
Execution and Design of an Anti HPIV-1 Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses: An Immunoinformatic Approach

 , ,
, ,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viral Proteins’ Sequence Retrieval
2.2. Antigenic Protein Identification
2.3. Physicochemical Analysis
2.4. Two-Dimensional (2D) and 3D Structural Analysis
2.5. B-Cell-Specific Epitopes Identification via IEDB Tools
2.6. Predicting T-Cells Restricted Epitopes
2.7. Selected Epitopes’ Population Coverage
2.8. Profiling of Predicted Epitopes
2.9. Vaccine Design and Profiling
2.10. Disulfide Engineering of Vaccine
2.11. Three-Dimensional (3D) Structural Analysis
2.12. Molecular Docking and Dynamics
2.13. Codon Improvement and Cloning
2.14. Immune Simulation
3. Results
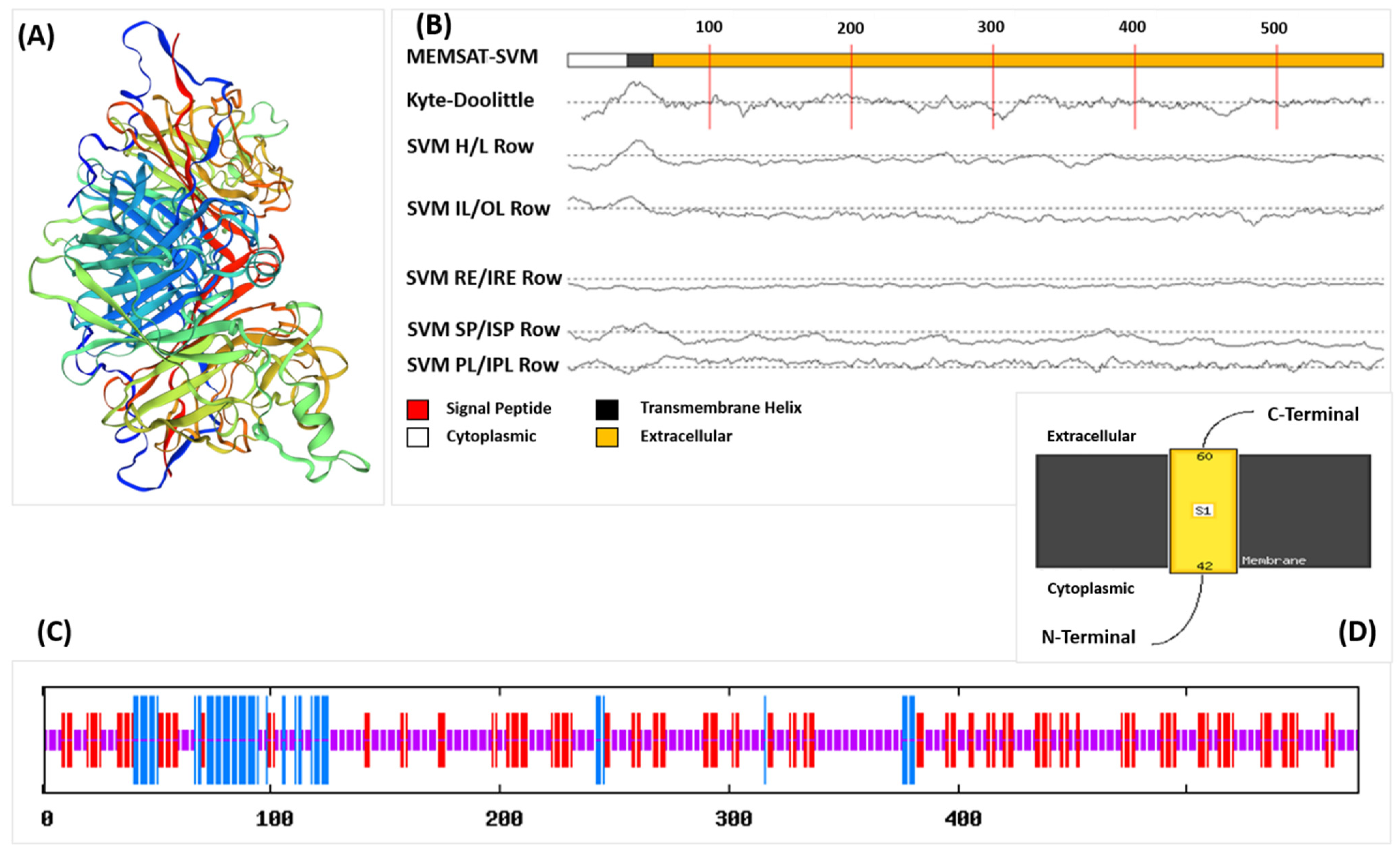
3.1. Antigenicity, Structural and Physicochemical Analysis of Viral HN Protein
3.2. B-Cell-Restricted Epitope Predictions
3.3. Predicting T-Cells Restricted Epitopes
3.4. Filtration of Predicted Epitopes
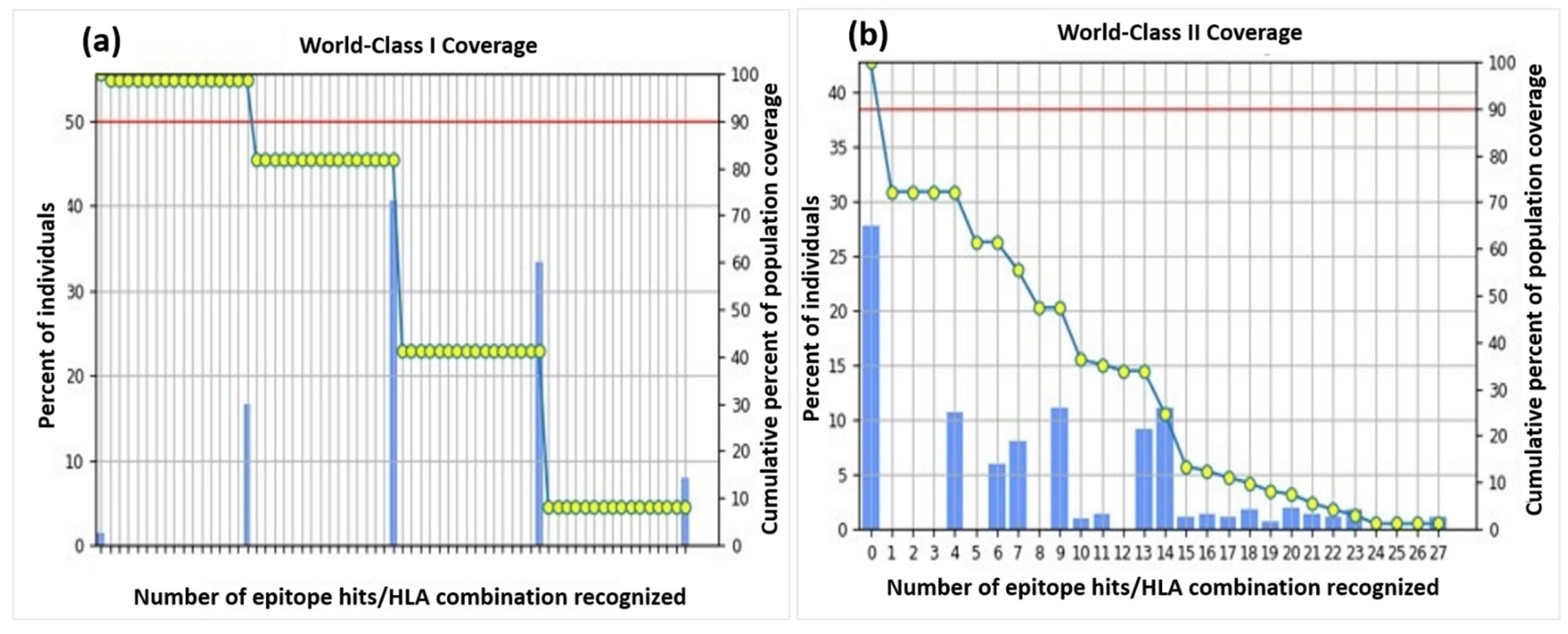
3.5. Population-Coverage Analysis
3.6. Assembly of a Multi-Epitope Vaccine
3.7. Quality Check Analysis and Profiling of Designed Vaccine
3.8. Physicochemical Properties
3.9. Prediction of Secondary and Tertiary Structure
3.10. Tertiary Structure Refinement and Validation
3.11. Vaccine Stability by Disulfide Engineering
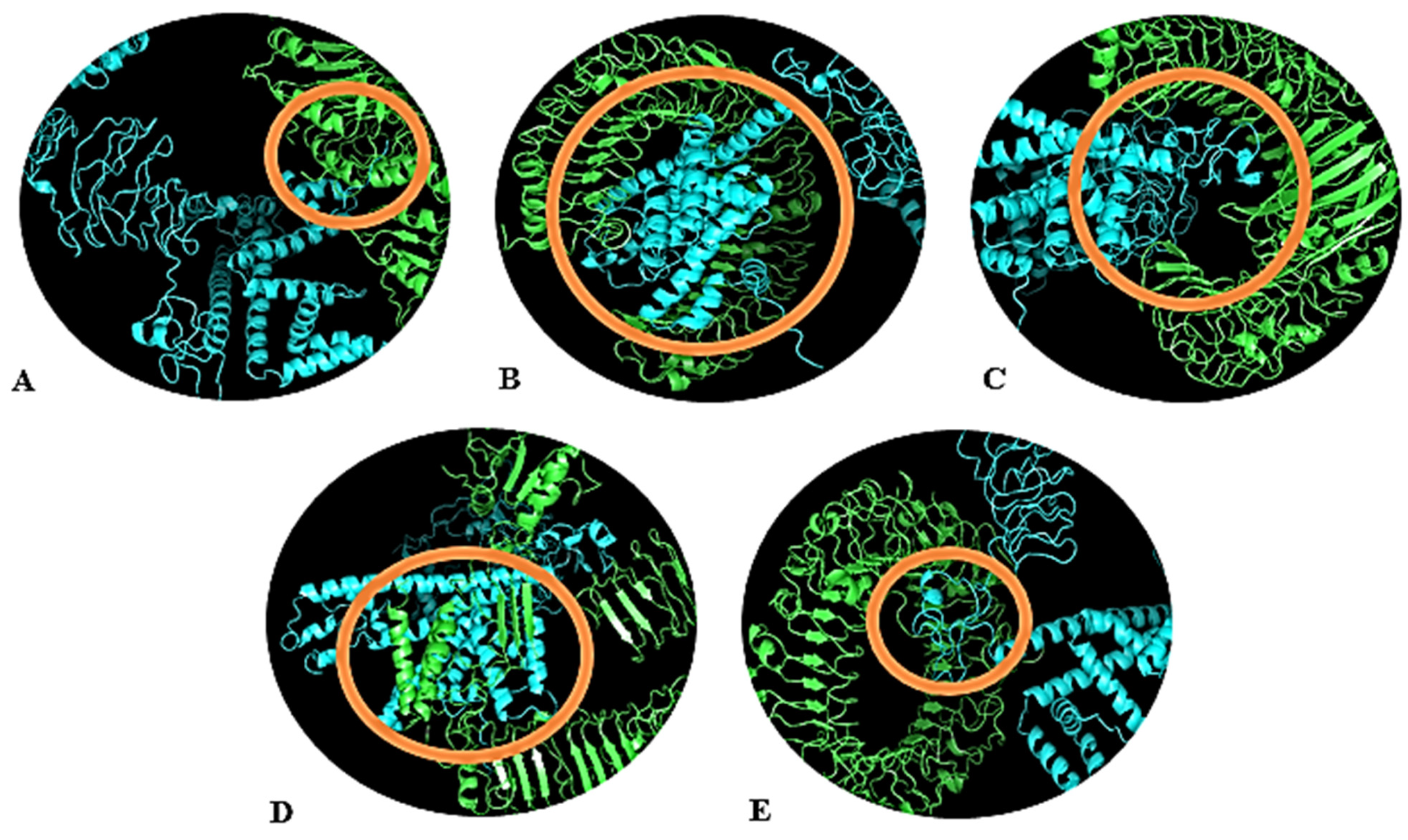
3.12. Molecular Docking of Designed Vaccine with Ligand Binding TLRs
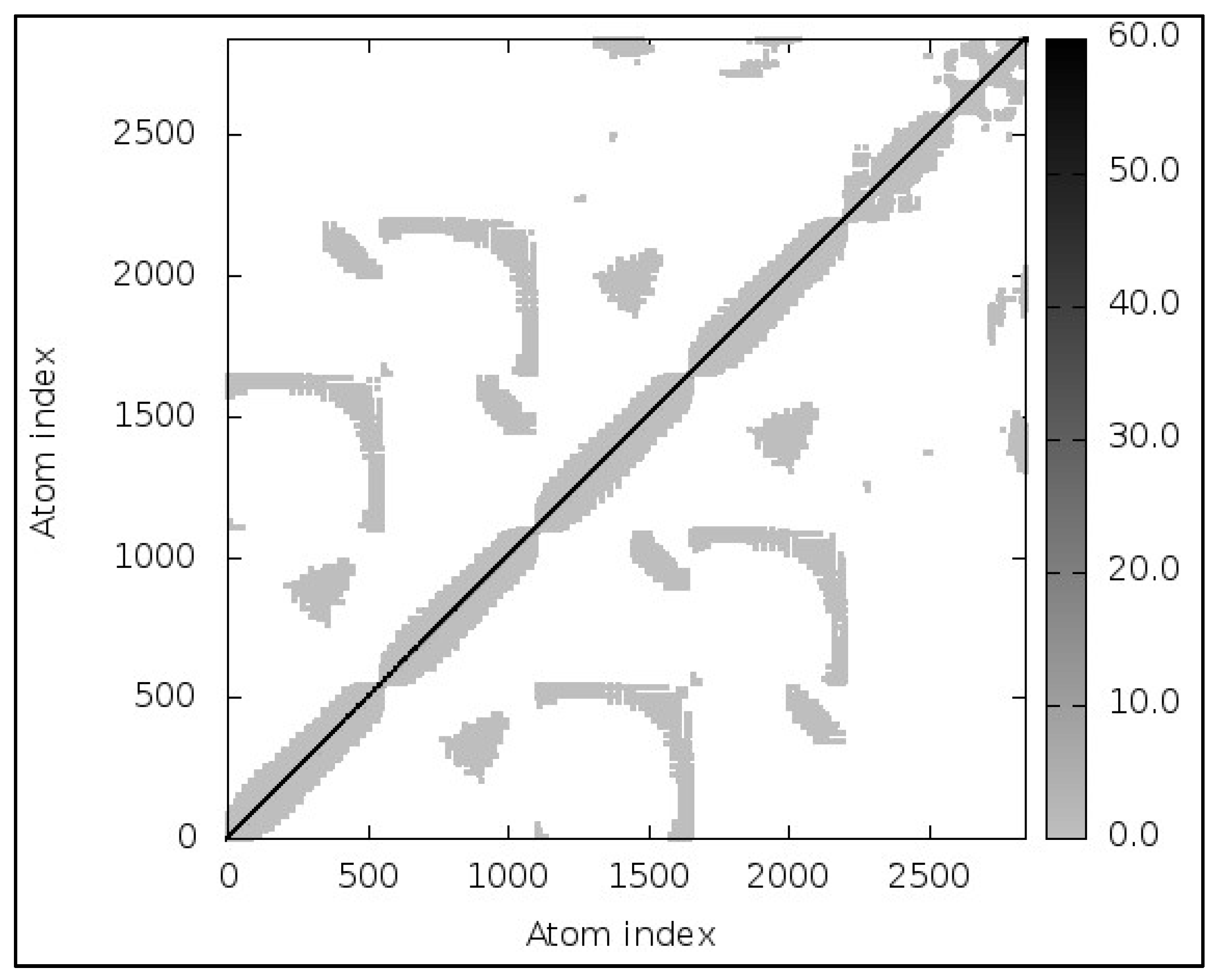
3.13. Molecular Dynamic Simulations
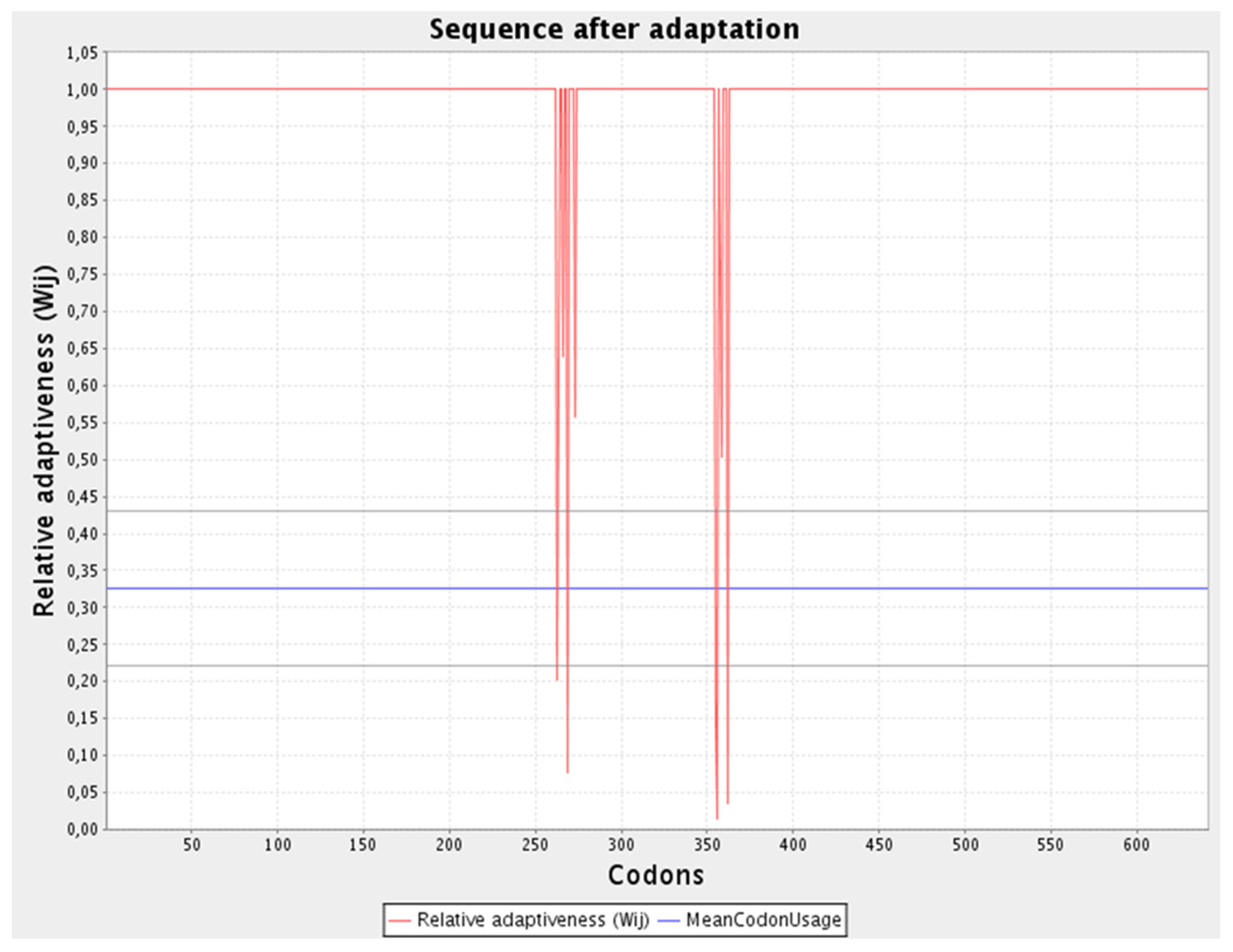
3.14. Adaptation and Enhancement of Codon along with In-Silico Cloning
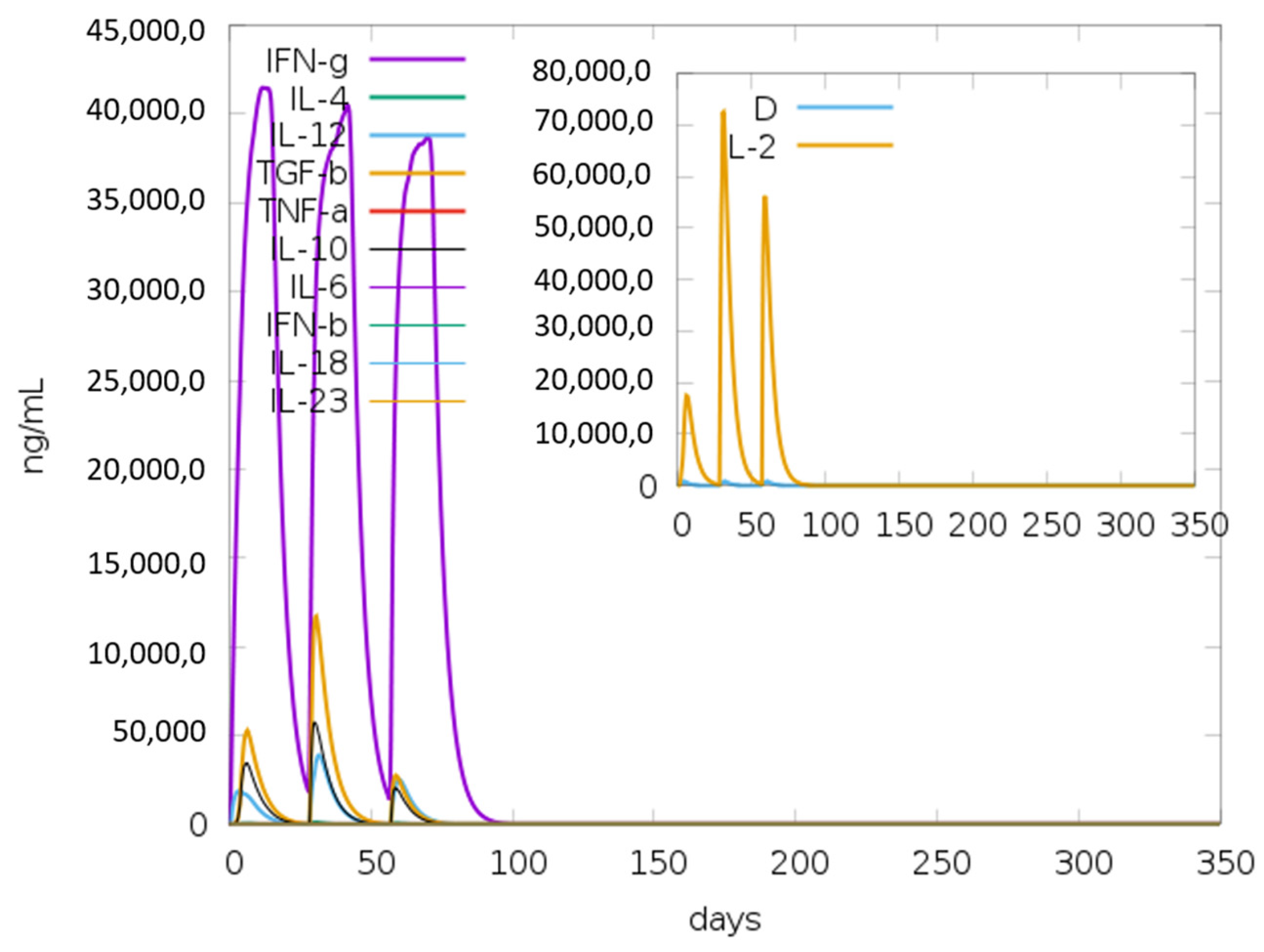
3.15. Immune Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Branche, A.R.; Falsey, A.R. Parainfluenza virus infection. Semin. Respir. Crit. Care Med. 2016, 37, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Coronel, E.C.; Murti, K.G.; Takimoto, T.; Portner, A. Human parainfluenza virus type 1 matrix and nucleoprotein genes transiently expressed in mammalian cells induce the release of virus-like particles containing nucleocapsid-like structures. J. Virol. 