
Designing a Multi-Epitope Vaccine against Toxoplasma gondii: An Immunoinformatics Approach
 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrieval of the Parasite Protein Sequences
2.2. Linear B Lymphocyte Epitope Prediction
2.3. Prediction of Cytotoxic T-Lymphocytes Epitopes
2.4. Prediction of Helper T-Lymphocyte Epitopes
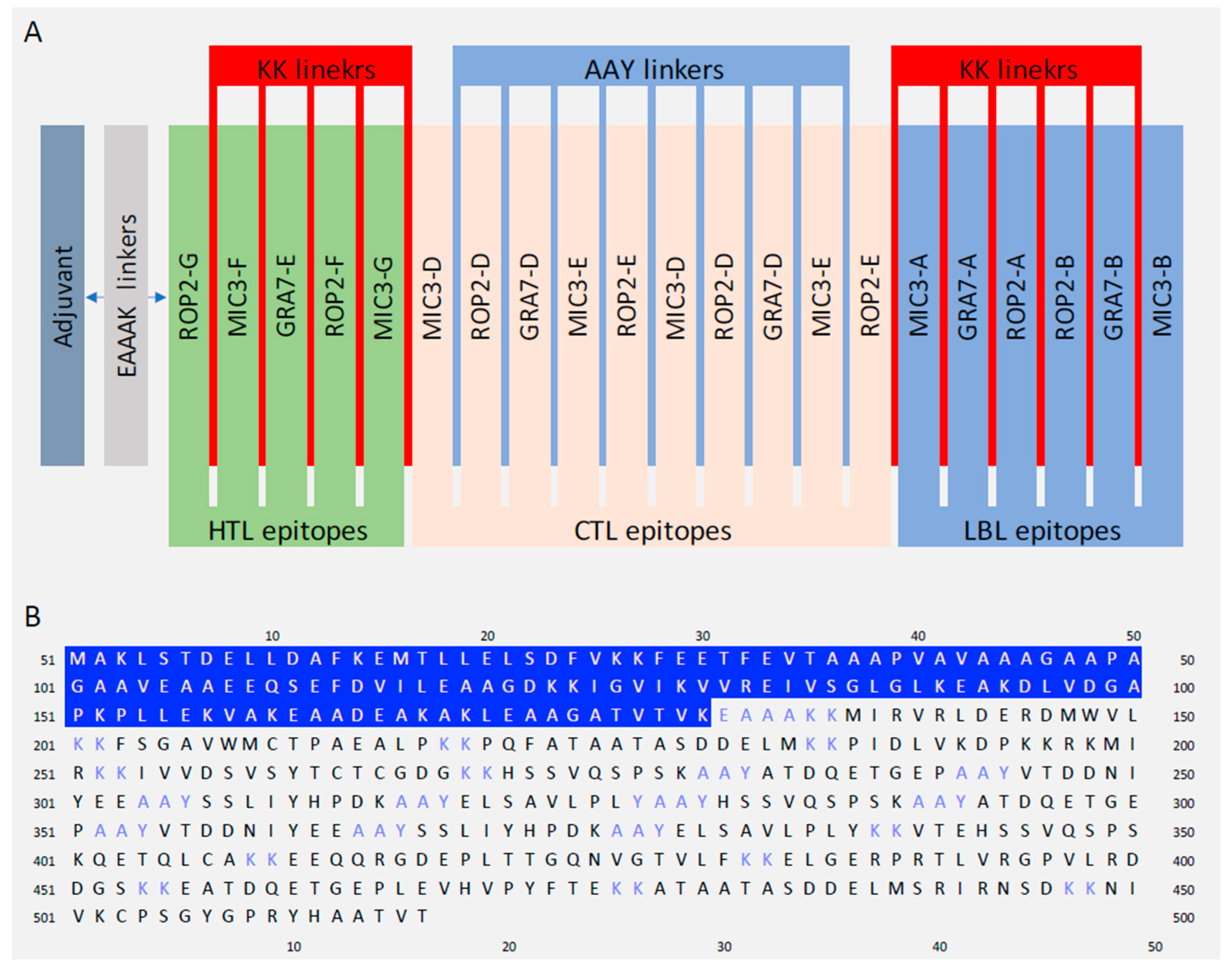
2.5. Designing of Multi-Epitope Vaccine Construct
2.6. Physiochemical Properties, Allergenicity, Solubility, and Antigenicity Prediction of the Designed Vaccine
2.7. Predicting Secondary Structure
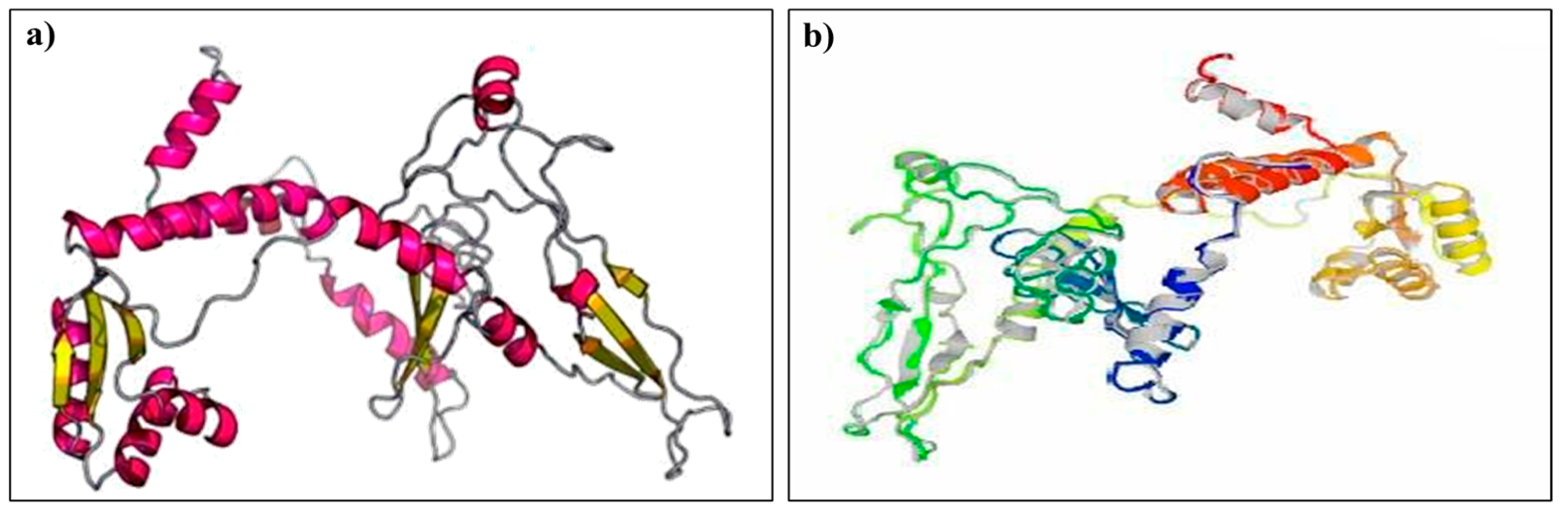
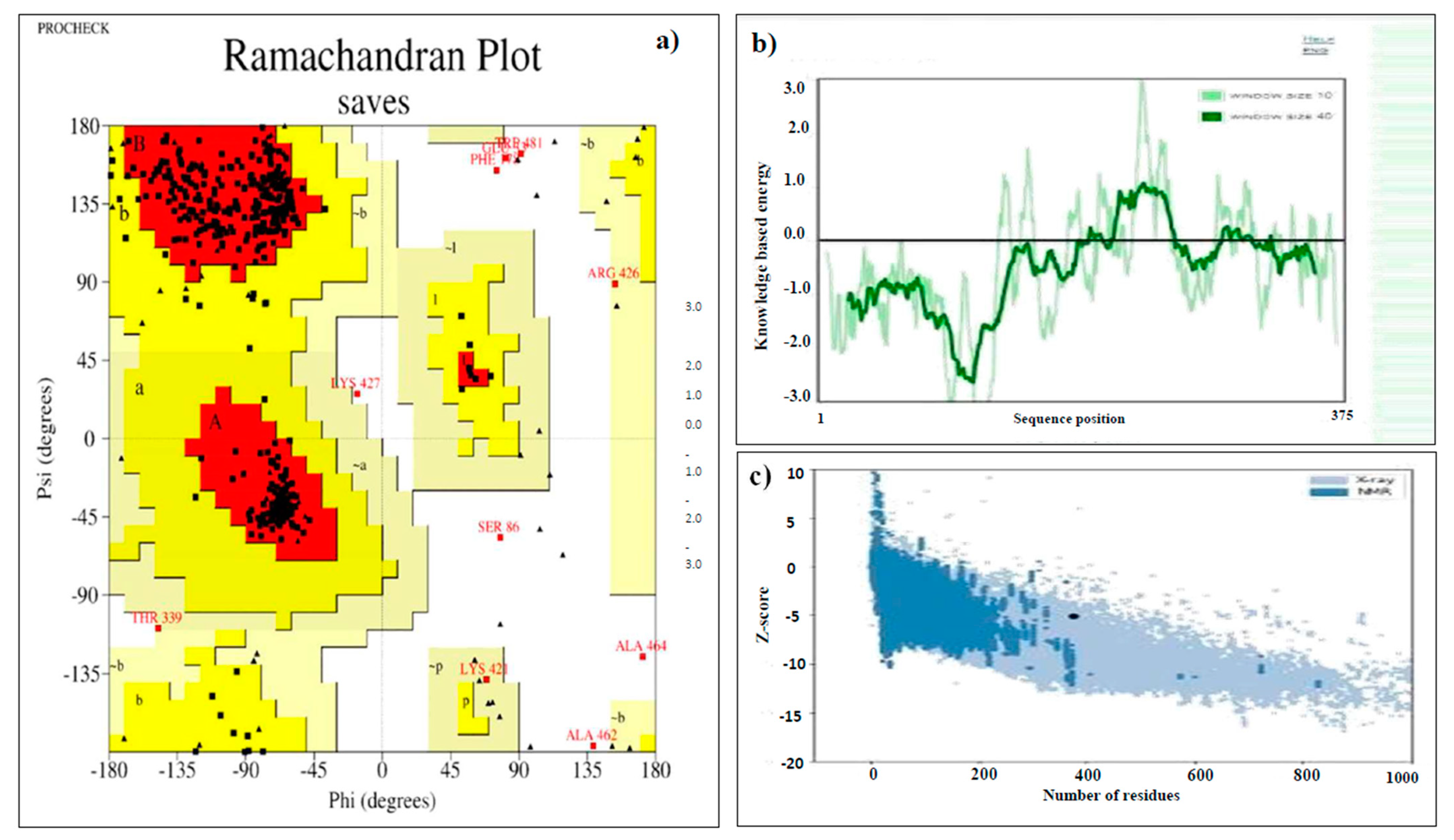
2.8. Predicting the Tertiary Structure, Refinement, and Validation of the Designed Vaccine
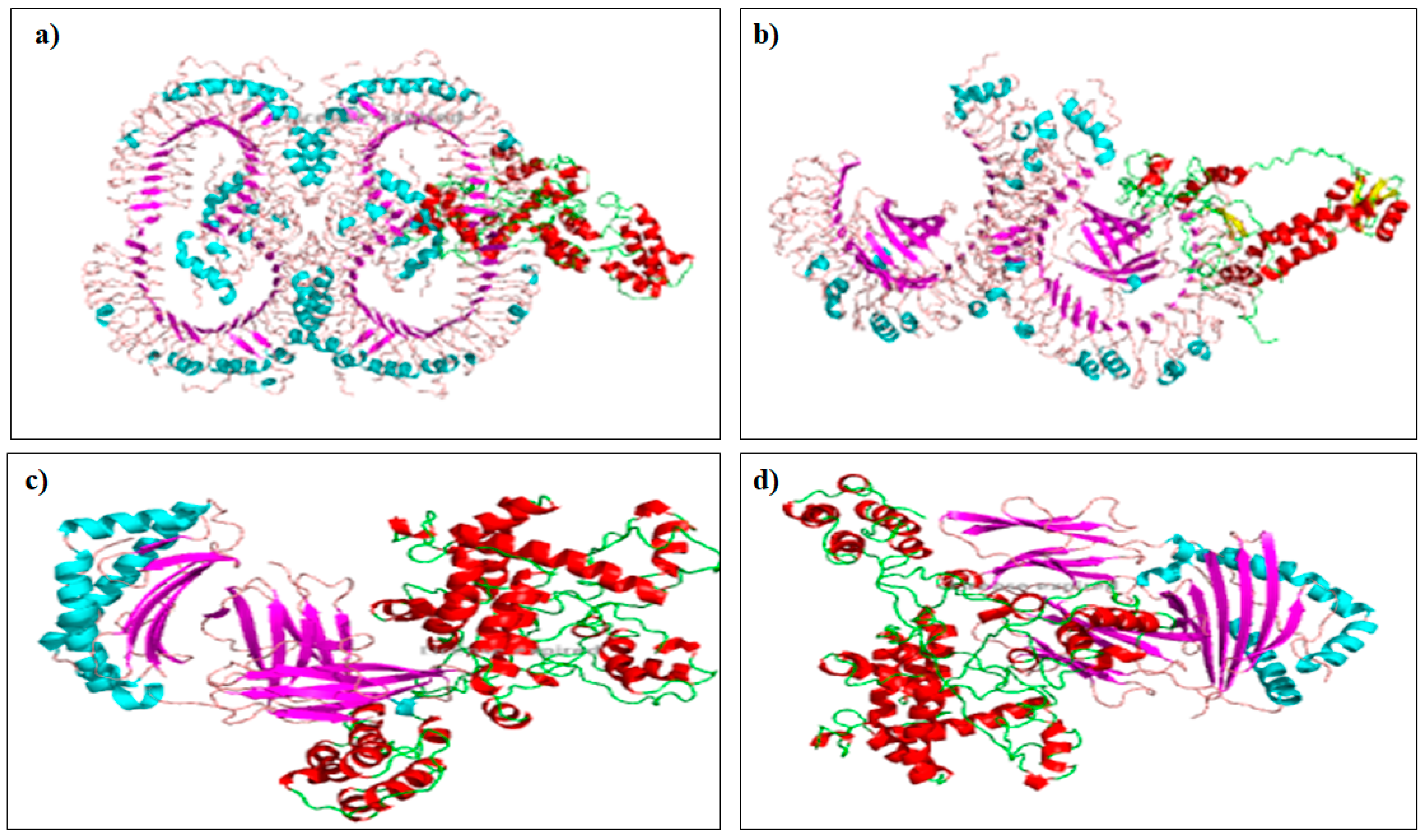
2.9. Molecular Docking with Toll-Like Receptor (TLR)
2.10. Immune Simulation
2.11. Molecular Dynamics Simulation
2.12. Codon Optimization and In Silico Cloning
3. Results
3.1. Protein Retrieval and Antigenic Prediction
3.2. Linear B Lymphocyte Epitope Prediction
3.3. Cytotoxic T Lymphocyte Epitopes Prediction
3.4. Helper T Lymphocyte Epitopes Prediction
3.5. Designing Multiepitope Vaccine
3.6. Physiochemical Properties, Antigenicity, Allergenicity, and Solubility Prediction of the Designed Vaccine
3.7. Secondary Structure Prediction
3.8. Tertiary Structure Prediction, Refinement, and Validation
3.9. Molecular Docking with Toll-Like Receptor (TLR)
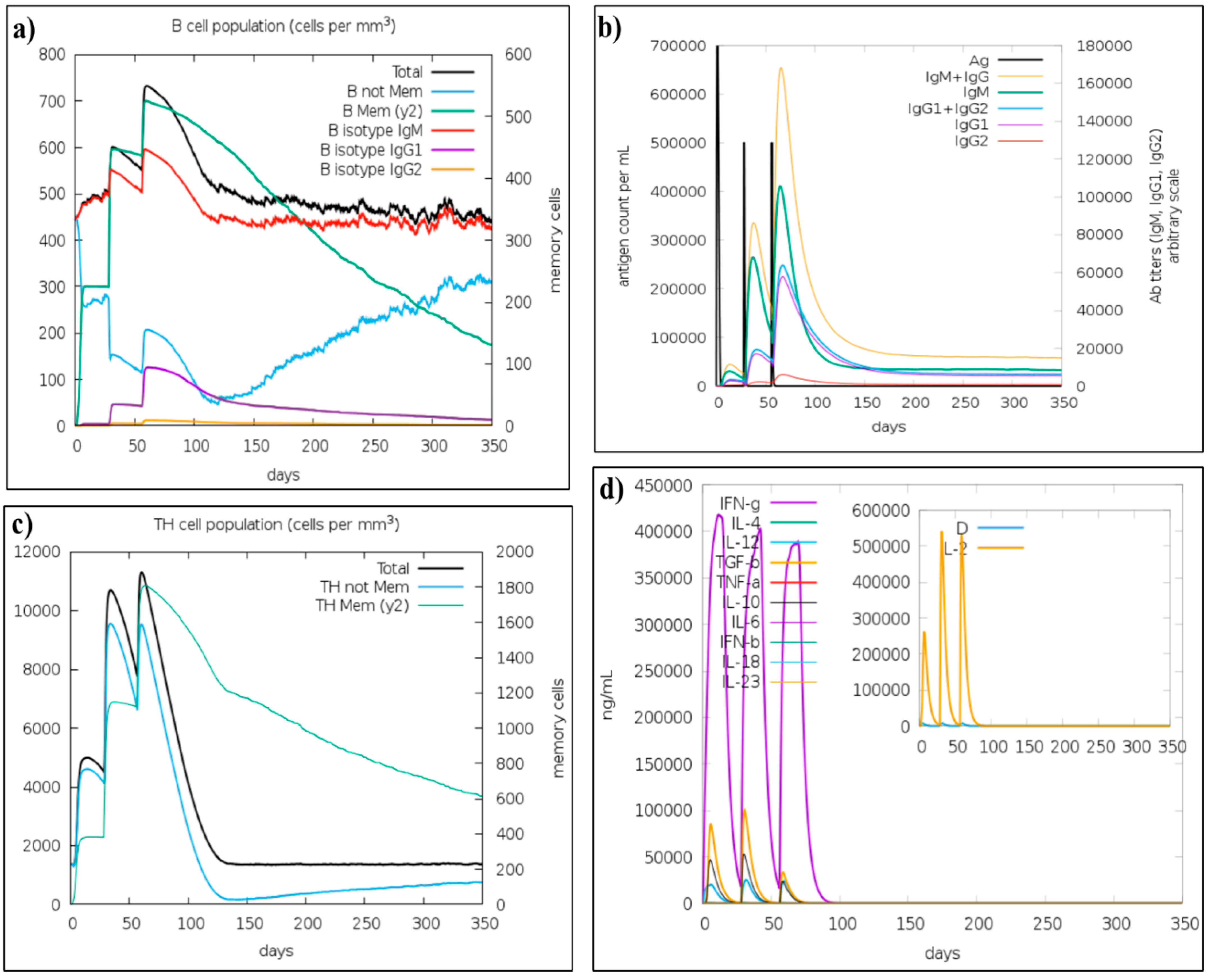
3.10. Immune Simulation
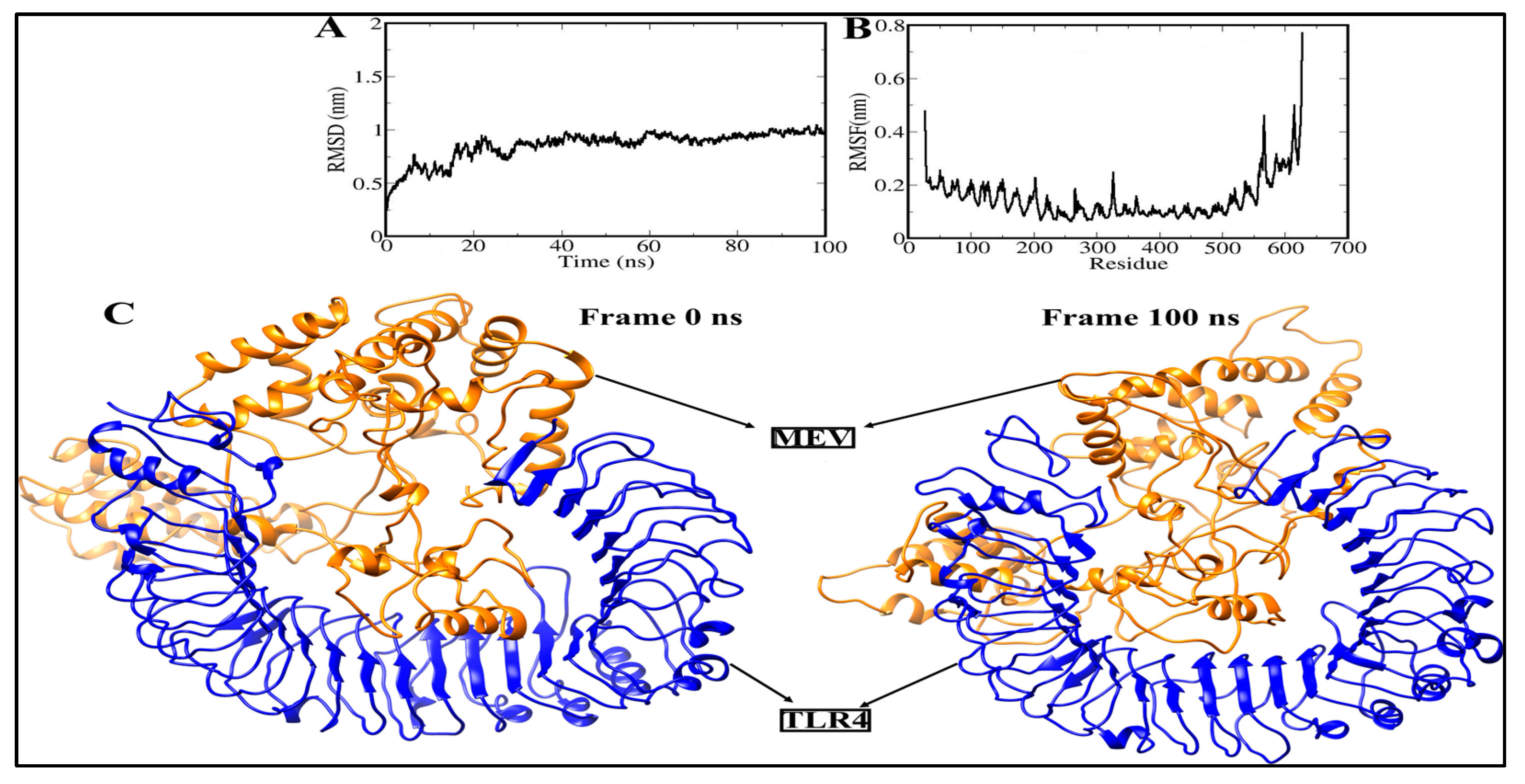
3.11. Molecular Dynamics Simulation
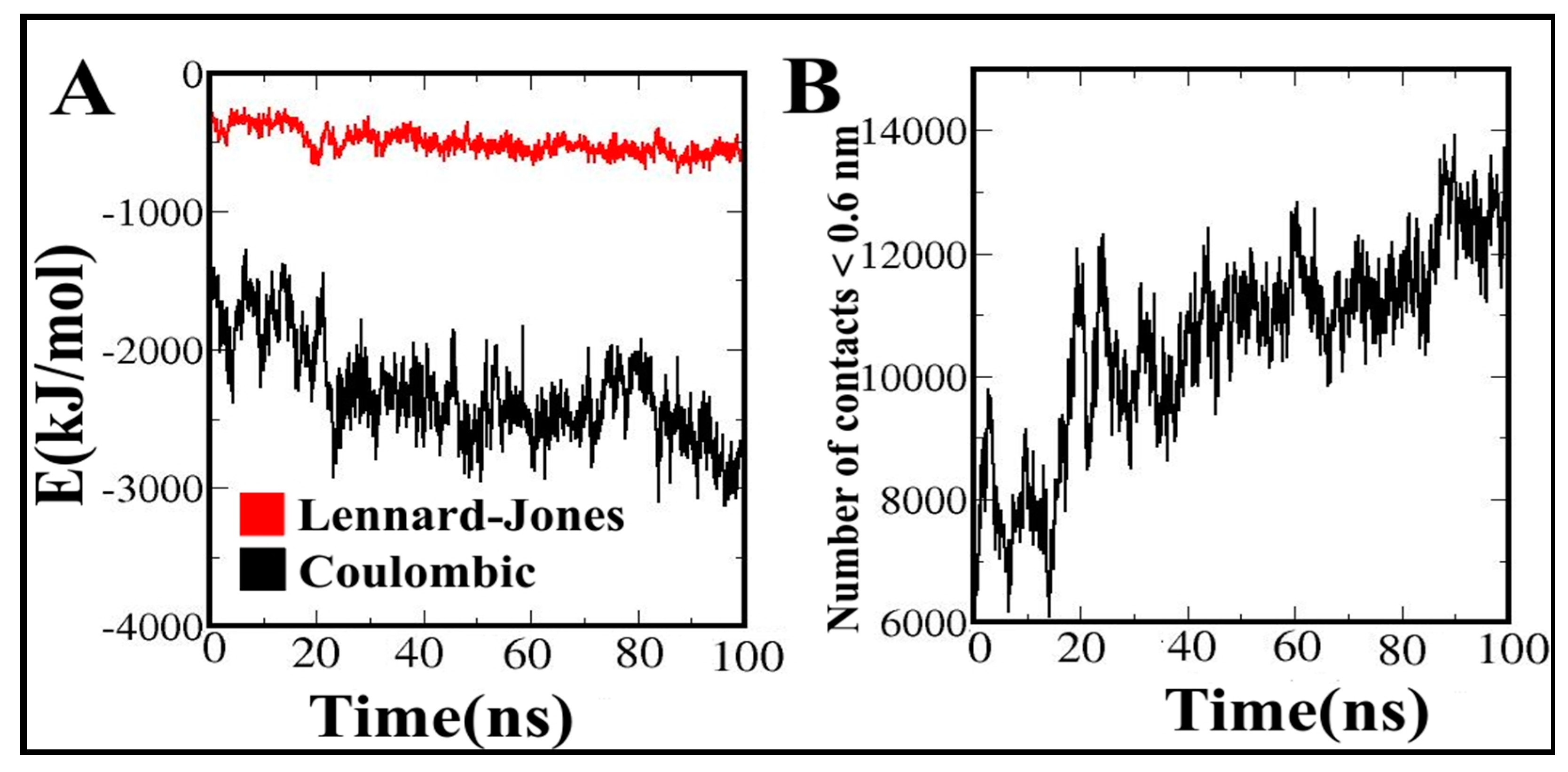
3.12. Contribution of Energy Components to the Vaccine Binding along MD Simulations
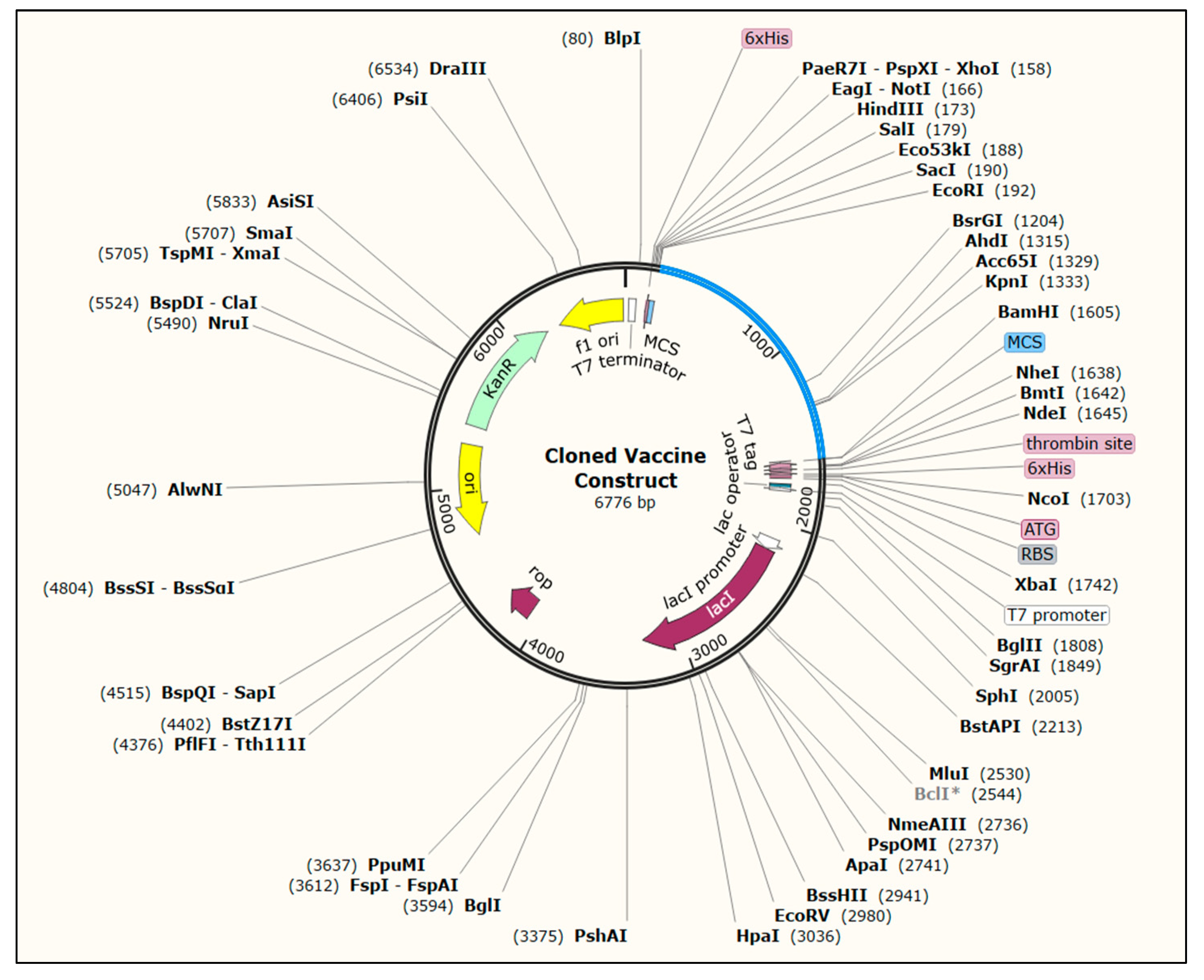
3.13. Codon Optimization and In Silico Cloning
