machinery. Biochem. J. 2023, 480, 283–296. [Google Scholar] [CrossRef]
- Schiffrin, B.; Machin, J.M.; Karamanos, T.K.; Zhuravleva, A.; Brockwell, D.J.; Radford, S.E.; Calabrese, A.N. Dynamic interplay between the periplasmic chaperone SurA and the BAM complex in outer membrane protein folding. Commun. Biol. 2022, 5, 560. [Google Scholar] [CrossRef]
- Figaj, D.; Ambroziak, P.; Rzepka, I.; Skórko-Glonek, J. SurA-like and Skp-like Proteins as Important Virulence Determinants of the Gram Negative Bacterial Pathogens. Int. J. Mol. Sci. 2023, 24, 295. [Google Scholar] [CrossRef] [PubMed]
- Dautin, N. Folding Control in the Path of Type 5 Secretion. Toxins 2021, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Meuskens, I.; Saragliadis, A.; Leo, J.C.; Linke, D. Type V Secretion Systems: An Overview of Passenger Domain Functions. Front. Microbiol. 2019, 10, 1163. [Google Scholar] [CrossRef] [PubMed]
- Klauser, T.; Pohlner, J.; Meyer, T.F. The secretion pathway of IgA protease-type proteins in gram-negative bacteria. Bioessays 1993, 15, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Loveless, B.J.; Saier, M.H., Jr. A novel family of channel-forming, autotransporting, bacterial virulence factors. Mol. Membr. Biol. 1997, 14, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Chang, P.C.; Kuo, C.H.; Tzeng, C.S.; Wang, W.C. Characterization of the C-terminal domain of Helicobacter pylori vacuolating toxin and its relationship with extracellular toxin production. Biochem. Biophys. Res. Commun. 1998, 250, 397–402. [Google Scholar] [CrossRef]
- Fischer, W.; Buhrdorf, R.; Gerland, E.; Haas, R. Outer membrane targeting of passenger proteins by the vacuolating cytotoxin autotransporter of Helicobacter pylori. Infect. Immun. 2001, 69, 6769–6775. [Google Scholar] [CrossRef]
- Nguyen, V.Q.; Caprioli, R.M.; Cover, T.L. Carboxy-terminal proteolytic processing of Helicobacter pylori vacuolating toxin. Infect. Immun. 2001, 69, 543–546. [Google Scholar] [CrossRef]
- Bumann, D.; Aksu, S.; Wendland, M.; Janek, K.; Zimny-Arndt, U.; Sabarth, N.; Meyer, T.F.; Jungblut, P.R. Proteome analysis of secreted proteins of the gastric pathogen Helicobacter pylori. Infect. Immun. 2002, 70, 3396–3403. [Google Scholar] [CrossRef]
- Smith, T.G.; Lim, J.M.; Weinberg, M.V.; Wells, L.; Hoover, T.R. Direct analysis of the extracellular proteome from two strains of Helicobacter pylori. Proteomics 2007, 7, 2240–2245. [Google Scholar] [CrossRef]
- Voss, B.J.; Gaddy, J.A.; McDonald, W.H.; Cover, T.L. Analysis of surface-exposed outer membrane proteins in Helicobacter pylori. J. Bacteriol. 2014, 196, 2455–2471. [Google Scholar] [CrossRef] [PubMed]
- Snider, C.A.; Voss, B.J.; McDonald, W.H.; Cover, T.L. Growth phase-dependent composition of the Helicobacter pylori exoproteome. J. Proteom. 2016, 130, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Weeks, D.L.; Shin, J.M.; Scott, D.R.; Young, M.K.; Sachs, G. Proteins released by Helicobacter pylori in vitro. J. Bacteriol. 2002, 184, 6155–6162. [Google Scholar] [CrossRef] [PubMed]
- Dautin, N.; Barnard, T.J.; Anderson, D.E.; Bernstein, H.D. Cleavage of a bacterial autotransporter by an evolutionarily convergent autocatalytic mechanism. EMBO J. 2007, 26, 1942–1952. [Google Scholar] [CrossRef]
- Tajima, N.; Kawai, F.; Park, S.Y.; Tame, J.R. A novel intein-like autoproteolytic mechanism in autotransporter proteins. J. Mol. Biol. 2010, 402, 645–656. [Google Scholar] [CrossRef]
- Rossetto, O.; de Bernard, M.; Pellizzari, R.; Vitale, G.; Caccin, P.; Schiavo, G.; Montecucco, C. Bacterial toxins with intracellular protease activity. Clin. Chim. Acta 2000, 291, 189–199. [Google Scholar] [CrossRef]
- Reyrat, J.M.; Lanzavecchia, S.; Lupetti, P.; de Bernard, M.; Pagliaccia, C.; Pelicic, V.; Charrel, M.; Ulivieri, C.; Norais, N.; Ji, X.; et al. 3D imaging of the 58 kDa cell binding subunit of the Helicobacter pylori cytotoxin. J. Mol. Biol. 1999, 290, 459–470. [Google Scholar] [CrossRef]
- Vinion-Dubiel, A.D.; McClain, M.S.; Czajkowsky, D.M.; Iwamoto, H.; Ye, D.; Cao, P.; Schraw, W.; Szabo, G.; Blanke, S.R.; Shao, Z.; et al. A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA-induced cell vacuolation. J. Biol. Chem. 1999, 274, 37736–37742. [Google Scholar] [CrossRef]
- Sabarth, N.; Hurvitz, R.; Schmidt, M.; Zimny-Arndt, U.; Jungblut, P.R.; Meyer, T.F.; Bumann, D. Identification of Helicobacter pylori surface proteins by selective proteinase K digestion and antibody phage display. J. Microbiol. Methods 2005, 62, 345–349. [Google Scholar] [CrossRef]
- Ilver, D.; Barone, S.; Mercati, D.; Lupetti, P.; Telford, J.L. Helicobacter pylori toxin VacA is transferred to host cells via a novel contact-dependent mechanism. Cell Microbiol. 2004, 6, 167–174. [Google Scholar] [CrossRef]
- Jarzab, M.; Posselt, G.; Meisner-Kober, N.; Wessler, S. Helicobacter pylori-Derived Outer Membrane Vesicles (OMVs): Role in Bacterial Pathogenesis? Microorganisms 2020, 8, 1328. [Google Scholar] [CrossRef] [PubMed]
- Sommi, P.; Ricci, V.; Fiocca, R.; Necchi, V.; Romano, M.; Telford, J.L.; Solcia, E.; Ventura, U. Persistence of Helicobacter pylori VacA toxin and vacuolating potential in cultured gastric epithelial cells. Am. J. Physiol. 1998, 275, G681–G688. [Google Scholar] [CrossRef] [PubMed]
- Keenan, J.; Day, T.; Neal, S.; Cook, B.; Perez-Perez, G.; Allardyce, R.; Bagshaw, P. A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiol. Lett. 2000, 182, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.; Torres, L.; Espinosa, M.; Fierros-Zarate, G.; Maldonado, V.; Melendez-Zajgla, J. External membrane vesicles from Helicobacter pylori induce apoptosis in gastric epithelial cells. FEMS Microbiol. Lett. 2006, 260, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, E.; Brown, P.A.; Smith, S.M.; Botting, C.H.; Yamaoka, Y.Y.; Terres, A.M.; Kelleher, D.P.; Windle, H.J. Proteomic and functional characterization of the outer membrane vesicles from the gastric pathogen Helicobacter pylori. Proteom. Clin. Appl. 2009, 3, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, A.; Vallstrom, A.; Petzold, K.; Tegtmeyer, N.; Schleucher, J.; Carlsson, S.; Haas, R.; Backert, S.; Wai, S.N.; Grobner, G.; et al. Biochemical and functional characterization of Helicobacter pylori vesicles. Mol. Microbiol. 2010, 77, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Letley, D.; Rhead, J.; Atherton, J.; Robinson, K. Helicobacter pylori membrane vesicles stimulate innate pro- and anti-inflammatory responses and induce apoptosis in Jurkat T cells. Infect. Immun. 2014, 82, 1372–1381. [Google Scholar] [CrossRef]
- Turner, L.; Praszkier, J.; Hutton, M.L.; Steer, D.; Ramm, G.; Kaparakis-Liaskos, M.; Ferrero, R.L. Increased Outer Membrane Vesicle Formation in a Helicobacter pylori tolB Mutant. Helicobacter 2015, 20, 269–283. [Google Scholar] [CrossRef]
