
Design of Multi-Epitope Vaccine for Staphylococcus saprophyticus: Pan-Genome and Reverse Vaccinology Approach

 , , , ,
, , , ,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Research Methodology
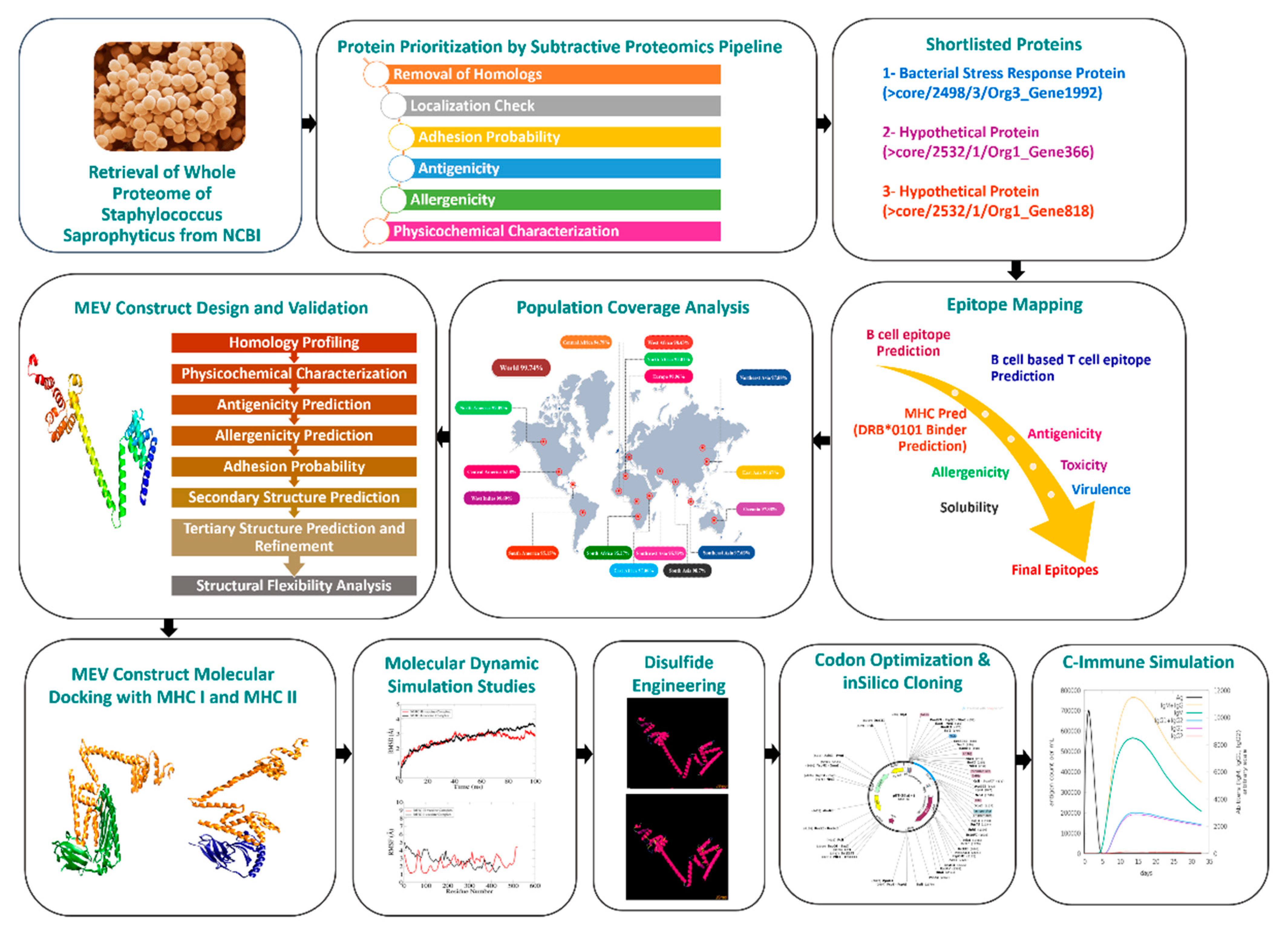
2.1. Extraction and Analysis of the Whole Proteome
2.2. Sub-Cellular Localization
2.3. Identification of Potential Vaccine Candidates by Reverse Vaccinology Approach
2.4. Prediction and Processing of Epitopes
2.5. Population Coverage and Epitope Conservation
2.6. Multiple-Epitope Vaccine Designing and Processing
2.7. Primary and Secondary Structure (SS) Analysis
2.8. Tertiary Structure (TS) Prediction and Validation
2.9. Estimation of Structural Flexibility
2.10. Molecular Docking Studies
2.11. Molecular Dynamics Simulation Analysis
2.12. Calculation of Binding Free Energies
2.13. Disulfide Engineering
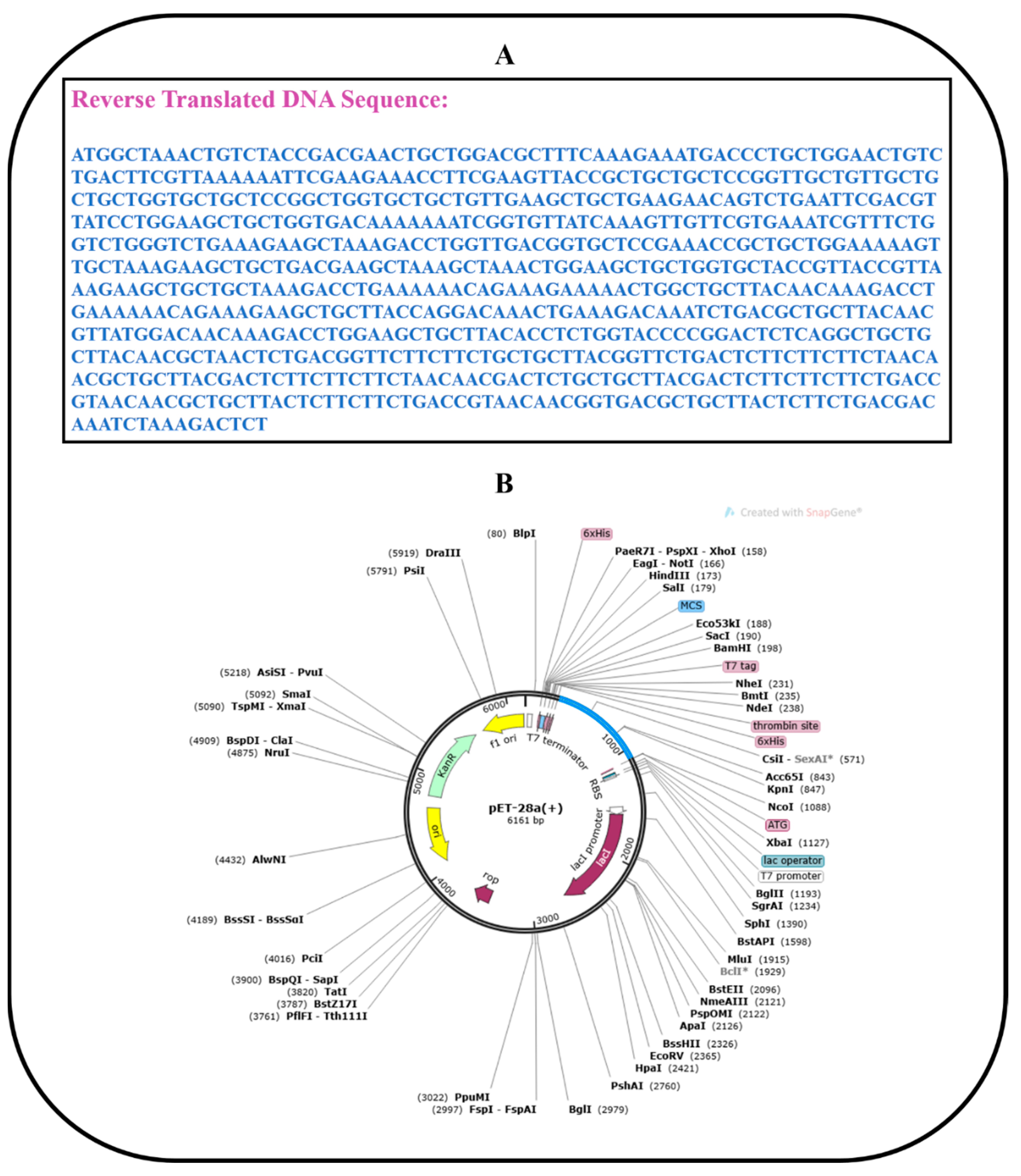
2.14. Codon Optimization and In Silico Cloning
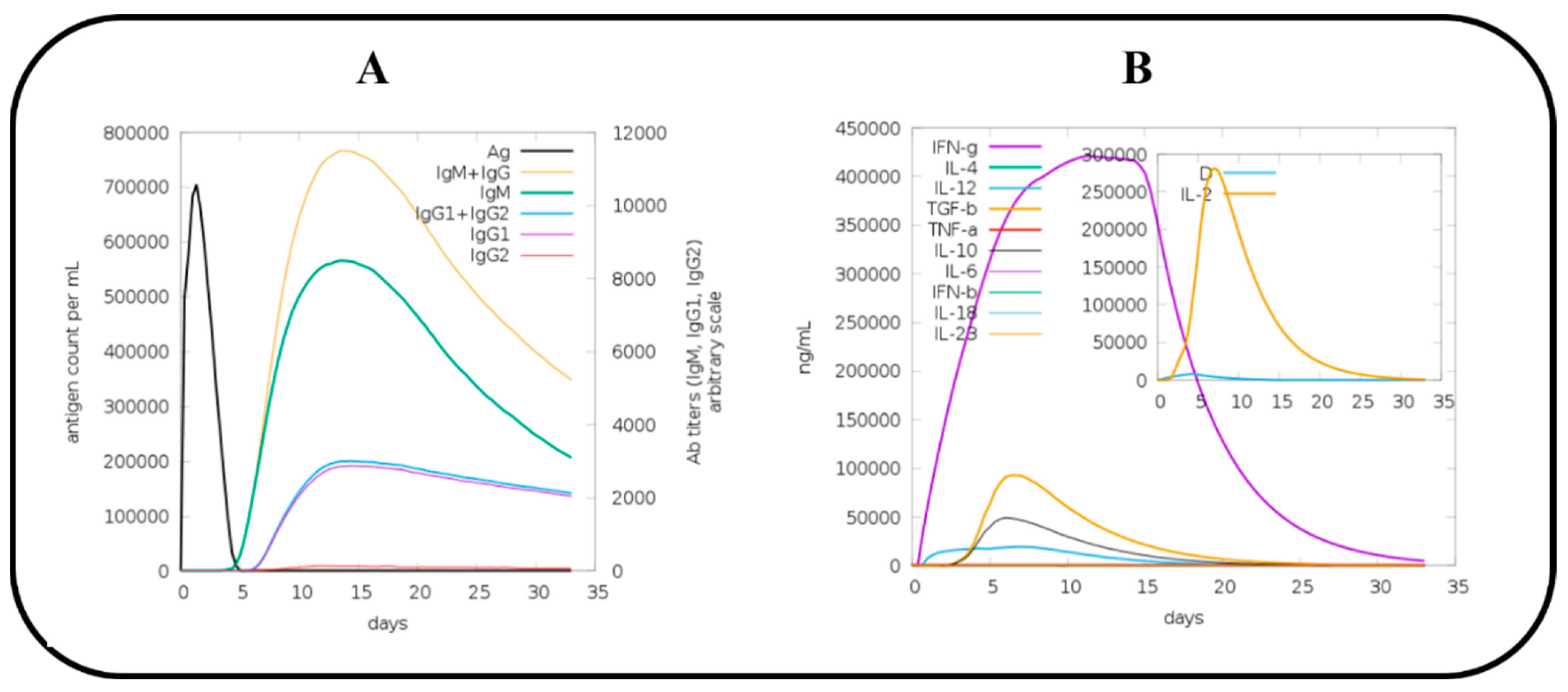
2.15. C Immune Simulation (IS)
3. Results
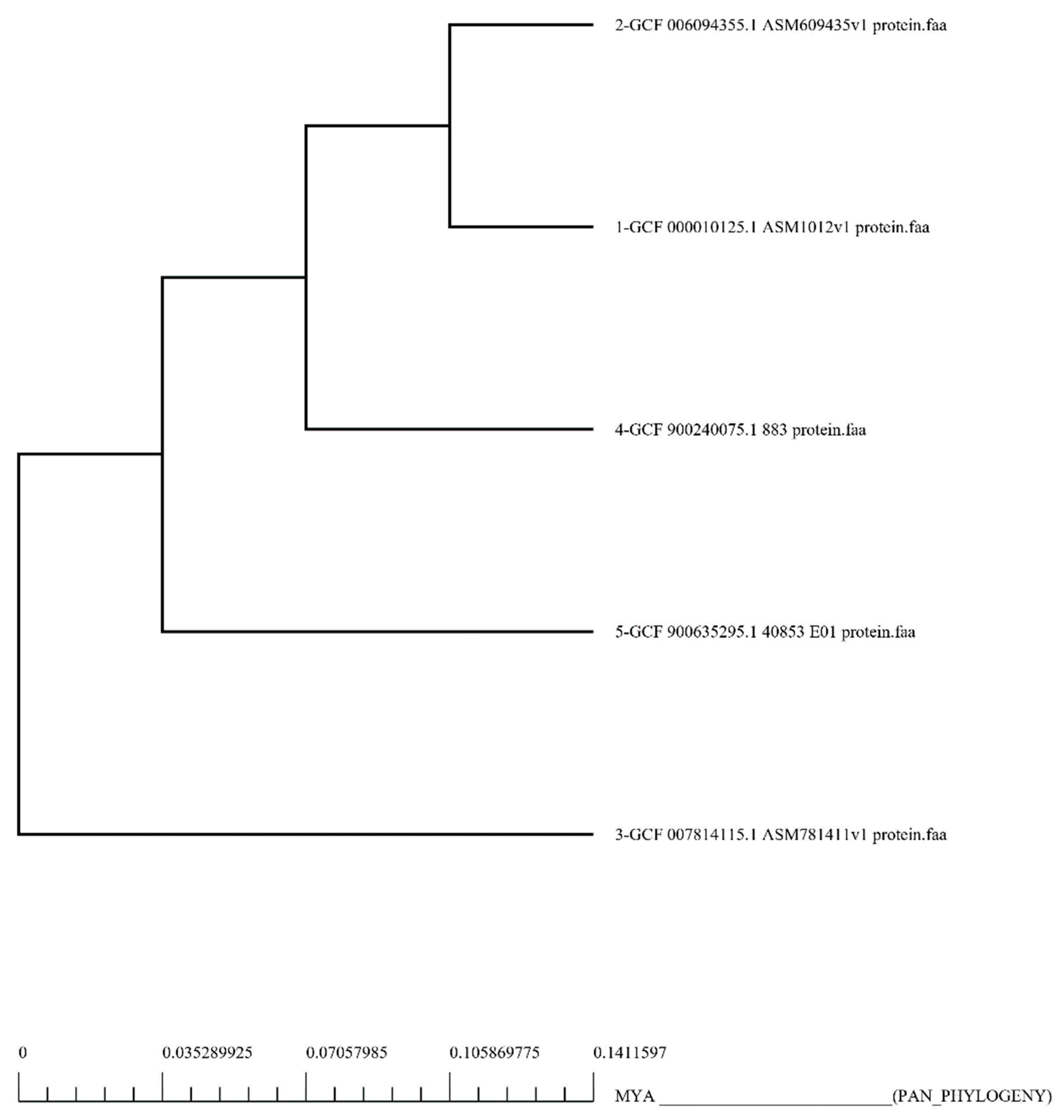
3.1. Extraction and Analysis of the Whole Proteome
3.2. Sub-Cellular Localization
3.3. Identification of Potential Vaccine Targets via Reverse Vaccinology
3.4. Epitope Prioritization
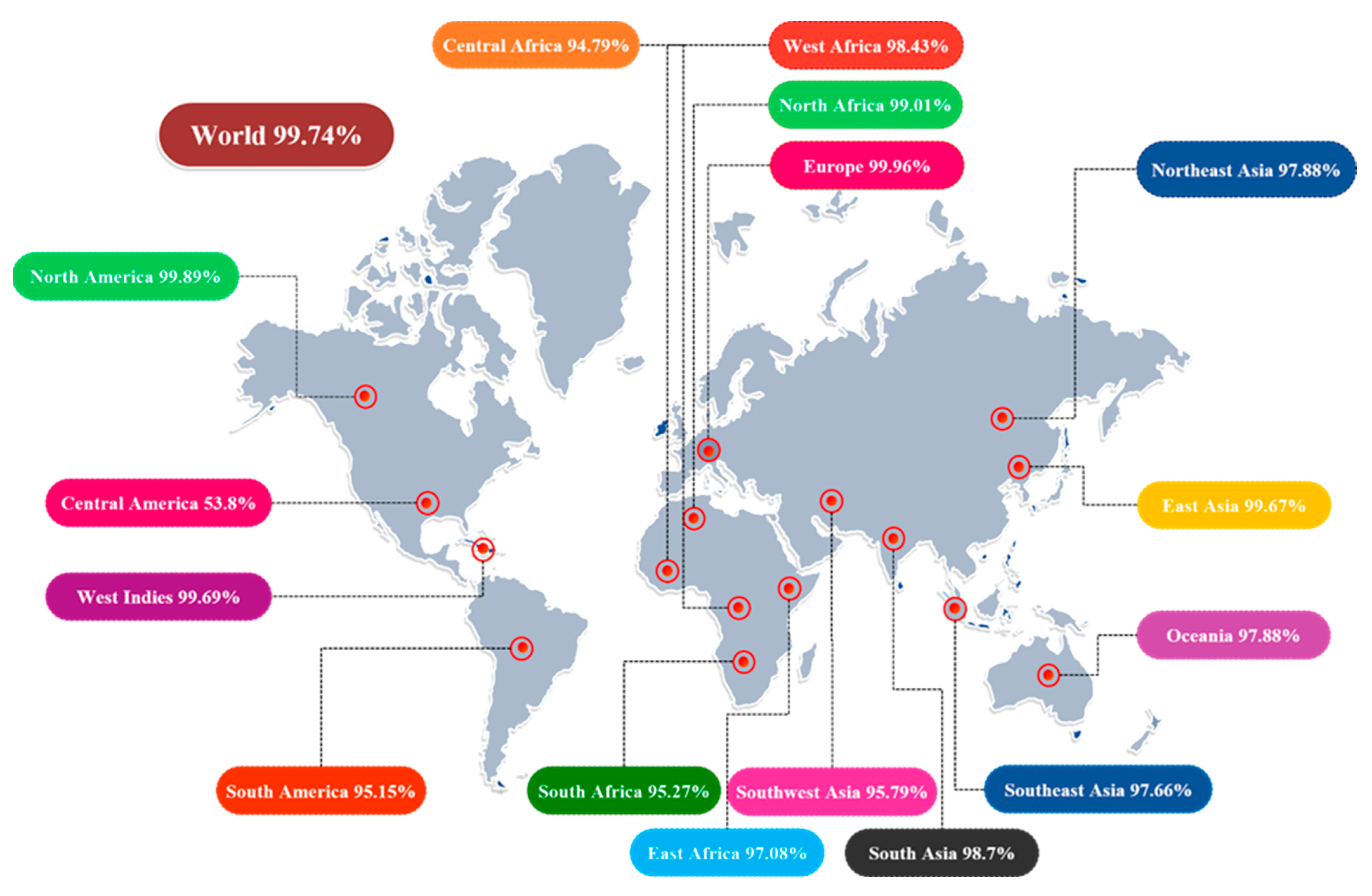
3.5. Population Coverage Analysis
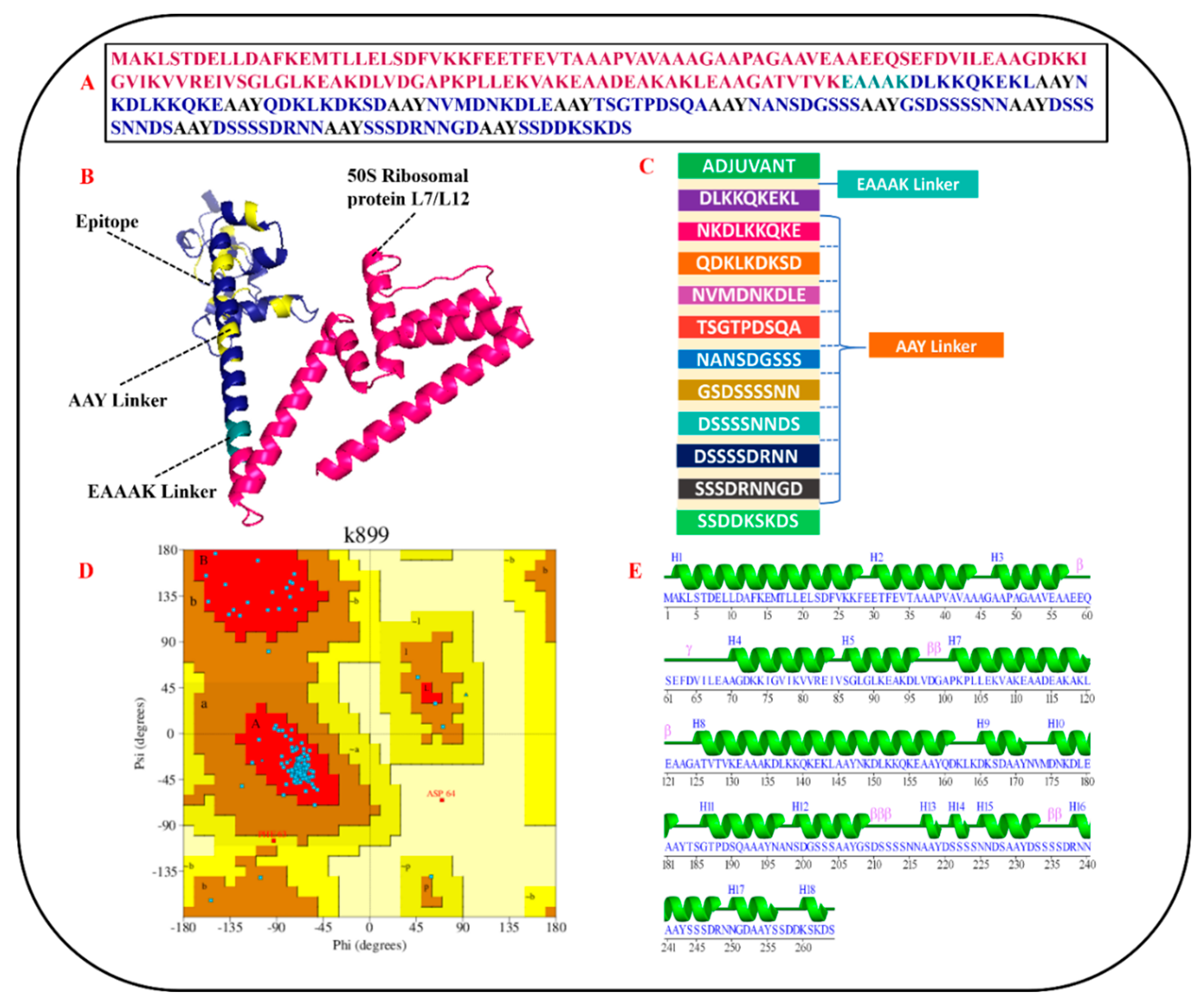
3.6. Designing of MEPVC and Post-Processing
3.7. Profiling of Immunogenic Potential and Physiochemical Characteristics
3.8. MEV Structure Prediction and Validation
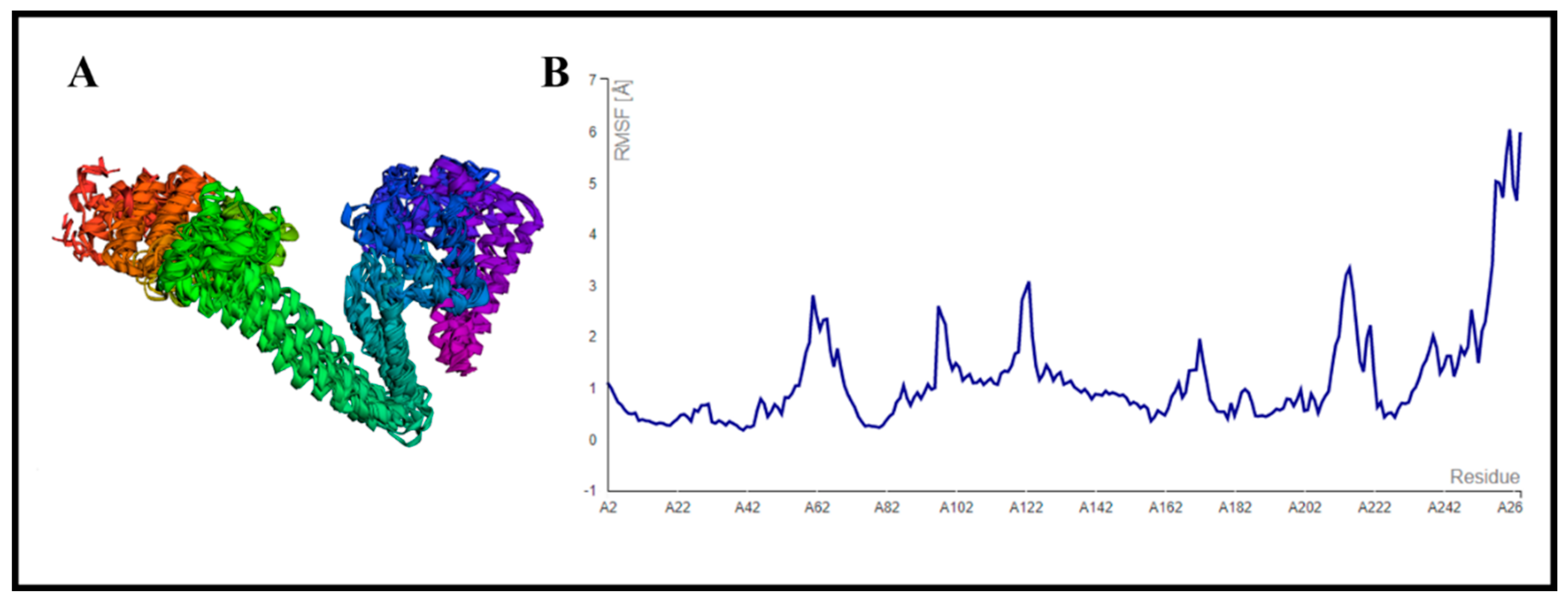
3.9. CABS-Flex Analysis
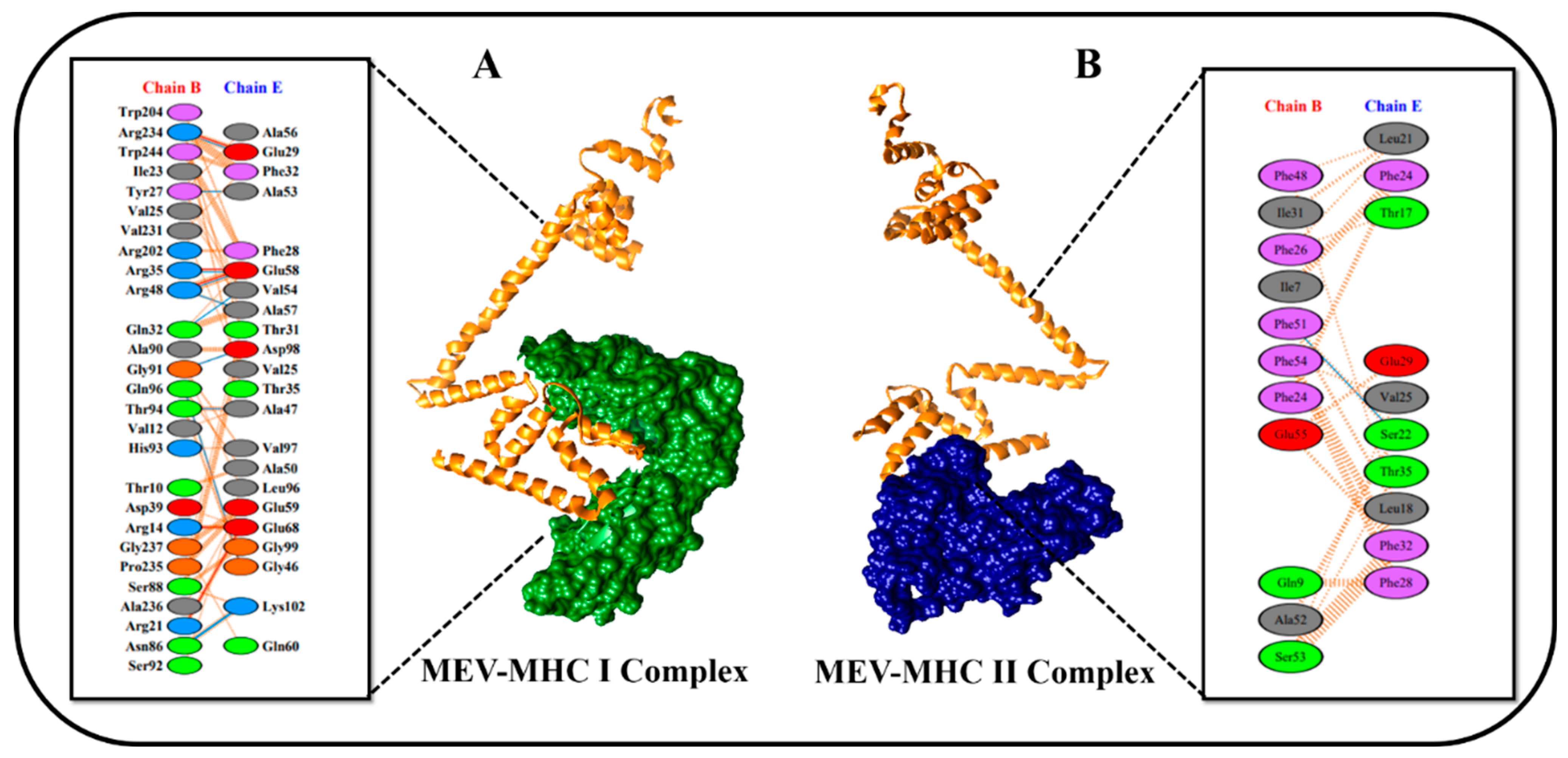
3.10. Molecular Docking Studies
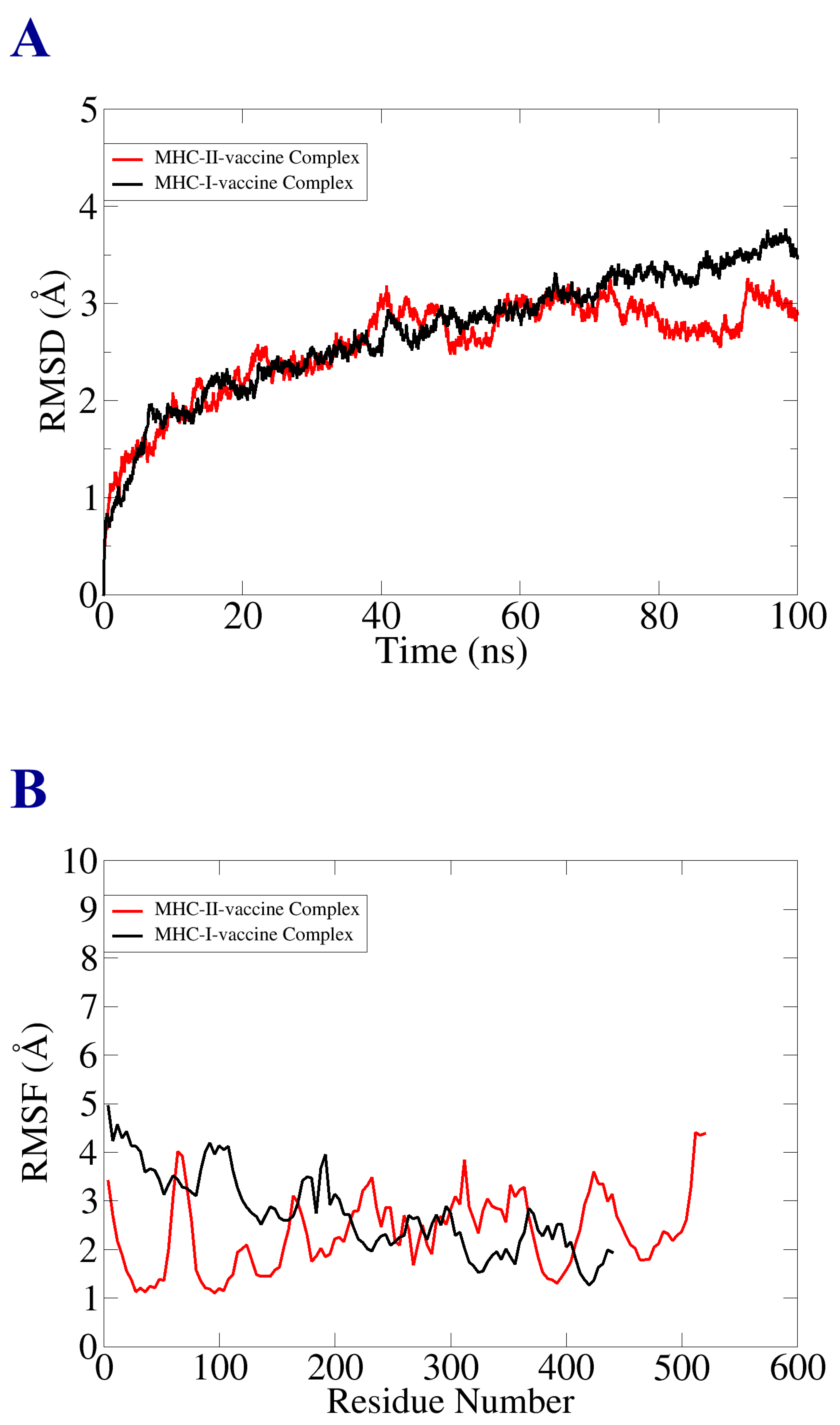
3.11. Molecular Dynamic Simulation
3.12. Binding Free Energies Estimation (MM/GBSA Analysis)
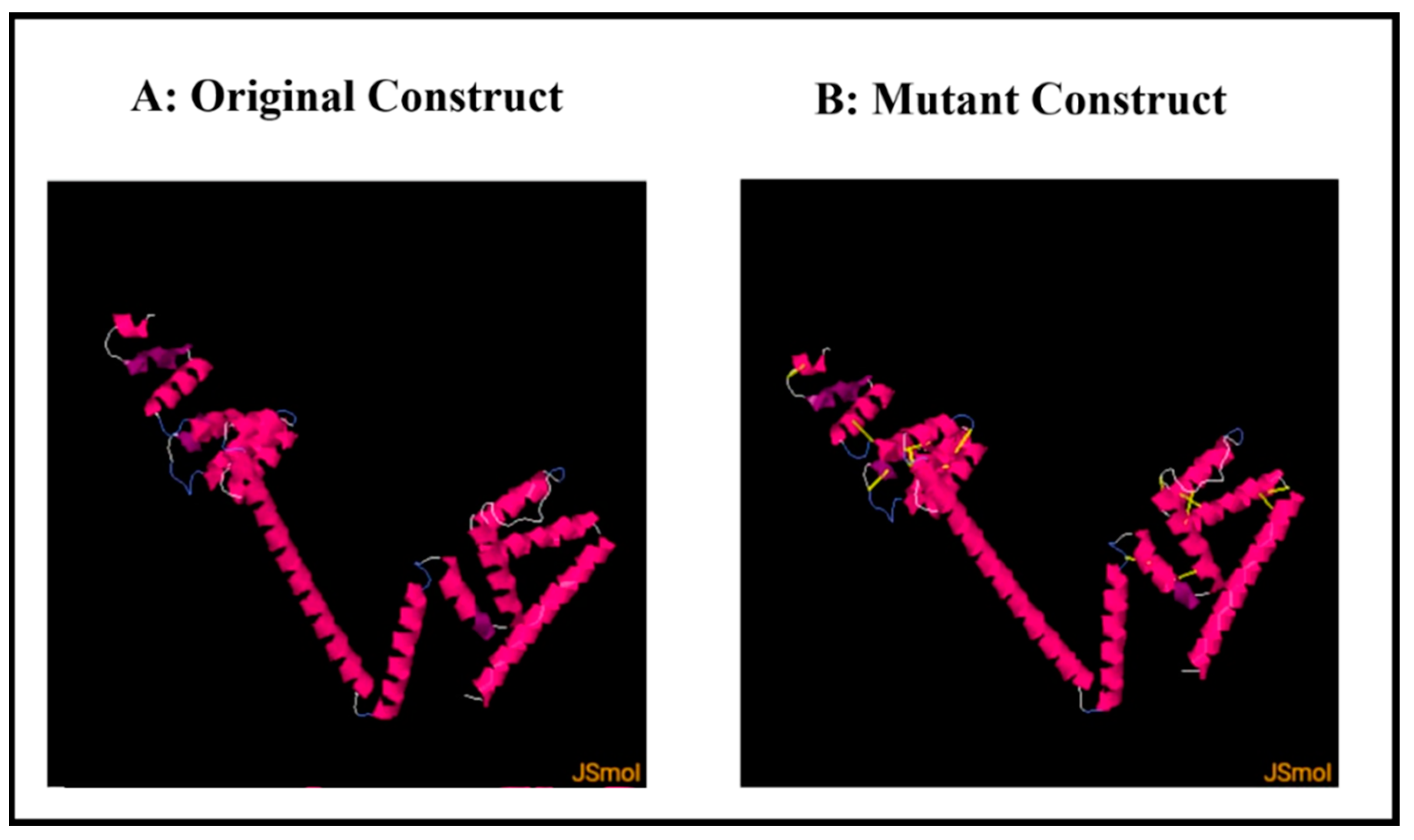
3.13. Disulfide Engineering
3.14. Codon Optimization and In Silico Cloning
3.15. Immune Simulation (IS) of MEV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kline, K.A.; Lewis, A.L. Gram-Positive Uropathogens, Polymicrobial Urinary Tract Infection, and the Emerging Microbiota of the Urinary Tract. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.R.; Raad, I. Fatal Staphylococcus Saprophyticus Native Valve Endocarditis in an Intravenous Drug Addict. J. Infect. Dis. 1990, 162, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Glimåker, M.; Granert, C.; Krook, A. Septicemia Caused by Staphylococcus Saprophyticus. Scand. J. Infect. Dis. 1988, 20, 347–348. [Google Scholar] [CrossRef]