1999, 73, 7035–7038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, R.; Kolakofsky, D. Paramyxoviridae: The viruses and their replication. In Fields Virology, 3rd ed.; Lippincott-Raven Press: Philadelphia, PA, USA, 1996; pp. 1177–1204. [Google Scholar]
- Vilchez, R.A.; McCurry, K.; Dauber, J.; Iacono, A.; Keenan, R.; Zeevi, A.; Griffith, B.; Kusne, S. The epidemiology of parainfluenza virus infection in lung transplant recipients. Clin. Infect. Dis. 2001, 33, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Hasham, K.; Ahmed, N.; Zeshan, B. Circulating microRNAs in oncogenic viral infections: Potential diagnostic biomarkers. SN Appl. Sci. 2020, 2, 442. [Google Scholar] [CrossRef] [Green Version]
- Beck, E.T.; He, J.; Nelson, M.I.; Bose, M.E.; Fan, J.; Kumar, S.; Henrickson, K.J. Genome sequencing and phylogenetic analysis of 39 human parainfluenza virus type 1 strains isolated from 1997–2010. PLoS ONE 2012, 7, e46048. [Google Scholar] [CrossRef]
- Denny, F.W., Jr. The clinical impact of human respiratory virus infections. Am. J. Respir. Crit. Care Med. 1995, 152, S4. [Google Scholar] [CrossRef]
- Woo, P.C.; Young, K.; Tsang, K.W.; Ooi, C.G.; Peiris, M.; Yuen, K.-Y. Adult croup: A rare but more severe condition. Respiration 2000, 67, 684–688. [Google Scholar] [CrossRef]
- Bailly, B.; Dirr, L.; El-Deeb, I.M.; Altmeyer, R.; Guillon, P.; von Itzstein, M. A dual drug regimen synergistically blocks human parainfluenza virus infection. Sci. Rep. 2016, 6, 24138. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.G.; Porotto, M.; Palermo, L.M.; Cunha, L.F.; Greengard, O.; Moscona, A. Adaptation of human parainfluenza virus to airway epithelium reveals fusion properties required for growth in host tissue. MBio 2012, 3, e00137-12. [Google Scholar] [CrossRef] [Green Version]