- Choi, H.I.; Choi, J.P.; Seo, J.; Kim, B.J.; Rho, M.; Han, J.K.; Kim, J.G. Helicobacter pylori-derived extracellular vesicles increased in the gastric juices of gastric adenocarcinoma patients and induced inflammation mainly via specific targeting of gastric epithelial cells. Exp. Mol. Med. 2017, 49, e330. [Google Scholar] [CrossRef]
- Turner, L.; Bitto, N.J.; Steer, D.L.; Lo, C.; D’Costa, K.; Ramm, G.; Shambrook, M.; Hill, A.F.; Ferrero, R.L.; Kaparakis-Liaskos, M. Helicobacter pylori Outer Membrane Vesicle Size Determines Their Mechanisms of Host Cell Entry and Protein Content. Front. Immunol. 2018, 9, 1466. [Google Scholar] [CrossRef]
- Liu, Q.; Li, X.; Zhang, Y.; Song, Z.; Li, R.; Ruan, H.; Huang, X. Orally-administered outer-membrane vesicles from Helicobacter pylori reduce H. pylori infection via Th2-biased immune responses in mice. Pathog. Dis. 2019, 77, ftz050. [Google Scholar] [CrossRef] [PubMed]
- Zavan, L.; Bitto, N.J.; Johnston, E.L.; Greening, D.W.; Kaparakis-Liaskos, M. Helicobacter pylori Growth Stage Determines the Size, Protein Composition, and Preferential Cargo Packaging of Outer Membrane Vesicles. Proteomics 2019, 19, e1800209. [Google Scholar] [CrossRef] [PubMed]
- Chew, Y.; Chung, H.Y.; Lin, P.Y.; Wu, D.C.; Huang, S.K.; Kao, M.C. Outer Membrane Vesicle Production by Helicobacter pylori Represents an Approach for the Delivery of Virulence Factors CagA, VacA and UreA into Human Gastric Adenocarcinoma (AGS) Cells. Int. J. Mol. Sci. 2021, 22, 3942. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.; Pinto, V.; Fernandes, T.; Malheiro, A.R.; Osório, H.; Figueiredo, C.; Leite, M. Isolation Method and Characterization of Outer Membranes Vesicles of Helicobacter pylori Grown in a Chemically Defined Medium. Front. Microbiol. 2021, 12, 654193. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Li, X.; Wang, J.; Wang, Y.; Zhang, C.; Dai, S.; Wang, X.; Deng, X.; Zhao, L.; Shan, B. Outer Membrane Vesicles Secreted by Helicobacter pylori Transmitting Gastric Pathogenic Virulence Factors. ACS Omega 2022, 7, 240–258. [Google Scholar] [CrossRef] [PubMed]
- Saberi, S.; Esmaeili, M.; Saghiri, R.; Shekari, F.; Mohammadi, M. Assessment of the mixed origin of the gastric epithelial extracellular vesicles in acellular transfer of Helicobacter pylori toxins and a systematic review. Microb. Pathog. 2023, 177, 106024. [Google Scholar] [CrossRef] [PubMed]
- Palacios, E.; Lobos-González, L.; Guerrero, S.; Kogan, M.J.; Shao, B.; Heinecke, J.W.; Quest, A.F.G.; Leyton, L.; Valenzuela-Valderrama, M. Helicobacter pylori outer membrane vesicles induce astrocyte reactivity through nuclear factor-κappa B activation and cause neuronal damage in vivo in a murine model. J. Neuroinflammation 2023, 20, 66. [Google Scholar] [CrossRef] [PubMed]
- Garner, J.A.; Cover, T.L. Binding and internalization of the Helicobacter pylori vacuolating cytotoxin by epithelial cells. Infect. Immun. 1996, 64, 4197–4203. [Google Scholar] [CrossRef]
- Cover, T.L.; Hanson, P.I.; Heuser, J.E. Acid-induced dissociation of VacA, the Helicobacter pylori vacuolating cytotoxin, reveals its pattern of assembly. J. Cell Biol. 1997, 138, 759–769. [Google Scholar] [CrossRef]
- Ye, D.; Willhite, D.C.; Blanke, S.R. Identification of the minimal intracellular vacuolating domain of the Helicobacter pylori vacuolating toxin. J. Biol. Chem. 1999, 274, 9277–9282. [Google Scholar] [CrossRef]
- Ye, D.; Blanke, S.R. Mutational analysis of the Helicobacter pylori vacuolating toxin amino terminus: Identification of amino acids essential for cellular vacuolation. Infect. Immun. 2000, 68, 4354–4357. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.J.; McClain, M.S.; Cover, T.L. Interactions between p-33 and p-55 domains of the Helicobacter pylori vacuolating cytotoxin (VacA). J. Biol. Chem. 2004, 279, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, M.; Panchal, H. In silico investigation and structural characterization of virulent factor and a metallo peptidase present in Helicobacter pylori strain J99. Interdiscip. Sci. 2012, 4, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Lupetti, P.; Heuser, J.E.; Manetti, R.; Massari, P.; Lanzavecchia, S.; Bellon, P.L.; Dallai, R.; Rappuoli, R.; Telford, J.L. Oligomeric and subunit structure of the Helicobacter pylori vacuolating cytotoxin. J. Cell Biol. 1996, 133, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Blanke, S.R. Functional complementation reveals the importance of intermolecular monomer interactions for Helicobacter pylori VacA vacuolating activity. Mol. Microbiol. 2002, 43, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Adrian, M.; Cover, T.L.; Dubochet, J.; Heuser, J.E. Multiple oligomeric states of the Helicobacter pylori vacuolating toxin demonstrated by cryo-electron microscopy. J. Mol. Biol. 2002, 318, 121–133. [Google Scholar] [CrossRef] [PubMed]
- El-Bez, C.; Adrian, M.; Dubochet, J.; Cover, T.L. High resolution structural analysis of Helicobacter pylori VacA toxin oligomers by cryo-negative staining electron microscopy. J. Struct. Biol. 2005, 151, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, S.; Bellon, P.L.; Lupetti, P.; Dallai, R.; Rappuoli, R.; Telford, J.L. Three-dimensional reconstruction of metal replicas of the Helicobacter pylori vacuolating cytotoxin. J. Struct. Biol. 1998, 121, 9–18. [Google Scholar] [CrossRef]
- Chambers, M.G.; Pyburn, T.M.; González-Rivera, C.; Collier, S.E.; Eli, I.; Yip, C.K.; Takizawa, Y.; Lacy, D.B.; Cover, T.L.; Ohi, M.D. Structural analysis of the oligomeric states of Helicobacter pylori VacA toxin. J. Mol. Biol. 2013, 425, 524–535. [Google Scholar] [CrossRef]
- Pyburn, T.M.; Foegeding, N.J.; González-Rivera, C.; McDonald, N.A.; Gould, K.L.; Cover, T.L.; Ohi, M.D. Structural organization of membrane-inserted hexamers formed by Helicobacter pylori VacA toxin. Mol. Microbiol. 2016, 102, 22–36. [Google Scholar] [CrossRef]
- Su, M.; Erwin, A.L.; Campbell, A.M.; Pyburn, T.M.; Salay, L.E.; Hanks, J.L.; Lacy, D.B.; Akey, D.L.; Cover, T.L.; Ohi, M.D. Cryo-EM Analysis Reveals Structural Basis of Helicobacter pylori VacA Toxin Oligomerization. J. Mol. Biol. 2019, 431, 1956–1965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, H.; Li, S.; Pintilie, G.D.; Mou, T.C.; Gao, Y.; Zhang, Q.; van den Bedem, H.; Schmid, M.F.; Au, S.W.N.; et al. Cryo-EM structures of Helicobacter pylori vacuolating cytotoxin A oligomeric assemblies at near-atomic resolution. Proc. Natl. Acad. Sci. USA 2019, 116, 6800–6805. [Google Scholar] [CrossRef] [PubMed]