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dubey, J.P. History of the discovery of the life cycle of Toxoplasma gondii. Int. J. Parasitol. 2009, 39, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Saadatnia, G.; Golkar, M. A review on human toxoplasmosis. Scand. J. Infect. Dis. 2012, 44, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.M.; Kim, K. Toxoplasma Gondii: The Model Apicomplexan. Perspectives and Methods; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Black, M.W.; Boothroyd, J.C. Lytic Cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev. 2000, 64, 607–623. [Google Scholar] [CrossRef]
- Wang, Z.-D.; Liu, H.-H.; Ma, Z.-X.; Ma, H.-Y.; Li, Z.-Y.; Yang, Z.-B.; Zhu, X.-Q.; Xu, B.; Wei, F.; Liu, Q. Toxoplasma gondii Infection in Immunocompromised Patients: A Systematic Review and Meta-Analysis. Front. Microbiol. 2017, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Abbas, I.; Villena, I.; Dubey, J.P. A review on toxoplasmosis in humans and animals from Egypt. Parasitology 2020, 147, 135–159. [Google Scholar] [CrossRef]
- Kolören, Z.; Dubey, J.P. A review of toxoplasmosis in humans and animals in Turkey. Parasitology 2019, 147, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Tenter, A.; Heckeroth, A.; Weiss, L. Toxoplasma gondii: From animals to humans. Int. J. Parasitol. 2000, 30, 1217–1258. [Google Scholar] [CrossRef]
- Roberts, C.W.; Roberts, F.; Lyons, R.E.; Kirisits, M.J.; Mui, E.J.; Finnerty, J.; Johnson, J.J.; Ferguson, D.J.; Coggins, J.R.; Krell, T. The shikimate pathway and its branches in apicomplexan parasites. J. Infect. Dis. 2002, 185, S25–S36. [Google Scholar] [CrossRef]
- Verma, R.; Khanna, P. Development of Toxoplasma gondii vaccine. Hum. Vaccines Immunother. 2013, 9, 291–293. [Google Scholar] [CrossRef]
- Mamaghani, A.J.; Fathollahi, A.; Arab-Mazar, Z.; Fathollahi, M.; Spotin, A.; Bashiri, H.; Bozorgomid, A. Toxoplasma gondii vaccine candidates: A concise review. Ir. J. Med. Sci. 2022, 191, 1–31. [Google Scholar] [CrossRef]