- Slobod, K.S.; Shenep, J.L.; Luján-Zilbermann, J.; Allison, K.; Brown, B.; Scroggs, R.A.; Portner, A.; Coleclough, C.; Hurwitz, J.L. Safety and immunogenicity of intranasal murine parainfluenza virus type 1 (Sendai virus) in healthy human adults. Vaccine 2004, 22, 3182–3186. [Google Scholar] [CrossRef]
- Rafeek, R.A.; Divarathna, M.V.; Noordeen, F. A review on disease burden and epidemiology of childhood parainfluenza virus infections in Asian countries. Rev. Med. Virol. 2021, 31, e2164. [Google Scholar] [CrossRef] [PubMed]
- Dorosti, H.; Eslami, M.; Negahdaripour, M.; Ghoshoon, M.B.; Gholami, A.; Heidari, R.; Dehshahri, A.; Erfani, N.; Nezafat, N.; Ghasemi, Y. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J. Biomol. Struct. Dyn. 2019, 37, 3524–3535. [Google Scholar] [CrossRef] [PubMed]
- Faisal, A.-R.M.; Imtiaz, S.H.; Zerin, T.; Rahman, T.; Shekhar, H.U. Computer aided epitope design as a peptide vaccine component against Lassa virus. Bioinformation 2017, 13, 417. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Almofti, Y.A.; Abd-Elrahman, K.A. Immunoinformatics approach for multiepitopes vaccine prediction against glycoprotein B of avian infectious laryngotracheitis virus. Adv. Bioinform. 2019, 2019, 1270485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaliamurthi, S.; Selvaraj, G.; Kaushik, A.C.; Gu, K.-R.; Wei, D.-Q. Designing of CD8+ and CD8+-overlapped CD4+ epitope vaccine by targeting late and early proteins of human papillomavirus. Biol. Targets Ther. 2018, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Urrutia-Baca, V.H.; Gomez-Flores, R.; De La Garza-Ramos, M.A.; Tamez-Guerra, P.; Lucio-Sauceda, D.G.; Rodríguez-Padilla, M.C. Immunoinformatics approach to design a novel epitope-based oral vaccine against Helicobacter pylori. J. Comput. Biol. 2019, 26, 1177–1190. [Google Scholar] [CrossRef] [Green Version]
- Maeda, D.L.N.F.; Batista, M.T.; Pereira, L.R.; de Jesus Cintra, M.; Amorim, J.H.; Mathias-Santos, C.; Pereira, S.A.; Boscardin, S.B.; Silva, S.D.R.; Faquim-Mauro, E.L.; et al. Adjuvant-Mediated Epitope Specificity and Enhanced Neutralizing Activity of Antibodies Targeting Dengue Virus Envelope Protein. Front. Immunol. 2017, 8, 1175. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2005; pp. 571–607. [Google Scholar]