- Gangwer, K.A.; Mushrush, D.J.; Stauff, D.L.; Spiller, B.; McClain, M.S.; Cover, T.L.; Lacy, D.B. Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc. Natl. Acad. Sci. USA 2007, 104, 16293–16298. [Google Scholar] [CrossRef] [PubMed]
- González-Rivera, C.; Campbell, A.M.; Rutherford, S.A.; Pyburn, T.M.; Foegeding, N.J.; Barke, T.L.; Spiller, B.W.; McClain, M.S.; Ohi, M.D.; Lacy, D.B.; et al. A Nonoligomerizing Mutant Form of Helicobacter pylori VacA Allows Structural Analysis of the p33 Domain. Infect. Immun. 2016, 84, 2662–2670. [Google Scholar] [CrossRef] [PubMed]
- Domańska, G.; Motz, C.; Meinecke, M.; Harsman, A.; Papatheodorou, P.; Reljic, B.; Dian-Lothrop, E.A.; Galmiche, A.; Kepp, O.; Becker, L.; et al. Helicobacter pylori VacA toxin/subunit p34: Targeting of an anion channel to the inner mitochondrial membrane. PLoS Pathog. 2010, 6, e1000878. [Google Scholar] [CrossRef] [PubMed]
- Seto, K.; Hayashi-Kuwabara, Y.; Yoneta, T.; Suda, H.; Tamaki, H. Vacuolation induced by cytotoxin from Helicobacter pylori is mediated by the EGF receptor in HeLa cells. FEBS Lett. 1998, 431, 347–350. [Google Scholar] [CrossRef]
- McClain, M.S.; Schraw, W.; Ricci, V.; Boquet, P.; Cover, T.L. Acid activation of Helicobacter pylori vacuolating cytotoxin (VacA) results in toxin internalization by eukaryotic cells. Mol. Microbiol. 2000, 37, 433–442. [Google Scholar] [CrossRef]
- Yahiro, K.; Wada, A.; Nakayama, M.; Kimura, T.; Ogushi, K.; Niidome, T.; Aoyagi, H.; Yoshino, K.; Yonezawa, K.; Moss, J.; et al. Protein-tyrosine phosphatase alpha, RPTP alpha, is a Helicobacter pylori VacA receptor. J. Biol. Chem. 2003, 278, 19183–19189. [Google Scholar] [CrossRef]
- De Guzman, B.B.; Hisatsune, J.; Nakayama, M.; Yahiro, K.; Wada, A.; Yamasaki, E.; Nishi, Y.; Yamazaki, S.; Azuma, T.; Ito, Y.; et al. Cytotoxicity and recognition of receptor-like protein tyrosine phosphatases, RPTPalpha and RPTPbeta, by Helicobacter pylori m2VacA. Cell Microbiol. 2005, 7, 1285–1293. [Google Scholar] [CrossRef]
- Yahiro, K.; Niidome, T.; Kimura, M.; Hatakeyama, T.; Aoyagi, H.; Kurazono, H.; Imagawa, K.; Wada, A.; Moss, J.; Hirayama, T. Activation of Helicobacter pylori VacA toxin by alkaline or acid conditions increases its binding to a 250-kDa receptor protein-tyrosine phosphatase beta. J. Biol. Chem. 1999, 274, 36693–36699. [Google Scholar] [CrossRef]
- Yahiro, K.; Satoh, M.; Nakano, M.; Hisatsune, J.; Isomoto, H.; Sap, J.; Suzuki, H.; Nomura, F.; Noda, M.; Moss, J.; et al. Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J. Biol. Chem. 2012, 287, 31104–31115. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Hisatsune, J.; Yamasaki, E.; Nishi, Y.; Wada, A.; Kurazono, H.; Sap, J.; Yahiro, K.; Moss, J.; Hirayama, T. Clustering of Helicobacter pylori VacA in lipid rafts, mediated by its receptor, receptor-like protein tyrosine phosphatase beta, is required for intoxication in AZ-521 Cells. Infect. Immun. 2006, 74, 6571–6580. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, A.; Shirasaka, D.; Yamamoto, S.; Ota, H.; Yahiro, K.; Fukada, M.; Shintani, T.; Wada, A.; Aoyama, N.; Hirayama, T.; et al. Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat. Genet. 2003, 33, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Sewald, X.; Gebert-Vogl, B.; Prassl, S.; Barwig, I.; Weiss, E.; Fabbri, M.; Osicka, R.; Schiemann, M.; Busch, D.H.; Semmrich, M.; et al. Integrin subunit CD18 Is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe 2008, 3, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Czajkowsky, D.M.; Iwamoto, H.; Cover, T.L.; Shao, Z. The vacuolating toxin from Helicobacter pylori forms hexameric pores in lipid bilayers at low pH. Proc. Natl. Acad. Sci. USA 1999, 96, 2001–2006. [Google Scholar] [CrossRef] [PubMed]
- Szabò, I.; Brutsche, S.; Tombola, F.; Moschioni, M.; Satin, B.; Telford, J.L.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 1999, 18, 5517–5527. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, H.; Czajkowsky, D.M.; Cover, T.L.; Szabo, G.; Shao, Z. VacA from Helicobacter pylori: A hexameric chloride channel. FEBS Lett. 1999, 450, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Tombola, F.; Carlesso, C.; Szabò, I.; de Bernard, M.; Reyrat, J.M.; Telford, J.L.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Helicobacter pylori vacuolating toxin forms anion-selective channels in planar lipid bilayers: Possible implications for the mechanism of cellular vacuolation. Biophys. J. 1999, 76, 1401–1409. [Google Scholar] [CrossRef]
- Tombola, F.; Oregna, F.; Brutsche, S.; Szabò, I.; Del Giudice, G.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Inhibition of the vacuolating and anion channel activities of the VacA toxin of Helicobacter pylori. FEBS Lett. 1999, 460, 221–225. [Google Scholar] [CrossRef]
- Ivie, S.E.; McClain, M.S.; Torres, V.J.; Algood, H.M.; Lacy, D.B.; Yang, R.; Blanke, S.R.; Cover, T.L. Helicobacter pylori VacA subdomain required for intracellular toxin activity and assembly of functional oligomeric complexes. Infect. Immun. 2008, 76, 2843–2851. [Google Scholar] [CrossRef]
- Genisset, C.; Galeotti, C.L.; Lupetti, P.; Mercati, D.; Skibinski, D.A.; Barone, S.; Battistutta, R.; de Bernard, M.; Telford, J.L. A Helicobacter pylori vacuolating toxin mutant that fails to oligomerize has a dominant negative phenotype. Infect. Immun. 2006, 74, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Debellis, L.; Papini, E.; Caroppo, R.; Montecucco, C.; Curci, S. Helicobacter pylori cytotoxin VacA increases alkaline secretion in gastric epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1440–G1448. [Google Scholar] [CrossRef] [PubMed]
- Schraw, W.; Li, Y.; McClain, M.S.; van der Goot, F.G.; Cover, T.L. Association of Helicobacter pylori vacuolating toxin (VacA) with lipid rafts. J. Biol. Chem. 2002, 277, 34642–34650. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.S.; Iwamoto, H.; Cao, P.; Vinion-Dubiel, A.D.; Li, Y.; Szabo, G.; Shao, Z.; Cover, T.L. Essential role of a GXXXG motif for membrane channel formation by Helicobacter pylori vacuolating toxin. J. Biol. Chem. 2003, 278, 12101–12108. [Google Scholar] [CrossRef] [PubMed]
- Caston, R.R.; Sierra, J.C.; Foegeding, N.J.; Truelock, M.D.; Campbell, A.M.; Frick-Cheng, A.E.; Bimczok, D.; Wilson, K.T.; McClain, M.S.; Cover, T.L. Functional Properties of Helicobacter pylori VacA Toxin m1 and m2 Variants. Infect. Immun. 2020, 88, e00032-20. [Google Scholar] [CrossRef] [PubMed]
- Palframan, S.L.; Kwok, T.; Gabriel, K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell Infect. Microbiol. 2012, 2, 92. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, N.C.; Monzo, P.; Kaddai, V.; Doye, A.; Ricci, V.; Boquet, P. Helicobacter pylori VacA cytotoxin: A probe for a clathrin-independent and Cdc42-dependent pinocytic pathway routed to late endosomes. Mol. Biol. Cell 2005, 16, 4852–4866. [Google Scholar] [CrossRef] [PubMed]