- Hovelius, B.; Colleen, S.; Mardh, P.-A. Urinary Tract Infections in Men Caused by Staphylococcus Saprophyticus. Scand. J. Infect. Dis. 1984, 16, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, C.A.; Hertz, C.S.; Sheagren, J.N. Staphylococcus Saprophyticus: Role in Urinary Tract Infections in Men. J. Urol. 1983, 130, 493–494. [Google Scholar] [CrossRef]
- Raz, R.; Colodner, R.; Kunin, C.M. Who Are You—Staphylococcus Saprophyticus? Clin. Infect. Dis. 2005, 40, 896–898. [Google Scholar] [CrossRef]
- Kuroda, M.; Yamashita, A.; Hirakawa, H.; Kumano, M.; Morikawa, K.; Higashide, M.; Maruyama, A.; Inose, Y.; Matoba, K.; Toh, H. Whole Genome Sequence of Staphylococcus Saprophyticus Reveals the Pathogenesis of Uncomplicated Urinary Tract Infection. Proc. Natl. Acad. Sci. USA 2005, 102, 13272–13277. [Google Scholar] [CrossRef] [Green Version]
- Fowler, J.E., Jr. Staphylococcus Saprophyticus as the Cause of Infected Urinary Calculus. Ann. Intern. Med. 1985, 102, 342–343. [Google Scholar] [CrossRef]
- Latham, R.H.; Running, K.; Stamm, W.E. Urinary Tract Infections in Young Adult Women Caused by Staphylococcus Saprophyticus. JAMA 1983, 250, 3063–3066. [Google Scholar] [CrossRef]
- Rupp, M.E.; Soper, D.E.; Archer, G.L. Colonization of the Female Genital Tract with Staphylococcus Saprophyticus. J. Clin. Microbiol. 1992, 30, 2975–2979. [Google Scholar] [CrossRef] [Green Version]
- Donati, C.; Rappuoli, R. Reverse Vaccinology in the 21st Century: Improvements over the Original Design. Ann. N. Y. Acad. Sci. 2013, 1285, 115–132. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Tosta, S.F.; Passos, M.S.; Kato, R.; Salgado, Á.; Xavier, J.; Jaiswal, A.K.; Soares, S.C.; Azevedo, V.; Giovanetti, M.; Tiwari, S. Multi-Epitope Based Vaccine against Yellow Fever Virus Applying Immunoinformatics Approaches. J. Biomol. Struct. Dyn. 2021, 39, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Dar, H.A.; Ismail, S.; Waheed, Y.; Ahmad, S.; Jamil, Z.; Aziz, H.; Hetta, H.F.; Muhammad, K. Designing a Multi-Epitope Vaccine against Mycobacteroides Abscessus by Pangenome-Reverse Vaccinology. Sci. Rep. 2021, 11, 11197. [Google Scholar] [CrossRef] [PubMed]