- Sarkar, B.; Ullah, M.A.; Araf, Y.; Rahman, M.S. Engineering a novel subunit vaccine against SARS-CoV-2 by exploring immunoinformatics approach. Inform. Med. Unlocked 2020, 21, 100478. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Dodangeh, S.; Fasihi-Ramandi, M.; Daryani, A.; Valadan, R.; Sarvi, S. In silico analysis and expression of a novel chimeric antigen as a vaccine candidate against Toxoplasma gondii. Microb. Pathog. 2019, 132, 275–281. [Google Scholar] [CrossRef]
- Naveed, M.; Bukhari, B.; Afzal, N.; Sadia, H.; Meer, B.; Riaz, T.; Ali, U.; Ahmed, N. Geographical, Molecular, and Computational Analysis of Migraine-Causing Genes. J. Comput. Biophys. Chem. 2021, 20, 391–403. [Google Scholar] [CrossRef]
- Jabbar, B.; Rafique, S.; Salo-Ahen, O.M.; Ali, A.; Munir, M.; Idrees, M.; Mirza, M.U.; Vanmeert, M.; Shah, S.Z.; Jabbar, I. Antigenic peptide prediction from E6 and E7 oncoproteins of HPV types 16 and 18 for therapeutic vaccine design using immunoinformatics and MD simulation analysis. Front. Immunol. 2018, 9, 3000. [Google Scholar] [CrossRef] [PubMed]
- Hajissa, K.; Zakaria, R.; Suppian, R.; Mohamed, Z. Epitope-based vaccine as a universal vaccination strategy against Toxoplasma gondii infection: A mini-review. J. Adv. Vet. Anim. Res. 2019, 6, 174. [Google Scholar] [CrossRef]
- Zhang, N.-Z.; Chen, J.; Wang, M.; Petersen, E.; Zhu, X.-Q. Vaccines against Toxoplasma gondii: New developments and perspectives. Expert Rev. Vaccines 2013, 12, 1287–1299. [Google Scholar] [CrossRef]
- Dodangeh, S.; Fasihi-Ramandi, M.; Daryani, A.; Valadan, R.; Asgarian-Omran, H.; Hosseininejad, Z.; Chegeni, T.N.; Pagheh, A.S.; Javidnia, J.; Sarvi, S. Protective efficacy by a novel multi-epitope vaccine, including MIC3, ROP8, and SAG1, against acute Toxoplasma gondii infection in BALB/c mice. Microb. Pathog. 2021, 153, 104764. [Google Scholar] [CrossRef]
- Foroutan, M.; Ghaffarifar, F.; Sharifi, Z.; Dalimi, A. Vaccination with a novel multi-epitope ROP8 DNA vaccine against acute Toxoplasma gondii infection induces strong B and T cell responses in mice. Comp. Immunol. Microbiol. Infect. Dis. 2020, 69, 101413. [Google Scholar] [CrossRef]