- Boquet, P.; Ricci, V. Intoxication strategy of Helicobacter pylori VacA toxin. Trends Microbiol. 2012, 20, 165–174. [Google Scholar] [CrossRef]
- Ulhuq, F.R.; Mariano, G. Bacterial pore-forming toxins. Microbiology 2022, 168, 001154. [Google Scholar] [CrossRef]
- Yahiro, K.; Wada, A.; Yamasaki, E.; Nakayama, M.; Nishi, Y.; Hisatsune, J.; Morinaga, N.; Sap, J.; Noda, M.; Moss, J.; et al. Essential domain of receptor tyrosine phosphatase beta (RPTPbeta) for interaction with Helicobacter pylori vacuolating cytotoxin. J. Biol. Chem. 2004, 279, 51013–51021. [Google Scholar] [CrossRef]
- Satoh, K.; Hirayama, T.; Takano, K.; Suzuki-Inoue, K.; Sato, T.; Ohta, M.; Nakagomi, J.; Ozaki, Y. VacA, the vacuolating cytotoxin of Helicobacter pylori, binds to multimerin 1 on human platelets. Thromb. J. 2013, 11, 23. [Google Scholar] [CrossRef]
- Utt, M.; Danielsson, B.; Wadström, T. Helicobacter pylori vacuolating cytotoxin binding to a putative cell surface receptor, heparan sulfate, studied by surface plasmon resonance. FEMS Immunol. Med. Microbiol. 2001, 30, 109–113. [Google Scholar] [CrossRef]
- Gupta, V.R.; Patel, H.K.; Kostolansky, S.S.; Ballivian, R.A.; Eichberg, J.; Blanke, S.R. Sphingomyelin functions as a novel receptor for Helicobacter pylori VacA. PLoS Pathog. 2008, 4, e1000073. [Google Scholar] [CrossRef]
- Gupta, V.R.; Wilson, B.A.; Blanke, S.R. Sphingomyelin is important for the cellular entry and intracellular localization of Helicobacter pylori VacA. Cell Microbiol. 2010, 12, 1517–1533. [Google Scholar] [CrossRef]
- Roche, N.; Ilver, D.; Angström, J.; Barone, S.; Telford, J.L.; Teneberg, S. Human gastric glycosphingolipids recognized by Helicobacter pylori vacuolating cytotoxin VacA. Microbes Infect. 2007, 9, 605–614. [Google Scholar] [CrossRef]
- Molinari, M.; Galli, C.; de Bernard, M.; Norais, N.; Ruysschaert, J.M.; Rappuoli, R.; Montecucco, C. The acid activation of Helicobacter pylori toxin VacA: Structural and membrane binding studies. Biochem. Biophys. Res. Commun. 1998, 248, 334–340. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Geisse, N.A.; Cover, T.L.; Henderson, R.M.; Edwardson, J.M. Targeting of Helicobacter pylori vacuolating toxin to lipid raft membrane domains analysed by atomic force microscopy. Biochem. J. 2004, 381, 911–917. [Google Scholar] [CrossRef]
- Gauthier, N.C.; Ricci, V.; Gounon, P.; Doye, A.; Tauc, M.; Poujeol, P.; Boquet, P. Glycosylphosphatidylinositol-anchored proteins and actin cytoskeleton modulate chloride transport by channels formed by the Helicobacter pylori vacuolating cytotoxin VacA in HeLa cells. J. Biol. Chem. 2004, 279, 9481–9489. [Google Scholar] [CrossRef]
- Kuo, C.H.; Wang, W.C. Binding and internalization of Helicobacter pylori VacA via cellular lipid rafts in epithelial cells. Biochem. Biophys. Res. Commun. 2003, 303, 640–644. [Google Scholar] [CrossRef]
- Lai, C.H.; Chang, Y.C.; Du, S.Y.; Wang, H.J.; Kuo, C.H.; Fang, S.H.; Fu, H.W.; Lin, H.H.; Chiang, A.S.; Wang, W.C. Cholesterol depletion reduces Helicobacter pylori CagA translocation and CagA-induced responses in AGS cells. Infect. Immun. 2008, 76, 3293–3303. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.K.; Willhite, D.C.; Patel, R.M.; Ye, D.; Williams, C.L.; Torres, E.M.; Marty, K.B.; MacDonald, R.A.; Blanke, S.R. Plasma membrane cholesterol modulates cellular vacuolation induced by the Helicobacter pylori vacuolating cytotoxin. Infect. Immun. 2002, 70, 4112–4123. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Galmiche, A.; Doye, A.; Necchi, V.; Solcia, E.; Boquet, P. High cell sensitivity to Helicobacter pylori VacA toxin depends on a GPI-anchored protein and is not blocked by inhibition of the clathrin-mediated pathway of endocytosis. Mol. Biol. Cell 2000, 11, 3897–3909. [Google Scholar] [CrossRef] [PubMed]
- Escribá, P.V.; González-Ros, J.M.; Goñi, F.M.; Kinnunen, P.K.; Vigh, L.; Sánchez-Magraner, L.; Fernández, A.M.; Busquets, X.; Horváth, I.; Barceló-Coblijn, G. Membranes: A meeting point for lipids, proteins and therapies. J. Cell Mol. Med. 2008, 12, 829–875. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.; Mengist, H.M.; Shi, C.; Zhang, C.; Wang, B.; Li, T.; Huang, Y.; Xu, Y.; Jin, T. Structural Basis of the Pore-Forming Toxin/Membrane Interaction. Toxins 2021, 13, 128. [Google Scholar] [CrossRef] [PubMed]
- de Bernard, M.; Papini, E.; de Filippis, V.; Gottardi, E.; Telford, J.; Manetti, R.; Fontana, A.; Rappuoli, R.; Montecucco, C. Low pH activates the vacuolating toxin of Helicobacter pylori, which becomes acid and pepsin resistant. J. Biol. Chem. 1995, 270, 23937–23940. [Google Scholar] [CrossRef] [PubMed]
- Reyrat, J.M.; Charrel, M.; Pagliaccia, C.; Burroni, D.; Lupetti, P.; de Bernard, M.; Ji, X.; Norais, N.; Papini, E.; Dallai, R.; et al. Characterisation of a monoclonal antibody and its use to purify the cytotoxin of Helicobacter pylori. FEMS Microbiol. Lett. 1998, 165, 79–84. [Google Scholar] [CrossRef]
- Moll, G.; Papini, E.; Colonna, R.; Burroni, D.; Telford, J.; Rappuoli, R.; Montecucco, C. Lipid interaction of the 37-kDa and 58-kDa fragments of the Helicobacter pylori cytotoxin. Eur. J. Biochem. 1995, 234, 947–952. [Google Scholar] [CrossRef]
- Pagliaccia, C.; Wang, X.M.; Tardy, F.; Telford, J.L.; Ruysschaert, J.M.; Cabiaux, V. Structure and interaction of VacA of Helicobacter pylori with a lipid membrane. Eur. J. Biochem. 2000, 267, 104–109. [Google Scholar] [CrossRef]
- Caso, G.C.; McClain, M.S.; Erwin, A.L.; Truelock, M.D.; Campbell, A.M.; Leasure, C.S.; Nagel, M.; Schey, K.L.; Lacy, D.B.; Ohi, M.D.; et al. Functional Properties of Oligomeric and Monomeric Forms of Helicobacter pylori VacA Toxin. Infect. Immun. 2021, 89, e0034821. [Google Scholar] [CrossRef]
- McClain, M.S.; Cao, P.; Cover, T.L. Amino-terminal hydrophobic region of Helicobacter pylori vacuolating cytotoxin (VacA) mediates transmembrane protein dimerization. Infect. Immun. 2001, 69, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.S.; Czajkowsky, D.M.; Torres, V.J.; Szabo, G.; Shao, Z.; Cover, T.L. Random mutagenesis of Helicobacter pylori vacA to identify amino acids essential for vacuolating cytotoxic activity. Infect. Immun. 2006, 74, 6188–6195. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.J.; Ivie, S.E.; McClain, M.S.; Cover, T.L. Functional properties of the p33 and p55 domains of the Helicobacter pylori vacuolating cytotoxin. J. Biol. Chem. 2005, 280, 21107–21114. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.J.; McClain, M.S.; Cover, T.L. Mapping of a domain required for protein-protein interactions and inhibitory activity of a Helicobacter pylori dominant-negative VacA mutant protein. Infect. Immun. 2006, 74, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Yahiro, K.; Hirayama, T.; Moss, J.; Noda, M. New Insights into VacA Intoxication Mediated through Its Cell Surface Receptors. Toxins 2016, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.S.; Cao, P.; Iwamoto, H.; Vinion-Dubiel, A.D.; Szabo, G.; Shao, Z.; Cover, T.L. A 12-amino-acid segment, present in type s2 but not type s1 Helicobacter pylori VacA proteins, abolishes cytotoxin activity and alters membrane channel formation. J. Bacteriol. 2001, 183, 6499–6508. [Google Scholar] [CrossRef]