- Shahid, F.; Zaheer, T.; Ashraf, S.T.; Shehroz, M.; Anwer, F.; Naz, A.; Ali, A. Chimeric Vaccine Designs against Acinetobacter Baumannii Using Pan Genome and Reverse Vaccinology Approaches. Sci. Rep. 2021, 11, 13213. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an Ultra-Fast Pan-Genome Analysis Pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A Web Server for Clustering and Comparing Biological Sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J. PSORTb 3.0: Improved Protein Subcellular Localization Prediction with Refined Localization Subcategories and Predictive Capabilities for All Prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- He, Y.; Xiang, Z.; Mobley, H.L. Vaxign: The First Web-Based Vaccine Design Program for Reverse Vaccinology and Applications for Vaccine Development. J. Biomed. Biotechnol. 2010, 2010, 297505. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular Mimicry as a Mechanism of Autoimmune Disease. Clin. Rev. Allergy Immunol. 2012, 42, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachdeva, G.; Kumar, K.; Jain, P.; Ramachandran, S. SPAAN: A Software Program for Prediction of Adhesins and Adhesin-like Proteins Using Neural Networks. Bioinformatics 2005, 21, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wizemann, T.M.; Adamou, J.E.; Langermann, S. Adhesins as Targets for Vaccine Development. Emerg. Infect. Dis. 1999, 5, 395. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP-a Server for in Silico Prediction of Allergens. BMC Bioinformatics. 2013, 14, S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ProtParam, E. ExPASy-ProtParam Tool; SIB: Lausanne, Switzerland, 2017. [Google Scholar]
- Adji, A.; Niode, N.J.; Memah, V.V.; Posangi, J.; Wahongan, G.J.; Ophinni, Y.; Idroes, R.; Mahmud, S.; Emran, T.B.; Nainu, F.; et al. Designing an epitope vaccine against Dermatophagoides pteronyssinus: An in silico study. Acta Trop. 2021, 222, 106028. [Google Scholar] [CrossRef] [PubMed]
- Bachmair, A.; Finley, D.; Varshavsky, A. In Vivo Half-Life of a Protein Is a Function of Its Amino-Terminal Residue. Science 1986, 234, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Ikai, A. Thermostability and Aliphatic Index of Globular Proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar]
- Guruprasad, K.; Reddy, B.B.; Pandit, M.W. Correlation between Stability of a Protein and Its Dipeptide Composition: A Novel Approach for Predicting in Vivo Stability of a Protein from Its Primary Sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of Putative Vaccine Candidates against Helicobacter Pylori Exploiting Exoproteome and Secretome: A Reverse Vaccinology Based Approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Wadood, A.; Jamal, A.; Riaz, M.; Khan, A.; Uddin, R.; Jelani, M.; Azam, S.S. Subtractive Genome Analysis for in Silico Identification and Characterization of Novel Drug Targets in Streptococcus Pneumonia Strain JJA. Microb. Pathog. 