- Verhelst, D.; De Craeye, S.; Dorny, P.; Melkebeek, V.; Goddeeris, B.; Cox, E.; Jongert, E. IFN-γ expression and infectivity of Toxoplasma infected tissues are associated with an antibody response against GRA7 in experimentally infected pigs. Vet. Parasitol. 2011, 179, 14–21. [Google Scholar] [CrossRef]
- Daryani, A.; Kalani, H.; Sharif, M.; Ziaei, H.; Sarvi, S.; Ahmadpour, E. Toxoplasma gondii: A review of excretory secretory antigens. J. Maz. Univ. Med. Sci. 2013, 22, 220–232. [Google Scholar]
- Mehla, K.; Ramana, J. Identification of epitope-based peptide vaccine candidates against enterotoxigenic Escherichia coli: A comparative genomics and immunoinformatics approach. Mol. BioSystems 2016, 12, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery Consortium; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef]
- Wang, P.; Sidney, J.; Kim, Y.; Sette, A.; Lund, O.; Nielsen, M.; Peters, B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010, 11, 568. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Springer Nature: Berlin, Germany, 2005; pp. 571–607. [Google Scholar]
- Abass, O.A.; Timofeev, V.I.; Sarkar, B.; Onobun, D.O.; Ogunsola, S.O.; Aiyenuro, A.E.; Aborode, A.T.; Aigboje, A.E.; Omobolanle, B.N.; Imolele, A.G.; et al. Immunoinformatics analysis to design novel epitope based vaccine candidate targeting the glycoprotein and nucleoprotein of Lassa mammarenavirus (LASMV) using strains from Nigeria. J. Biomol. Struct. Dyn. 2021, 1–20. [Google Scholar] [CrossRef]
- Shey, R.A.; Ghogomu, S.M.; Shintouo, C.M.; Nkemngo, F.N.; Nebangwa, D.N.; Esoh, K.; Yaah, N.E.; Manka’aFri, M.; Nguve, J.E.; Ngwese, R.A. Computational Design and Preliminary Serological Analysis of a Novel Multi-Epitope Vaccine Candidate Against Onchocerciasis and Related Filarial Diseases. Pathogens 2021, 10, 99. [Google Scholar] [CrossRef]