- González-Rivera, C.; Algood, H.M.; Radin, J.N.; McClain, M.S.; Cover, T.L. The intermediate region of Helicobacter pylori VacA is a determinant of toxin potency in a Jurkat T cell assay. Infect. Immun. 2012, 80, 2578–2588. [Google Scholar] [CrossRef]
- Pagliaccia, C.; de Bernard, M.; Lupetti, P.; Ji, X.; Burroni, D.; Cover, T.L.; Papini, E.; Rappuoli, R.; Telford, J.L.; Reyrat, J.M. The m2 form of the Helicobacter pylori cytotoxin has cell type-specific vacuolating activity. Proc. Natl. Acad. Sci. USA 1998, 95, 10212–10217. [Google Scholar] [CrossRef]
- Wang, W.C.; Wang, H.J.; Kuo, C.H. Two distinctive cell binding patterns by vacuolating toxin fused with glutathione S-transferase: One high-affinity m1-specific binding and the other lower-affinity binding for variant m forms. Biochemistry 2001, 40, 11887–11896. [Google Scholar] [CrossRef]
- Atherton, J.C.; Peek, R.M., Jr.; Tham, K.T.; Cover, T.L.; Blaser, M.J. Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology 1997, 112, 92–99. [Google Scholar] [CrossRef]
- Ji, X.; Fernandez, T.; Burroni, D.; Pagliaccia, C.; Atherton, J.C.; Reyrat, J.M.; Rappuoli, R.; Telford, J.L. Cell specificity of Helicobacter pylori cytotoxin is determined by a short region in the polymorphic midregion. Infect. Immun. 2000, 68, 3754–3757. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, D.A.; Genisset, C.; Barone, S.; Telford, J.L. The cell-specific phenotype of the polymorphic vacA midregion is independent of the appearance of the cell surface receptor protein tyrosine phosphatase beta. Infect. Immun. 2006, 74, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Letley, D.P.; Rhead, J.L.; Twells, R.J.; Dove, B.; Atherton, J.C. Determinants of non-toxicity in the gastric pathogen Helicobacter pylori. J. Biol. Chem. 2003, 278, 26734–26741. [Google Scholar] [CrossRef] [PubMed]
- Korona-Glowniak, I.; Cichoz-Lach, H.; Siwiec, R.; Andrzejczuk, S.; Glowniak, A.; Matras, P.; Malm, A. Antibiotic Resistance and Genotypes of Helicobacter pylori Strains in Patients with Gastroduodenal Disease in Southeast Poland. J. Clin. Med. 2019, 8, 71. [Google Scholar] [CrossRef]
- Jouimyi, M.R.; Bounder, G.; Essaidi, I.; Boura, H.; Badre, W.; Benomar, H.; Zerouali, K.; Lebrazi, H.; Kettani, A.; Maachi, F. Association of Helicobacter pylori vacA polymorphisms with the risk of gastric precancerous lesions in a Moroccan population. J. Infect. Dev. Ctries. 2021, 15, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Keikha, M.; Ali-Hassanzadeh, M.; Karbalaei, M. Association of Helicobacter pylori vacA genotypes and peptic ulcer in Iranian population: A systematic review and meta-analysis. BMC Gastroenterol. 2020, 20, 266. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.; Zambon, C.F.; Letley, D.P.; Stranges, A.; Marchet, A.; Rhead, J.L.; Schiavon, S.; Guariso, G.; Ceroti, M.; Nitti, D.; et al. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology 2008, 135, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Louw, J.A.; Kidd, M.S.; Kummer, A.F.; Taylor, K.; Kotze, U.; Hanslo, D. The relationship between Helicobacter pylori infection, the virulence genotypes of the infecting strain and gastric cancer in the African setting. Helicobacter 2001, 6, 268–273. [Google Scholar] [CrossRef]
- Phan, T.N.; Santona, A.; Tran, V.H.; Tran, T.N.H.; Le, V.A.; Cappuccinelli, P.; Rubino, S.; Paglietti, B. Genotyping of Helicobacter pylori shows high diversity of strains circulating in central Vietnam. Infect. Genet. Evol. 2017, 52, 19–25. [Google Scholar] [CrossRef]
- Memon, A.A.; Hussein, N.R.; Miendje Deyi, V.Y.; Burette, A.; Atherton, J.C. Vacuolating cytotoxin genotypes are strong markers of gastric cancer and duodenal ulcer-associated Helicobacter pylori strains: A matched case-control study. J. Clin. Microbiol. 2014, 52, 2984–2989. [Google Scholar] [CrossRef]
- Chang, W.L.; Yeh, Y.C.; Sheu, B.S. The impacts of H. pylori virulence factors on the development of gastroduodenal diseases. J. Biomed. Sci. 2018, 25, 68. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.; Machado, J.C.; Pharoah, P.; Seruca, R.; Sousa, S.; Carvalho, R.; Capelinha, A.F.; Quint, W.; Caldas, C.; van Doorn, L.J.; et al. Helicobacter pylori and interleukin 1 genotyping: An opportunity to identify high-risk individuals for gastric carcinoma. J. Natl. Cancer Inst. 2002, 94, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Njenga, P.; Njau, A.; Moloo, Z.; Revathi, G.; Tshibangu, E.; Yamaoka, Y. Pattern and trends of Helicobacter pylori genotypes in gastric cancer: A Kenyan 8-year study. Front. Med. 2023, 10, 1119513. [Google Scholar] [CrossRef]
- Lima, V.P.; Silva-Fernandes, I.J.; Alves, M.K.; Rabenhorst, S.H. Prevalence of Helicobacter pylori genotypes (vacA, cagA, cagE and virB11) in gastric cancer in Brazilian’s patients: An association with histopathological parameters. Cancer Epidemiol. 2011, 35, e32–e37. [Google Scholar] [CrossRef] [PubMed]
- El Khadir, M.; Boukhris Alaoui, S.; Benajah, D.A.; Ibrahimi, S.A.; Chbani, L.; El Abkari, M.; Bennani, B. VacA genotypes and cagA-EPIYA-C motifs of Helicobacter pylori and gastric histopathological lesions. Int. J. Cancer 2020, 147, 3206–3214. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.M.; Machado, J.C.; Letley, D.; Atherton, J.C.; Pardo, M.L.; Gonzalez, C.A.; Carneiro, F.; Figueiredo, C. A novel method for genotyping the Helicobacter pylori vacA intermediate region directly in gastric biopsy specimens. J. Clin. Microbiol. 2012, 50, 3983–3989. [Google Scholar] [CrossRef] [PubMed]
- Douraghi, M.; Talebkhan, Y.; Zeraati, H.; Ebrahimzadeh, F.; Nahvijoo, A.; Morakabati, A.; Ghafarpour, M.; Esmaili, M.; Bababeik, M.; Oghalaie, A.; et al. Multiple gene status in Helicobacter pylori strains and risk of gastric cancer development. Digestion 2009, 80, 200–207. [Google Scholar] [CrossRef]
- Miehlke, S.; Kirsch, C.; Agha-Amiri, K.; Günther, T.; Lehn, N.; Malfertheiner, P.; Stolte, M.; Ehninger, G.; Bayerdörffer, E. The Helicobacter pylori vacA s1, m1 genotype and cagA is associated with gastric carcinoma in Germany. Int. J. Cancer 2000, 87, 322–327. [Google Scholar] [CrossRef]