2018, 115, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Combating Tigecycline Resistant Acinetobacter Baumannii: A Leap Forward towards Multi-Epitope Based Vaccine Discovery. Eur. J. Pharm. Sci. 2019, 132, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A. The Immune Epitope Database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery Consortium; Raghava, G.P. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Gupta, D. VirulentPred: A SVM Based Prediction Method for Virulent Proteins in Bacterial Pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef] [Green Version]
- Deléage, G. ALIGNSEC: Viewing Protein Secondary Structure Predictions within Large Multiple Sequence Alignments. Bioinformatics 2017, 33, 3991–3992. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A Protein Structure and Structural Feature Prediction Server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [Green Version]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J. Galaxy: A Platform for Interactive Large-Scale Genome Analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.R.; Won, J.; Heo, L.; Seok, C. GalaxyRefine2: Simultaneous Refinement of Inaccurate Local Regions and Overall Protein Structure. Nucleic Acids Res. 2019, 47, W451–W455. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., III; De Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure Validation by Cα Geometry: ϕ, ψ and Cβ Deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Kuriata, A.; Gierut, A.M.; Oleniecki, T.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. CABS-Flex 2.0: A Web Server for Fast Simulations of Flexibility of Protein Structures. Nucleic Acids Res. 2018, 46, W338–W343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comeau, S.R.; Kozakov, D.; Brenke, R.; Shen, Y.; Beglov, D.; Vajda, S. ClusPro: Performance in CAPRI Rounds 6–11 and the New Server. Proteins Struct. Funct. Bioinform. 2007, 69, 781–785. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T., III; Darden, T.; Duke, R.; Gohlke, H. The FF14SB Force Field. Amber 2014, 14, 29–31. [Google Scholar]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A Web-Based Tool for Disulfide Engineering in Proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to Its Potential Expression Host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Castiglione, F. C-Immsim 10.1 Server. PLoS Pathog. 2012. Available online: http://www.cbs.dtu.dk/services/C-ImmSim-10.1/ (accessed on 8 April 2022).