- Obaidullah, A.J.; Alanazi, M.M.; Alsaif, N.A.; Albassam, H.; Almehizia, A.A.; Alqahtani, A.M.; Mahmud, S.; Sami, S.A.; Emran, T.B. Immunoinformatics-guided design of a multi-epitope vaccine based on the structural proteins of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2021, 11, 18103–18121. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Xu, J. RaptorX: Exploiting structure information for protein alignment by statistical inference. Proteins Struct. Funct. Bioinform. 2011, 79, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F.J.P.o. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef]
- Dodangeh, S.; Daryani, A.; Sharif, M.; Aghayan, S.A.; Pagheh, A.S.; Sarvi, S.; Rezaei, F. A systematic review on efficiency of microneme proteins to induce protective immunity against Toxoplasma gondii. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Yaseen, A.R.; Khalid, H.; Ali, U.; Rabaan, A.A.; Garout, M.; Halwani, M.A.; Al Mutair, A.; Alhumaid, S.; Al Alawi, Z.; et al. Execution and Design of an Anti HPIV-1 Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses: An Immunoinformatic Approach. Vaccines 2022, 10, 869. [Google Scholar] [CrossRef] [PubMed]
- Majidiani, H.; Dalimi, A.; Ghaffarifar, F.; Pirestani, M. Multi-epitope vaccine expressed in Leishmania tarentolae confers protective immunity to Toxoplasma gondii in BALB/c mice. Microb Pathoge. 2021, 155, 104925. [Google Scholar] [CrossRef] [PubMed]
- Forouharmehr, A.J.M.P. Engineering an efficient poly-epitope vaccine against Toxoplasma gondii infection: A computational vaccinology study. Microb. Pathog. 2021, 152, 104646. [Google Scholar] [CrossRef] [PubMed]
- Hajissa, K.; Zakaria, R.; Suppian, R.; Mohamed, Z. Immunogenicity of multiepitope vaccine candidate against Toxoplasma gondii infection in BALB/c mice. Iran. J. Parasitol. 2018, 13, 215–224. [Google Scholar] [PubMed]
- Sanches, R.C.; Tiwari, S.; Ferreira, L.C.; Oliveira, F.M.; Lopes, M.D.; Passos, M.J.; Maia, E.H.; Taranto, A.G.; Kato, R.; Azevedo, V.A.; et al. Immunoinformatics design of multi-epitope peptide-based vaccine against Schistosoma mansoni using transmembrane proteins as a target. Front. Immunol. 2021, 12, 621706. [Google Scholar] [CrossRef]