- Buchan, D.W.; Jones, D.T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dar, H.A.; Zaheer, T.; Shehroz, M.; Ullah, N.; Naz, K.; Muhammad, S.A.; Zhang, T.; Ali, A. Immunoinformatics-aided design and evaluation of a potential multi-epitope vaccine against Klebsiella pneumoniae. Vaccines 2019, 7, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.A.; Flower, D.R. Bioinformatic approach for identifying parasite and fungal candidate subunit vaccines. Open Vaccine J. 2008, 1, 4. [Google Scholar] [CrossRef]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Khan, S.; Ali, A.; Akbar, H.; Sayaf, A.M.; Khan, A.; Wei, D.-Q. Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci. Rep. 2019, 9, 13321. [Google Scholar] [CrossRef]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xu, Y.; Jin, X.; Shi, Z.; Wu, M.; Xu, N.; Yu, X.; Deng, S.; Zhang, K.; Zhang, L. Retrospective study on the effectiveness of a prevention strategy in a dental hospital during the COVID-19 pandemic. Clin. Oral Investig. 2021, 25, 5815–5822. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.C.; Davis, I.W.; Arendall III, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.R.; Won, J.; Heo, L.; Seok, C. GalaxyRefine2: Simultaneous refinement of inaccurate local regions and overall protein structure. Nucleic Acids Res. 2019, 47, W451–W455. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]
- Hajissa, K.; Zakaria, R.; Suppian, R.; Mohamed, Z. Epitope-based vaccine as a universal vaccination strategy against Toxoplasma gondii infection: A mini-Review. J. Adv. Vet. Anim. Res. 2019, 6, 174. [Google Scholar] [CrossRef]
- Rizvi, A.; Hussain, N.; Anjum, A.A.; Ahmed, N.; Naeem, A.; Khan, M.; Altaf, I. Effect of cell density on the biological titer and yield of 146S fraction of foot-and-mouth disease virus O in cell suspension. J. Virol. Methods 2022, 300, 114379. [Google Scholar] [CrossRef]