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.; Lipman, D.J.; Ostell, J. GenBank. Nucleic Acids Res. 1993, 21, 2963–2965. [Google Scholar] [CrossRef]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Arico, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capecchi, B. Identification of Vaccine Candidates against Serogroup B Meningococcus by Whole-Genome Sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, F.A.; Oettgen, H.C. Adaptive Immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Baseer, S.; Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Towards a Peptide-Based Vaccine against Shigella Sonnei: A Subtractive Reverse Vaccinology Based Approach. Biologicals 2017, 50, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhmmad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and Immuno-Proteomics Analysis of Acinetobacter Baumannii Strains Revealed the Core Peptide Vaccine Targets. BMC Genom. 2016, 17, 732. [Google Scholar] [CrossRef] [Green Version]
- Naz, K.; Naz, A.; Ashraf, S.T.; Rizwan, M.; Ahmad, J.; Baumbach, J.; Ali, A. PanRV: Pangenome-Reverse Vaccinology Approach for Identifications of Potential Vaccine Candidates in Microbial Pangenome. BMC Bioinform. 2019, 20, 123. [Google Scholar] [CrossRef]
- Rashid, M.I.; Naz, A.; Ali, A.; Andleeb, S. Prediction of Vaccine Candidates against Pseudomonas Aeruginosa: An Integrated Genomics and Proteomics Approach. Genomics 2017, 109, 274–283. [Google Scholar] [CrossRef]
- Chung, E.H. Vaccine Allergies. Clin. Exp. Vaccine Res. 2014, 3, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Cai, Y.; Chen, J.; Ye, X.; Mao, S.; Zhu, S.; Xue, X.; Chen, S.; Zhang, L. Evaluation of Tandem Chlamydia Trachomatis MOMP Multi-Epitopes Vaccine in BALB/c Mice Model. Vaccine 2017, 35, 3096–3103. [Google Scholar] [CrossRef]
- Lennerz, V.; Gross, S.; Gallerani, E.; Sessa, C.; Mach, N.; Boehm, S.; Hess, D.; Von Boehmer, L.; Knuth, A.; Ochsenbein, A.F. Immunologic Response to the Survivin-Derived Multi-Epitope Vaccine EMD640744 in Patients with Advanced Solid Tumors. Cancer Immunol. Immunother. 2014, 63, 381–394. [Google Scholar] [CrossRef]