- Winter, J.A.; Letley, D.P.; Cook, K.W.; Rhead, J.L.; Zaitoun, A.A.; Ingram, R.J.; Amilon, K.R.; Croxall, N.J.; Kaye, P.V.; Robinson, K.; et al. A role for the vacuolating cytotoxin, VacA, in colonization and Helicobacter pylori-induced metaplasia in the stomach. J. Infect. Dis. 2014, 210, 954–963. [Google Scholar] [CrossRef]
- Kim, S.; Chamberlain, A.K.; Bowie, J.U. Membrane channel structure of Helicobacter pylori vacuolating toxin: Role of multiple GXXXG motifs in cylindrical channels. Proc. Natl. Acad. Sci. USA 2004, 101, 5988–5991. [Google Scholar] [CrossRef]
- de Bernard, M.; Burroni, D.; Papini, E.; Rappuoli, R.; Telford, J.; Montecucco, C. Identification of the Helicobacter pylori VacA toxin domain active in the cell cytosol. Infect. Immun. 1998, 66, 6014–6016. [Google Scholar] [CrossRef]
- Campello, S.; Tombola, F.; Cabrini, G.; Zoratti, M. The vacuolating toxin of Helicobacter pylori mimicks the CFTR-mediated chloride conductance. FEBS Lett. 2002, 532, 237–240. [Google Scholar] [CrossRef]
- Tombola, F.; Pagliaccia, C.; Campello, S.; Telford, J.L.; Montecucco, C.; Papini, E.; Zoratti, M. How the loop and middle regions influence the properties of Helicobacter pylori VacA channels. Biophys. J. 2001, 81, 3204–3215. [Google Scholar] [CrossRef]
- Tombola, F.; Morbiato, L.; Del Giudice, G.; Rappuoli, R.; Zoratti, M.; Papini, E. The Helicobacter pylori VacA toxin is a urea permease that promotes urea diffusion across epithelia. J. Clin. Investig. 2001, 108, 929–937. [Google Scholar] [CrossRef]
- Tombola, F.; Del Giudice, G.; Papini, E.; Zoratti, M. Blockers of VacA provide insights into the structure of the pore. Biophys. J. 2000, 79, 863–873. [Google Scholar] [CrossRef]
- Ricci, V.; Sommi, P.; Fiocca, R.; Romano, M.; Solcia, E.; Ventura, U. Helicobacter pylori vacuolating toxin accumulates within the endosomal-vacuolar compartment of cultured gastric cells and potentiates the vacuolating activity of ammonia. J. Pathol. 1997, 183, 453–459. [Google Scholar] [CrossRef]
- Willhite, D.C.; Blanke, S.R. Helicobacter pylori vacuolating cytotoxin enters cells, localizes to the mitochondria, and induces mitochondrial membrane permeability changes correlated to toxin channel activity. Cell Microbiol. 2004, 6, 143–154. [Google Scholar] [CrossRef]
- Ahmed, A.A.Q.; Besio, R.; Xiao, L.; Forlino, A. Outer Membrane Vesicles (OMVs) as Biomedical Tools and Their Relevance as Immune-Modulating Agents against H. pylori Infections: Current Status and Future Prospects. Int. J. Mol. Sci. 2023, 24, 8542. [Google Scholar] [CrossRef]
- Ricci, V.; Chiozzi, V.; Necchi, V.; Oldani, A.; Romano, M.; Solcia, E.; Ventura, U. Free-soluble and outer membrane vesicle-associated VacA from Helicobacter pylori: Two forms of release, a different activity. Biochem. Biophys. Res. Commun. 2005, 337, 173–178. [Google Scholar] [CrossRef]
- Heczko, U.; Smith, V.C.; Mark Meloche, R.; Buchan, A.M.; Finlay, B.B. Characteristics of Helicobacter pylori attachment to human primary antral epithelial cells. Microbes Infect. 2000, 2, 1669–1676. [Google Scholar] [CrossRef]
- Parker, H.; Chitcholtan, K.; Hampton, M.B.; Keenan, J.I. Uptake of Helicobacter pylori outer membrane vesicles by gastric epithelial cells. Infect. Immun. 2010, 78, 5054–5061. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, A.; Nygard Skalman, L.; Obi, I.; Lundmark, R.; Arnqvist, A. Uptake of Helicobacter pylori vesicles is facilitated by clathrin-dependent and clathrin-independent endocytic pathways. mBio 2014, 5, e00979-14. [Google Scholar] [CrossRef] [PubMed]
- Kaparakis, M.; Turnbull, L.; Carneiro, L.; Firth, S.; Coleman, H.A.; Parkington, H.C.; Le Bourhis, L.; Karrar, A.; Viala, J.; Mak, J.; et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol. 2010, 12, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Utsch, C.; Haas, R. VacA’s Induction of VacA-Containing Vacuoles (VCVs) and Their Immunomodulatory Activities on Human T Cells. Toxins 2016, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, P.; Nabi, I.R. Regulation of raft-dependent endocytosis. J. Cell Mol. Med. 2007, 11, 644–653. [Google Scholar] [CrossRef]
- Sewald, X.; Jiménez-Soto, L.; Haas, R. PKC-dependent endocytosis of the Helicobacter pylori vacuolating cytotoxin in primary T lymphocytes. Cell Microbiol. 2011, 13, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Hotchin, N.A.; Cover, T.L.; Akhtar, N. Cell vacuolation induced by the VacA cytotoxin of Helicobacter pylori is regulated by the Rac1 GTPase. J. Biol. Chem. 2000, 275, 14009–14012. [Google Scholar] [CrossRef]
- Oldani, A.; Cormont, M.; Hofman, V.; Chiozzi, V.; Oregioni, O.; Canonici, A.; Sciullo, A.; Sommi, P.; Fabbri, A.; Ricci, V.; et al. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 2009, 5, e1000603. [Google Scholar] [CrossRef]
- Gauthier, N.C.; Monzo, P.; Gonzalez, T.; Doye, A.; Oldani, A.; Gounon, P.; Ricci, V.; Cormont, M.; Boquet, P. Early endosomes associated with dynamic F-actin structures are required for late trafficking of H. pylori VacA toxin. J. Cell Biol. 2007, 177, 343–354. [Google Scholar] [CrossRef]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-independent pathways of endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a016758. [Google Scholar] [CrossRef]
- Elkin, S.R.; Lakoduk, A.M.; Schmid, S.L. Endocytic pathways and endosomal trafficking: A primer. Wien. Med. Wochenschr. 2016, 166, 196–204. [Google Scholar] [CrossRef]
- Sabharanjak, S.; Sharma, P.; Parton, R.G.; Mayor, S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell 2002, 2, 411–423. [Google Scholar] [CrossRef]
- Ricci, V.; Romano, M.; Boquet, P. Molecular cross-talk between Helicobacter pylori and human gastric mucosa. World J. Gastroenterol. 2011, 17, 1383–1399. [Google Scholar] [CrossRef]
- Papini, E.; de Bernard, M.; Milia, E.; Bugnoli, M.; Zerial, M.; Rappuoli, R.; Montecucco, C. Cellular vacuoles induced by Helicobacter pylori originate from late endosomal compartments. Proc. Natl. Acad. Sci. USA 1994, 91, 9720–9724. [Google Scholar] [CrossRef]
- Papini, E.; Gottardi, E.; Satin, B.; de Bernard, M.; Massari, P.; Telford, J.; Rappuoli, R.; Sato, S.B.; Montecucco, C. The vacuolar ATPase proton pump is present on intracellular vacuoles induced by Helicobacter pylori. J. Med. Microbiol. 1996, 45, 84–89. [Google Scholar] [CrossRef]
- Molinari, M.; Galli, C.; Norais, N.; Telford, J.L.; Rappuoli, R.; Luzio, J.P.; Montecucco, C. Vacuoles induced by Helicobacter pylori toxin contain both late endosomal and lysosomal markers. J. Biol. Chem. 1997, 272, 25339–25344. [Google Scholar] [CrossRef]
- Li, Y.; Wandinger-Ness, A.; Goldenring, J.R.; Cover, T.L. Clustering and redistribution of late endocytic compartments in response to Helicobacter pylori vacuolating toxin. Mol. Biol. Cell 2004, 15, 1946–1959. [Google Scholar] [CrossRef]