- Naveed, M.; Ali, U.; Karobari, M.I.; Ahmed, N.; Mohamed, R.N.; Abullais, S.S.; Kader, M.A.; Marya, A.; Messina, P.; Scardina, G.A. A Vaccine Construction against COVID-19-Associated Mucormycosis Contrived with Immunoinformatics-Based Scavenging of Potential Mucoralean Epitopes. Vaccines 2022, 10, 664. [Google Scholar] [CrossRef]
- Hajissa, K.; Zakaria, R.; Suppian, R.; Mohamed, Z. Immunogenicity of multiepitope vaccine candidate against Toxoplasma gondii infection in BALB/c mice. Iran. J. Parasitol. 2018, 13, 215. [Google Scholar] [PubMed]
- Khazaei-Poul, Y.; Farhadi, S.; Ghani, S.; Ahmadizad, S.A.; Ranjbari, J. Monocyclic peptides: Types, synthesis and applications. Curr. Pharm. Biotechnol. 2021, 22, 123–135. [Google Scholar] [CrossRef] [PubMed]
- St, I.; Salih, S.; M, B.; A, E.; S, K.; Mohammed, K.A.; Hamdi, A.; Hassan, M. In silico Prediction of Peptide based Vaccine against Fowlpox Virus (FPV). Immunome Res. 2018, 14, 1000154. [Google Scholar] [CrossRef]
- Zheng, J.; Lin, X.; Wang, X.; Zheng, L.; Lan, S.; Jin, S.; Ou, Z.; Wu, J. In Silico Analysis of Epitope-Based Vaccine Candidates against Hepatitis B Virus Polymerase Protein. Viruses 2017, 9, 112. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Shantier, S.W.; Mustafa, M.I.; Osman, H.K.; Elmansi, H.E.; Osman, I.-A.A.; Mohammed, R.A.; Abdelrhman, F.A.; Elnnewery, M.E.; Yousif, E.M.; et al. Epitope-Based Peptide Vaccine against Glycoprotein G of Nipah Henipavirus Using Immunoinformatics Approaches. J. Immunol. Res. 2020, 2020, 2567957. [Google Scholar] [CrossRef] [Green Version]
- Naveed, M.; Tehreem, S.; Arshad, S.; Bukhari, S.A.; Shabbir, M.A.; Essa, R.; Ali, N.; Zaib, S.; Khan, A.; Al-Harrasi, A.; et al. Design of a novel multiple epitope-based vaccine: An immunoinformatics approach to combat SARS-CoV-2 strains. J. Infect. Public Health 2021, 14, 938–946. [Google Scholar] [CrossRef]
- Gao, Q.; Bao, L.; Mao, H.; Wang, L.; Xu, K.; Yang, M.; Li, Y.; Zhu, L.; Wang, N.; Lv, Z.; et al. Development of an inactivated vaccine candidate for SARS-CoV-2. Science 2020, 369, 77–81. [Google Scholar] [CrossRef]