- Bibi, S.; Ullah, I.; Zhu, B.; Adnan, M.; Liaqat, R.; Kong, W.-B.; Niu, S. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Front. Immunol. 2021, 11, 1249. [Google Scholar] [CrossRef]
- Naveed, M.; Ali, U.; Karobari, M.I.; Ahmed, N.; Mohamed, R.N.; Abullais, S.S.; Kader, M.A.; Marya, A.; Messina, P.; Scardina, G.A. A Vaccine Construction against COVID-19-Associated Mucormycosis Contrived with Immunoinformatics-Based Scavenging of Potential Mucoralean Epitopes. Vaccines 2022, 10, 664. [Google Scholar] [CrossRef]
- Enayatkhani, M.; Hasaniazad, M.; Faezi, S.; Gouklani, H.; Davoodian, P.; Ahmadi, N.; Einakian, M.A.; Karmostaji, A.; Ahmadi, K. Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: An in silico study. J. Biomol. Struct. Dyn. 2021, 39, 2857–2872. [Google Scholar] [CrossRef]
- Patra, P.; Mondal, N.; Patra, B.C.; Bhattacharya, M. Epitope-based vaccine designing of Nocardia asteroides targeting the virulence factor mce-family protein by immunoinformatics approach. Int. J. Pept. Res. Ther. 2020, 26, 1165–1176. [Google Scholar] [CrossRef]
- Shamriz, S.; Ofoghi, H.; Moazami, N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016, 76, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Khan, S.; Ali, A.; Akbar, H.; Sayaf, A.M.; Khan, A.; Wei, D.-Q. Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci. Rep. 2019, 9, 13321. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Nguyen, M.T. Recent advances of vaccine adjuvants for infectious diseases. Immune Netw. 2015, 15, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Irvine, D.J. Guiding Principles in the Design of Molecular Bioconjugates for Vaccine Applications. Bioconjugate Chem. 2015, 26, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Position | Name | LBC Epitopes | Antigenicity | Allergenicity | Toxicity | Human Homology |
|---|---|---|---|---|---|---|---|
| ROP2 | 56–75 | ROP2_A | ELGERPRTLVRGPVLRDDGS | 0.6399 | Non-allergen | Non-toxic | Non-human homology |
| 80–99 | ROP2_B | EATDQETGEPLEVHVPYFTE | 0.7908 | Non-allergen | Non-toxic | Non-human homology | |