- Yamasaki, E.; Wada, A.; Kumatori, A.; Nakagawa, I.; Funao, J.; Nakayama, M.; Hisatsune, J.; Kimura, M.; Moss, J.; Hirayama, T. Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J. Biol. Chem. 2006, 281, 11250–11259. [Google Scholar] [CrossRef]
- Terebiznik, M.R.; Vazquez, C.L.; Torbicki, K.; Banks, D.; Wang, T.; Hong, W.; Blanke, S.R.; Colombo, M.I.; Jones, N.L. Helicobacter pylori VacA toxin promotes bacterial intracellular survival in gastric epithelial cells. Infect. Immun. 2006, 74, 6599–6614. [Google Scholar] [CrossRef]
- de Bernard, M.; Moschioni, M.; Habermann, A.; Griffiths, G.; Montecucco, C. Cell vacuolization induced by Helicobacter pylori VacA cytotoxin does not depend on late endosomal SNAREs. Cell Microbiol. 2002, 4, 11–18. [Google Scholar] [CrossRef]
- Papini, E.; Bugnoli, M.; De Bernard, M.; Figura, N.; Rappuoli, R.; Montecucco, C. Bafilomycin A1 inhibits Helicobacter pylori-induced vacuolization of HeLa cells. Mol. Microbiol. 1993, 7, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Papini, E.; de Bernard, M.; Bugnoli, M.; Milia, E.; Rappuoli, R.; Montecucco, C. Cell vacuolization induced by Helicobacter pylori: Inhibition by bafilomycins A1, B1, C1 and D. FEMS Microbiol. Lett. 1993, 113, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Reddy, L.Y.; Blaser, M.J. Effects of ATPase inhibitors on the response of HeLa cells to Helicobacter pylori vacuolating toxin. Infect. Immun. 1993, 61, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Genisset, C.; Puhar, A.; Calore, F.; de Bernard, M.; Dell’Antone, P.; Montecucco, C. The concerted action of the Helicobacter pylori cytotoxin VacA and of the v-ATPase proton pump induces swelling of isolated endosomes. Cell Microbiol. 2007, 9, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Morbiato, L.; Tombola, F.; Campello, S.; Del Giudice, G.; Rappuoli, R.; Zoratti, M.; Papini, E. Vacuolation induced by VacA toxin of Helicobacter pylori requires the intracellular accumulation of membrane permeant bases, Cl(−) and water. FEBS Lett. 2001, 508, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C. Protein toxins and membrane transport. Curr. Opin. Cell Biol. 1998, 10, 530–536. [Google Scholar] [CrossRef]
- Cover, T.L.; Puryear, W.; Perez-Perez, G.I.; Blaser, M.J. Effect of urease on HeLa cell vacuolation induced by Helicobacter pylori cytotoxin. Infect. Immun. 1991, 59, 1264–1270. [Google Scholar] [CrossRef]
- Cover, T.L.; Vaughn, S.G.; Cao, P.; Blaser, M.J. Potentiation of Helicobacter pylori vacuolating toxin activity by nicotine and other weak bases. J. Infect. Dis. 1992, 166, 1073–1078. [Google Scholar] [CrossRef]
- Necchi, V.; Sommi, P.; Ricci, V.; Solcia, E. In vivo accumulation of Helicobacter pylori products, NOD1, ubiquitinated proteins and proteasome in a novel cytoplasmic structure. PLoS ONE 2010, 5, e9716. [Google Scholar] [CrossRef]
- Tricottet, V.; Bruneval, P.; Vire, O.; Camilleri, J.P.; Bloch, F.; Bonte, N.; Roge, J. Campylobacter-like organisms and surface epithelium abnormalities in active, chronic gastritis in humans: An ultrastructural study. Ultrastruct. Pathol. 1986, 10, 113–122. [Google Scholar] [CrossRef]
- Fiocca, R.; Luinetti, O.; Villani, L.; Chiaravalli, A.M.; Capella, C.; Solcia, E. Epithelial cytotoxicity, immune responses, and inflammatory components of Helicobacter pylori gastritis. Scand. J. Gastroenterol. Suppl. 1994, 205, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Holland, R.L.; Bosi, K.D.; Harpring, G.H.; Luo, J.; Wallig, M.; Phillips, H.; Blanke, S.R. Chronic in vivo exposure to Helicobacter pylori VacA: Assessing the efficacy of automated and long-term intragastric toxin infusion. Sci. Rep. 2020, 10, 9307. [Google Scholar] [CrossRef] [PubMed]
- Jeger, J.L. Endosomes, lysosomes, and the role of endosomal and lysosomal biogenesis in cancer development. Mol. Biol. Rep. 2020, 47, 9801–9810. [Google Scholar] [CrossRef] [PubMed]
- Papini, E.; Satin, B.; Bucci, C.; de Bernard, M.; Telford, J.L.; Manetti, R.; Rappuoli, R.; Zerial, M.; Montecucco, C. The small GTP binding protein rab7 is essential for cellular vacuolation induced by Helicobacter pylori cytotoxin. EMBO J. 1997, 16, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Ohnishi, H.; Wada, A.; Hirayama, T.; Ohno, H.; Ueda, N.; Yasuda, H.; Iiri, T.; Wada, Y.; Futai, M.; et al. Involvement of syntaxin 7 in human gastric epithelial cell vacuolation induced by the Helicobacter pylori-produced cytotoxin VacA. J. Biol. Chem. 2003, 278, 25585–25590. [Google Scholar] [CrossRef] [PubMed]
- Mashima, H.; Suzuki, J.; Hirayama, T.; Yoshikumi, Y.; Ohno, H.; Ohnishi, H.; Yasuda, H.; Fujita, T.; Omata, M. Involvement of vesicle-associated membrane protein 7 in human gastric epithelial cell vacuolation induced by Helicobacter pylori-produced VacA. Infect. Immun. 2008, 76, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C.; Papini, E.; de Bernard, M.; Zoratti, M. Molecular and cellular activities of Helicobacter pylori pathogenic factors. FEBS Lett. 1999, 452, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Sommi, P.; Boquet, P. 19—Helicobacter pylori vacuolating toxin. In The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th ed.; Alouf, J., Ladant, D., Popoff, M.R., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 515–557. [Google Scholar]
- Satin, B.; Norais, N.; Telford, J.; Rappuoli, R.; Murgia, M.; Montecucco, C.; Papini, E. Effect of Helicobacter pylori vacuolating toxin on maturation and extracellular release of procathepsin D and on epidermal growth factor degradation. J. Biol. Chem. 1997, 272, 25022–25028. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Q.L.; Cheng, D.D.; Xu, W.T.; Lu, N.H. Adhesion and Invasion of Gastric Mucosa Epithelial Cells by Helicobacter pylori. Front. Cell Infect. Microbiol. 2016, 6, 159. [Google Scholar] [CrossRef]
- Capurro, M.I.; Greenfield, L.K.; Prashar, A.; Xia, S.; Abdullah, M.; Wong, H.; Zhong, X.Z.; Bertaux-Skeirik, N.; Chakrabarti, J.; Siddiqui, I.; et al. VacA generates a protective intracellular reservoir for Helicobacter pylori that is eliminated by activation of the lysosomal calcium channel TRPML1. Nat. Microbiol. 2019, 4, 1411–1423. [Google Scholar] [CrossRef]