- Oyarzun, P.; Ellis, J.J.; Gonzalez-Galarza, F.F.; Jones, A.R.; Middleton, D.; Boden, M.; Kobe, B. A Bioinformatics Tool for Epitope-Based Vaccine Design That Accounts for Human Ethnic Diversity: Application to Emerging Infectious Diseases. Vaccine 2015, 33, 1267–1273. [Google Scholar] [CrossRef]
- Naz, S.; Ahmad, S.; Abbasi, S.W.; Ismail, S.; Waseem, S.; ul Qamar, M.T.; Ali, Z. Identification of Immunodominant Epitopes in Allelic Variants VK210 and VK247 of Pakistani Based Plasmodium Vivax Circumsporozoite Immunogen. Infect. Genet. Evol. 2021, 96, 105120. [Google Scholar] [CrossRef]
- Velders, M.P.; Weijzen, S.; Eiben, G.L.; Elmishad, A.G.; Kloetzel, P.-M.; Higgins, T.; Ciccarelli, R.B.; Evans, M.; Man, S.; Smith, L. Defined Flanking Spacers and Enhanced Proteolysis Is Essential for Eradication of Established Tumors by an Epitope String DNA Vaccine. J. Immunol. 2001, 166, 5366–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Sun, S.; Hu, Z.; Zhou, F.; Yin, M.; Xiao, C.; Zhang, J. Epitope DNA Vaccines against Tuberculosis: Spacers and Ubiquitin Modulates Cellular Immune Responses Elicited by Epitope DNA Vaccine. Scand. J. Immunol. 2004, 60, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, B.; Zhang, T.; Jia, J.; Lu, J.; Chen, Z.; Ni, Z.; Tan, T. Effect of Linker Length and Flexibility on the Clostridium Thermocellum Esterase Displayed on Bacillus Subtilis Spores. Appl. Biochem. Biotechnol. 2017, 182, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Motamedi, M.J.; Amani, J.; Shahsavandi, S.; Salmanian, A.H. In Silico Design of Multimeric HN-F Antigen as a Highly Immunogenic Peptide Vaccine against Newcastle Disease Virus. Int. J. Pept. Res. Ther. 2014, 20, 179–194. [Google Scholar] [CrossRef]
- Kwissa, M.; Nakaya, H.I.; Oluoch, H.; Pulendran, B. Distinct TLR Adjuvants Differentially Stimulate Systemic and Local Innate Immune Responses in Nonhuman Primates. Blood J. Am. Soc. Hematol. 2012, 119, 2044–2055. [Google Scholar] [CrossRef]
- Mata-Haro, V.; Cekic, C.; Martin, M.; Chilton, P.M.; Casella, C.R.; Mitchell, T.C. The Vaccine Adjuvant Monophosphoryl Lipid A as a TRIF-Biased Agonist of TLR4. Science 2007, 316, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Bohannon, J.K.; Hernandez, A.; Enkhbaatar, P.; Adams, W.L.; Sherwood, E.R. The Immunobiology of TLR4 Agonists: From Endotoxin Tolerance to Immunoadjuvants. Shock Augusta Ga 2013, 40, 451. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Shin, S.J.; Lee, M.H.; Lee, M.-G.; Kang, T.H.; Park, W.S.; Soh, B.Y.; Park, J.H.; Shin, Y.K.; Kim, H.W. A Potential Protein Adjuvant Derived from Mycobacterium Tuberculosis Rv0652 Enhances Dendritic Cells-Based Tumor Immunotherapy. PLoS ONE 2014, 9, e104351. [Google Scholar] [CrossRef]