| GRA7 | 166–185 | GRA7_A | EEQQRGDEPLTTGQNVGTVL | 0.9203 | Non-allergen | Non-toxic | Non-human homology |
| 24–43 | GRA_B | ATAATASDDELMSRIRNSDF | 1.0281 | Non-allergen | Non-toxic | Non-human homology | |
| MIC3 | 58–77 | MIC_A | VTETHSSVQSPSKQETQLCA | 0.8003 | Non-allergen | Non-toxic | Non-human homology |
| 334–353 | MIC_B | NIVFKCPSGYHPRYHAHTVT | 0.6133 | Non-allergen | Non-toxic | Non-human homology |
| Protein | Position | Name | CTL Epitopes | C-Score | Immunogenicity | Antigenicity | Allergenicity | Toxicity | Conservancy | Human Homology |
|---|---|---|---|---|---|---|---|---|---|---|
| ROP2 | 269–277 | ROP2_D | ATDQETGEP | 0.7645 | + | 0.6994 | Non-allergen | Non-toxic | Conserved | Non-human homology |
| 545–553 | ROP2_E | ELSAVLPLY | 1.1063 | + | 0.5766 | Non-allergen | Non-toxic | Conserved | Non-human homology | |
| GRA7 | 111–119 | GRA7_D | VTDDNIYEE | 0.7539 | + | 0.6984 | Non-allergen | Non-toxic | Conserved | Non-human homology |
| MIC3 | 62–70 | MIC3_D | HSSVQSPSK | 1.2568 | + | 1.355 | Non-allergen | Non-toxic | Conserved | Non-human homology |
| 131–139 | MIC3_E | SSLIYHPDK | 0.8854 | + | 0.6068 | Non-allergen | Non-toxic | Conserved | Non-human homology |
| Protein | Position | Name | HTL Epitope | IFN γ | IL-4 | IL-10 | Antigenicity | Allergenicity | Toxicity | Conservancy | Human Homology |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ROP2 | 81–95 | ROP2_E | GSWLEQEAAEEVTPL | + | - | - | 0.8870 | Non-allergen | Non-toxic | conserved | Non-human homology |
| 325–339 | ROP2_F | PIDLVKDPKKRKMIR | + | + | + | 1.0777 | Non-allergen | Non-toxic | conserved | Non-human homology | |
| GRA7 | 336–360 | ROP2_G | KMIRVRLDERDMWVL | + | + | + | 1.0062 | Non-allergen | Non-toxic | conserved | Non-human homology |
| 21–35 | GRA7_E | PQFATAATASDDELM | + | - | - | 0.6795 | Non-allergen | Non-toxic | conserved | Non-human homology | |
| MIC3 | 14–28 | MIC3_E | FSGAVWMCTPAEALP | + | - | - | 0.5938 | Non-Allergen | Non-toxic | conserved | Non-human homology |