- Rahman, M.S.; Hoque, M.N.; Islam, M.R.; Akter, S.; Rubayet Ul Alam, A.S.M.; Siddique, M.A.; Saha, O.; Rahaman, M.M.; Sultana, M.; Crandall, K.A.; et al. Epitope-based chimeric peptide vaccine design against S, M and E proteins of SARS-CoV-2, the etiologic agent of COVID-19 pandemic: An in silico approach. PeerJ 2020, 8, e9572. [Google Scholar] [CrossRef]
- Choudhury, A.; Gupta, P.S.S.; Panda, S.K.; Rana, M.K.; Mukherjee, S. Designing AbhiSCoVac-A single potential vaccine for all ‘corona culprits’: Immunoinformatics and immune simulation approaches. J. Mol. Liq. 2022, 351, 118633. [Google Scholar] [CrossRef]
- Altmeyer, R.; Bailly, B. Inhibitors and Antiviral Drugs Designed to Target the Human Res. Microbes Infect 2014, 3, e62. [Google Scholar]
- Iorio, R.M.; Glickman, R.L.; Sheehan, J.P. Inhibition of fusion by neutralizing monoclonal antibodies to the haemagglutinin-neuraminidase glycoprotein of Newcastle disease virus. J. Gen. Virol. 1992, 73 Pt 5, 1167–1176. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, L.N.; Jiang, W.; Bhamidipati, K.; Millican, M.; Macaubas, C.; Hung, S.-C.; Mellins, E.D. The Other Function: Class II-Restricted Antigen Presentation by B Cells. Front. Immunol. 2017, 8, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir Ul Qamar, M.; Shokat, Z.; Muneer, I.; Ashfaq, U.A.; Javed, H.; Anwar, F.; Bari, A.; Zahid, B.; Saari, N. Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach. Vaccines 2020, 8, 288. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, B.; Ullah, M.A.; Araf, Y.; Das, S.; Rahman, M.H.; Moin, A.T. Designing novel epitope-based polyvalent vaccines against herpes simplex virus-1 and 2 exploiting the immunoinformatics approach. J. Biomol. Struct. Dyn. 2020, 39, 6585–6605. [Google Scholar] [CrossRef] [PubMed]