- He, Y.; Zhang, J.; Donahue, C.; Falo, L.D., Jr. Skin-Derived Dendritic Cells Induce Potent CD8+ T Cell Immunity in Recombinant Lentivector-Mediated Genetic Immunization. Immunity 2006, 24, 643–656. [Google Scholar] [CrossRef] [Green Version]
- Larregina, A.T.; Falo, L.D., Jr. Changing Paradigms in Cutaneous Immunology: Adapting with Dendritic Cells. J. Investig. Dermatol. 2005, 124, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppenheim, J.; Biragyn, A.; Kwak, L.; Yang, D. Roles of Antimicrobial Peptides Such as Defensins in Innate and Adaptive Immunity. Ann. Rheum. Dis. 2003, 62, ii17–ii21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Territo, M.C.; Ganz, T.; Selsted, M.; Lehrer, R. Monocyte-Chemotactic Activity of Defensins from Human Neutrophils. J. Clin. Investig. 1989, 84, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum: Summaries and Analyses of PDB Structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef]
- Alhamadsheh, M.M.; Musayev, F.; Komissarov, A.A.; Sachdeva, S.; Wright, H.T.; Scarsdale, N.; Florova, G.; Reynolds, K.A. Alkyl-CoA Disulfides as Inhibitors and Mechanistic Probes for FabH Enzymes. Chem. Biol. 2007, 14, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Dombkowski, A.A.; Sultana, K.Z.; Craig, D.B. Protein Disulfide Engineering. FEBS Lett. 2014, 588, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [Green Version]
- Alharbi, M.; Alshammari, A.; Alasmari, A.F.; Alharbi, S.M.; Tahir ul Qamar, M.; Ullah, A.; Ahmad, S.; Irfan, M.; Khalil, A.A.K. Designing of a Recombinant Multi-Epitopes Based Vaccine against Enterococcus Mundtii Using Bioinformatics and Immunoinformatics Approaches. Int. J. Environ. Res. Public Health 2022, 19, 3729. [Google Scholar] [CrossRef]
- Albekairi, T.H.; Alshammari, A.; Alharbi, M.; Alshammary, A.F.; Tahir ul Qamar, M.; Ullah, A.; Irfan, M.; Ahmad, S. Designing of a Novel Multi-Antigenic Epitope-Based Vaccine against E. Hormaechei: An Intergraded Reverse Vaccinology and Immunoinformatics Approach. Vaccines 2022, 10, 665. [Google Scholar] [CrossRef]