- Sit, W.Y.; Chen, Y.A.; Chen, Y.L.; Lai, C.H.; Wang, W.C. Cellular evasion strategies of Helicobacter pylori in regulating its intracellular fate. Semin. Cell Dev. Biol. 2020, 101, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.I.; Prashar, A.; Jones, N.L. MCOLN1/TRPML1 inhibition—A novel strategy used by Helicobacter pylori to escape autophagic killing and antibiotic eradication therapy in vivo. Autophagy 2020, 16, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Boquet, P.; Ricci, V.; Galmiche, A.; Gauthier, N.C. Gastric cell apoptosis and H. pylori: Has the main function of VacA finally been identified? Trends Microbiol. 2003, 11, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Blanke, S.R. Micro-managing the executioner: Pathogen targeting of mitochondria. Trends Microbiol. 2005, 13, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Galmiche, A.; Rassow, J.; Doye, A.; Cagnol, S.; Chambard, J.C.; Contamin, S.; de Thillot, V.; Just, I.; Ricci, V.; Solcia, E.; et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000, 19, 6361–6370. [Google Scholar] [CrossRef] [PubMed]
- Kuck, D.; Kolmerer, B.; Iking-Konert, C.; Krammer, P.H.; Stremmel, W.; Rudi, J. Vacuolating cytotoxin of Helicobacter pylori induces apoptosis in the human gastric epithelial cell line AGS. Infect. Immun. 2001, 69, 5080–5087. [Google Scholar] [CrossRef]
- Cover, T.L.; Krishna, U.S.; Israel, D.A.; Peek, R.M., Jr. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003, 63, 951–957. [Google Scholar]
- Cho, S.J.; Kang, N.S.; Park, S.Y.; Kim, B.O.; Rhee, D.K.; Pyo, S. Induction of apoptosis and expression of apoptosis related genes in human epithelial carcinoma cells by Helicobacter pylori VacA toxin. Toxicon 2003, 42, 601–611. [Google Scholar] [CrossRef]
- Manente, L.; Perna, A.; Buommino, E.; Altucci, L.; Lucariello, A.; Citro, G.; Baldi, A.; Iaquinto, G.; Tufano, M.A.; De Luca, A. The Helicobacter pylori’s protein VacA has direct effects on the regulation of cell cycle and apoptosis in gastric epithelial cells. J. Cell Physiol. 2008, 214, 582–587. [Google Scholar] [CrossRef]
- Lan, C.H.; Sheng, J.Q.; Fang, D.C.; Meng, Q.Z.; Fan, L.L.; Huang, Z.R. Involvement of VDAC1 and Bcl-2 family of proteins in VacA-induced cytochrome c release and apoptosis of gastric epithelial carcinoma cells. J. Dig. Dis. 2010, 11, 43–49. [Google Scholar] [CrossRef]
- Yahiro, K.; Akazawa, Y.; Nakano, M.; Suzuki, H.; Hisatune, J.; Isomoto, H.; Sap, J.; Noda, M.; Moss, J.; Hirayama, T. Helicobacter pylori VacA induces apoptosis by accumulation of connexin 43 in autophagic vesicles via a Rac1/ERK-dependent pathway. Cell Death Discov. 2015, 1, 15035. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Kim, J.S.; Kim, N.; Ko, S.H.; Jeon, J.I.; Kim, Y.J. Helicobacter pylori vacuolating cytotoxin induces apoptosis via activation of endoplasmic reticulum stress in dendritic cells. J. Gastroenterol. Hepatol. 2015, 30, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Calore, F.; Genisset, C.; Casellato, A.; Rossato, M.; Codolo, G.; Esposti, M.D.; Scorrano, L.; de Bernard, M. Endosome-mitochondria juxtaposition during apoptosis induced by H. pylori VacA. Cell Death Differ. 2010, 17, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Ganten, T.M.; Aravena, E.; Sykora, J.; Koschny, R.; Mohr, J.; Rudi, J.; Stremmel, W.; Walczak, H. Helicobacter pylori-induced apoptosis in T cells is mediated by the mitochondrial pathway independent of death receptors. Eur. J. Clin. Investig. 2007, 37, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Isomoto, H.; Nakayama, M.; Hisatsune, J.; Nishi, Y.; Nakashima, Y.; Matsushima, K.; Kurazono, H.; Nakao, K.; Hirayama, T.; et al. Helicobacter pylori VacA reduces the cellular expression of STAT3 and pro-survival Bcl-2 family proteins, Bcl-2 and Bcl-XL, leading to apoptosis in gastric epithelial cells. Dig. Dis. Sci. 2011, 56, 999–1006. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, J.S.; Lee, J.Y.; Sim, Y.S.; Kim, Y.J.; Oh, Y.K.; Yoon, H.J.; Kang, J.S.; Youn, J.; Kim, N.; et al. Dual effects of Helicobacter pylori vacuolating cytotoxin on human eosinophil apoptosis in early and late periods of stimulation. Eur. J. Immunol. 2010, 40, 1651–1662. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef]
- Jain, P.; Luo, Z.Q.; Blanke, S.R. Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc. Natl. Acad. Sci. USA 2011, 108, 16032–16037. [Google Scholar] [CrossRef]
- Foo, J.H.; Culvenor, J.G.; Ferrero, R.L.; Kwok, T.; Lithgow, T.; Gabriel, K. Both the p33 and p55 subunits of the Helicobacter pylori VacA toxin are targeted to mammalian mitochondria. J. Mol. Biol. 2010, 401, 792–798. [Google Scholar] [CrossRef]
- Kimura, M.; Goto, S.; Wada, A.; Yahiro, K.; Niidome, T.; Hatakeyama, T.; Aoyagi, H.; Hirayama, T.; Kondo, T. Vacuolating cytotoxin purified from Helicobacter pylori causes mitochondrial damage in human gastric cells. Microb. Pathog. 1999, 26, 45–52. [Google Scholar] [CrossRef]
- Wang, L.; Yi, J.; Yin, X.Y.; Hou, J.X.; Chen, J.; Xie, B.; Chen, G.; Wang, Q.F.; Wang, L.N.; Wang, X.Y.; et al. Vacuolating Cytotoxin A Triggers Mitophagy in Helicobacter pylori-Infected Human Gastric Epithelium Cells. Front. Oncol. 2022, 12, 881829. [Google Scholar] [CrossRef] [PubMed]
- Willhite, D.C.; Cover, T.L.; Blanke, S.R. Cellular vacuolation and mitochondrial cytochrome c release are independent outcomes of Helicobacter pylori vacuolating cytotoxin activity that are each dependent on membrane channel formation. J. Biol. Chem. 2003, 278, 48204–48209. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Dashwood, R.H.; Dashwood, M.M.; Zaidi, S.I.; Hewitt, S.M.; Green, W.R.; Lee, E.L.; Daremipouran, M.; Nouraie, M.; Malekzadeh, R.; et al. H. pylori-induced apoptosis in human gastric cancer cells mediated via the release of apoptosis-inducing factor from mitochondria. Helicobacter 2008, 13, 506–517. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Ref. |
|---|---|
| CCUG 17874 | [74] * |
| 60190, 84–183 | [75] * |
| 4767-C, 2074-Cd | [76] * |
| NCTC11637 | [77] * |
| CCUG 17875 | [78] * |
| 60190, SS1, SS1s1i1 | [79] * |
| 251 (and mutants: tolB, tolB+, pal, pal+, tolBpal, cagPAI) | [80] |
| HP99 | [81] * |
| 251 mutant cagPAI | [82] |
| B128_7.13 | [83] |
| 26695 | [84,85,86] |
| NCTC11637, H. pylori 400 (CGMCC 15126) | [87] |
| TN2 | [88] |
| 60190 | [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarzab, M.; Skorko-Glonek, J. There Are No Insurmountable Barriers: Passage of the Helicobacter pylori VacA Toxin from Bacterial Cytoplasm to Eukaryotic Cell Organelle. Membranes 2024, 14, 11. https://doi.org/10.3390/membranes14010011
Jarzab M, Skorko-Glonek J. There Are No Insurmountable Barriers: Passage of the Helicobacter pylori VacA Toxin from Bacterial Cytoplasm to Eukaryotic Cell Organelle. Membranes. 2024; 14(1):11. https://doi.org/10.3390/membranes14010011
Chicago/Turabian StyleJarzab, Miroslaw, and Joanna Skorko-Glonek. 2024. "There Are No Insurmountable Barriers: Passage of the Helicobacter pylori VacA Toxin from Bacterial Cytoplasm to Eukaryotic Cell Organelle" Membranes 14, no. 1: 11. https://doi.org/10.3390/membranes14010011