- Xagorari, A.; Chlichlia, K. Toll-like receptors and viruses: Induction of innate antiviral immune responses. Open Microbiol. J. 2008, 2, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Gibney, K.B.; Attwood, L.O.; Nicholson, S.; Tran, T.; Druce, J.; Healy, J.; Strachan, J.; Franklin, L.; Hall, R.; Cross, G.B. Emergence of attenuated measles illness among IgG-positive/IgM-negative measles cases: Victoria, Australia, 2008–2017. Clin. Infect. Dis. 2020, 70, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Pandey, R.K.; Khatoon, N.; Narula, A.; Mishra, A.; Prajapati, V.K. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 2017, 7, 9232. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
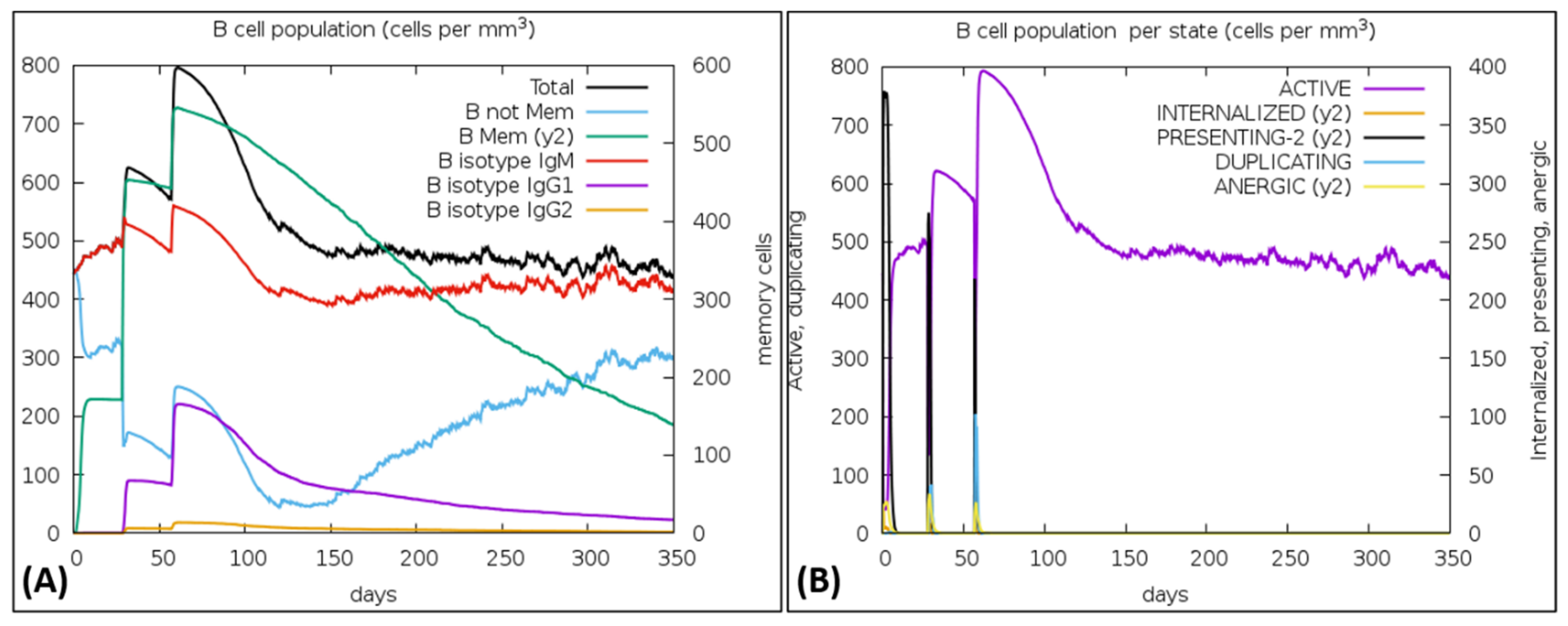{kind=link}
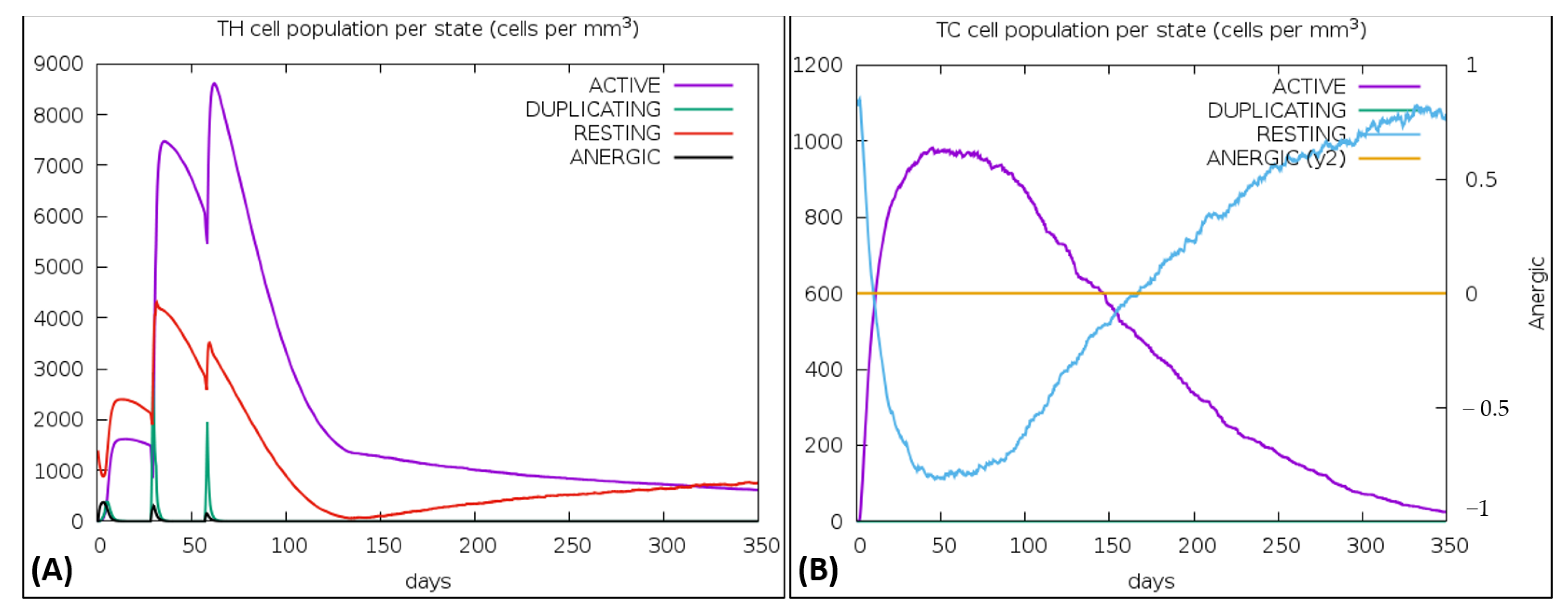{kind=link}
{kind=link}
| Molecule | MW (Da) | Instability Index | Half-Life | Theoretical pI | AA No. | GRAVY | Aliphatic Index |
|---|---|---|---|---|---|---|---|
| HN protein | 63,981.34 | 36.48 | 30 h | 8.17 | 575 | −0.161 | 94.94 |
| Vaccine | 69,661.70 | 33.32 | 3.5 h | 9.04 | 641 | −0.060 | 93.53 |
| Start | End | Peptide | Length | Antigenicity Score |
|---|---|---|---|---|
| 343 | 375 | TTPLQGDTKCVTNRCANVNQSVCNDALKITWLK | 33 | 0.5002 |
| 445 | 470 | MTIKWAPHEVLSRPGNQDCNWYNRCP | 26 | 0.8932 |
| 520 | 528 | RLKNVQLEA | 9 | 1.3398 |
| Start | End | Allele | Peptide | Length | Antigenicity Score |
|---|---|---|---|---|---|
| MHC-I EPITOPES | |||||
| 399 | 407 | HLA-A * 26:01 | ETIPITQNY | 9 | 0.9982 |
| 150 | 159 | HLA-B * 57:01 | ISPLDPHDFW | 10 | 0.9645 |
| 150 | 159 | HLA-B * 58:01 | ISPLDPHDFW | 10 | 0.9538 |
| 94 | 102 | HLA-A * 68:02 | EVISRTINI | 9 | 0.9457 |
| 247 | 256 | HLA-A * 68:01 | HTYDINDNRK | 10 | 0.9373 |
| 305 | 313 | HLA-A * 24:02 | RYKNEDITF | 9 | 0.9317 |
| 41 | 49 | HLA-B * 57:01 | TTMHTILSF | 9 | 0.9105 |
| 389 | 397 | HLA-A * 02:03 | YLSDRPKIV | 9 | 0.8992 |
| 305 | 313 | HLA-A * 23:01 | RYKNEDITF | 9 | 0.8926 |
| 364 | 373 | HLA-B * 57:01 | VCNDALKITW | 10 | 0.8925 |
| 2 | 11 | HLA-B * 44:03 | AEKGKTNSSY | 10 | 0.8767 |
| 2 | 11 | HLA-B * 44:02 | AEKGKTNSSY | 10 | 0.8757 |
| 150 | 159 | HLA-B * 53:01 | ISPLDPHDFW | 10 | 0.8588 |
| 367 | 375 | HLA-A * 68:01 | DALKITWLK | 9 | 0.8520 |
| 41 | 49 | HLA-B * 58:01 | TTMHTILSF | 9 | 0.8503 |
| 290 | 298 | HLA-A * 68:01 | LVFDILDLK | 9 | 0.8461 |