| 205–219 | MIC3_F | IVVDSVSYTCTCGDG | + | - | - | 1.1324 | Non-allergen | Non-toxic | conserved | Non-human homology |
| Characteristics | Finding | Remarks |
|---|---|---|
| Number of amino acids | 469 | Suitable |
| Molecular weight | 51,035.79 | high |
| Theoretical PI | 5.46 | Acidic |
| Chemical formula | C2257H3602N598O719S13 | - |
| Extinction coefficient (at 280 nm in H20) | 39,560 | - |
| Estimated half-life (mammalian reticulocytes, in vitro) | 30 h | - |
| Estimated half-life (yeast, in vivo) | >20 h | - |
| Estimated half-life (E. coli, in vivo) | >10 h | - |
| Instability index of vaccine | 37.53 | Stable |
| Aliphatic index of vaccine | 77.08 | Thermostable |
| Grand average of hydropathicity (GRAVY) | −0.439 | Hydrophilic |
| Antigenicity | 0.6182 | Antigenic |
| Allergenicity | No | Non-Allergen |
| Solubility | 0.903461 | Soluble |
| SOPMA Server | PSIPRED Server | |||
|---|---|---|---|---|
| Features | Amino Acids | Percentage | Amino Acids | Percentage |
| Alpha helix | 195 | 41.58 | 186 | 39.66 |
| Beta strand | 83 | 17.70 | 55 | 11.73 |
| Random coil | 191 | 40.72 | 241 | 51.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammed-Akanmu, M.; Mim, M.; Osman, A.Y.; Sheikh, A.M.; Behmard, E.; Rabaan, A.A.; Suppain, R.; Hajissa, K. Designing a Multi-Epitope Vaccine against Toxoplasma gondii: An Immunoinformatics Approach. Vaccines 2022, 10, 1389. https://doi.org/10.3390/vaccines10091389
Hammed-Akanmu M, Mim M, Osman AY, Sheikh AM, Behmard E, Rabaan AA, Suppain R, Hajissa K. Designing a Multi-Epitope Vaccine against Toxoplasma gondii: An Immunoinformatics Approach. Vaccines. 2022; 10(9):1389. https://doi.org/10.3390/vaccines10091389
Chicago/Turabian StyleHammed-Akanmu, Mutiat, Maria Mim, Abdinasir Yusuf Osman, Abdulrahman M. Sheikh, Esmaeil Behmard, Ali A. Rabaan, Rapeah Suppain, and Khalid Hajissa. 2022. "Designing a Multi-Epitope Vaccine against Toxoplasma gondii: An Immunoinformatics Approach" Vaccines 10, no. 9: 1389. https://doi.org/10.3390/vaccines10091389