| MHC-II EPITOPES | |||||
| 46 | 60 | HLA-DRB4 * 01:01 | ILSFIIMILCIDLII | 14 | 0.08 |
| 45 | 59 | HLA-DRB4 * 01:01 | TILSFIIMILCIDL | 14 | 0.08 |
| 96 | 110 | HLA-DRB4 * 01:01 | ISRTINIQSSVQSGI | 14 | 0.15 |
| 95 | 109 | HLA-DRB4 * 01:01 | VISRTINIQSSVQSG | 14 | 0.2 |
| 94 | 108 | HLA-DRB4 * 01:01 | EVISRTINIQSSVQS | 14 | 0.23 |
| 166 | 180 | HLA-DRB3 * 02:02 | PLLSNNPNISLLPGP | 14 | 0.46 |
| 44 | 58 | HLA-DRB4 * 01:01 | HTILSFIIMILCIDL | 14 | 0.76 |
| 514 | 528 | HLA-DRB4 * 01:01 | EIINMLRLKNVQLEA | 14 | 1.2 |
| 515 | 529 | HLA-DRB4 * 01:01 | IINMLRLKNVQLEAA | 14 | 1.2 |
| 516 | 530 | HLA-DRB4 * 01:01 | INMLRLKNVQLEAAY | 14 | 1.4 |
| 415 | 429 | HLA-DRB1 * 15:01 | LKLGKKIYIYTRSSG | 14 | 1.5 |
| 43 | 57 | HLA-DRB1 * 15:01 | MHTILSFIIMILCID | 14 | 1.6 |
| 43 | 57 | HLA-DRB4 * 01:01 | MHTILSFIIMILCID | 14 | 2.3 |
| 517 | 531 | HLA-DRB4 * 01:01 | NMLRLKNVQLEAAYT | 14 | 2.6 |
| 511 | 525 | HLA-DRB4 * 01:01 | NTSEIINMLRLKNVQ | 14 | 2.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naveed, M.; Yaseen, A.R.; Khalid, H.; Ali, U.; Rabaan, A.A.; Garout, M.; Halwani, M.A.; Al Mutair, A.; Alhumaid, S.; Al Alawi, Z.; et al. Execution and Design of an Anti HPIV-1 Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses: An Immunoinformatic Approach. Vaccines 2022, 10, 869. https://doi.org/10.3390/vaccines10060869
Naveed M, Yaseen AR, Khalid H, Ali U, Rabaan AA, Garout M, Halwani MA, Al Mutair A, Alhumaid S, Al Alawi Z, et al. Execution and Design of an Anti HPIV-1 Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses: An Immunoinformatic Approach. Vaccines. 2022; 10(6):869. https://doi.org/10.3390/vaccines10060869
Chicago/Turabian StyleNaveed, Muhammad, Allah Rakha Yaseen, Hira Khalid, Urooj Ali, Ali A. Rabaan, Mohamed Garout, Muhammad A. Halwani, Abbas Al Mutair, Saad Alhumaid, Zainab Al Alawi, and et al. 2022. "Execution and Design of an Anti HPIV-1 Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses: An Immunoinformatic Approach" Vaccines 10, no. 6: 869. https://doi.org/10.3390/vaccines10060869