- Ullah, A.; Ahmad, S.; Ismail, S.; Afsheen, Z.; Khurram, M.; Tahir ul Qamar, M.; AlSuhaymi, N.; Alsugoor, M.H.; Allemailem, K.S. Towards A Novel Multi-Epitopes Chimeric Vaccine for Simulating Strong Immune Responses and Protection against Morganella Morganii. Int. J. Environ. Res. Public. Health 2021, 18, 10961. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Shahid, F.; Khan, A.; Bhatti, S.; Ahmad, S.; Naz, A.; Almatroudi, A.; ul Qamar, M.T. Pan-Vaccinomics Approach towards a Universal Vaccine Candidate against WHO Priority Pathogens to Address Growing Global Antibiotic Resistance. Comput. Biol. Med. 2021, 136, 104705. [Google Scholar] [CrossRef] [PubMed]
- Czarniecki, C.W.; Sonnenfeld, G. Interferon-gamma and Resistance to Bacterial Infections. Apmis 1993, 101, 1–17. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Epitopes | B Start Site | B End Site | Percentile Score | MHC Pred (IC50) | Antigenicity | Allergenicity | Toxin Pred | Solubility | Virulent Pred |
|---|---|---|---|---|---|---|---|---|---|---|
| >core/2532/1/Org1_Gene366 (Hypothetical Protein) | DLKKQKEKL | 23 | 31 | 0.02 | 75.68 | 0.8910 | Non-Allergen | Non-Toxin | Soluble | 1.0606 (Virulent) |
| NKDLKKQKE | 21 | 29 | 13 | 37.24 | 1.3267 | Non-Allergen | Non-Toxin | Soluble | 1.0606 (Virulent) | |
| QDKLKDKSD | 35 | 43 | 51 | 21.98 | 0.9821 | Non-Allergen | Non-Toxin | Soluble | 1.0606 (Virulent) | |
| >core/2498/3/Org3_Gene1992 (Bacterial Stress Response Protein) | NVMDNKDLE | 16 | 24 | 6.8 | 15.92 | 0.9392 | Non-Allergen | Non-Toxin | Soluble | 1.0605 (Virulent) |
| >core/1222/1/Org1_Gene818 (Hypothetical Protein) | TSGTPDSQA | 181 | 189 | 5.3 | 30.83 | 1.3068 | Non-Allergen | Non-Toxin | Soluble | 1.0715 (Virulent) |
| NANSDGSSS | 98 | 106 | 17 | 34.67 | 2.5865 | Non-Allergen | Non-Toxin | Soluble | 1.0606 (Virulent) | |
| GSDSSSSNN | 87 | 95 | 23 | 60.39 | 2.3369 | Non-Allergen | Non-Toxin | Soluble | 1.0606 (Virulent) | |
| DSSSSNNDS | 89 | 97 | 35 | 42.66 | 1.8340 | Non-Allergen | Non-Toxin | Soluble | 1.0606 (Virulent) | |
| DSSSSDRNN | 67 | 75 | 37 | 88.1 | 1.9116 | Non-Allergen | Non-Toxin | Soluble | 1.0606 (Virulent) | |
| SSSDRNNGD | 69 | 77 | 31 | 96.16 | 1.6609 | Non-Allergen | Non-Toxin | Soluble | 1.0606 (Virulent) | |
| SSDDKSKDS | 29 | 37 | 37 | 10.07 | 2.4371 | Non-Allergen | Non-Toxin | Soluble | 1.0606 (Virulent) |
| Model | RMSD | MolProbity | Clash Score | Poor Rotamers | Rama Favored | GALAXY Energy |
|---|---|---|---|---|---|---|
| Initial | 0 | 2.602 | 81.5 | 1 | 96.6 | 7326.88 |
| MODEL 1 | 1.134 | 0.968 | 1 | 0 | 96.9 | −5724.21 |
| MODEL 2 | 1.153 | 0.733 | 0.5 | 0 | 97.7 | −5717.53 |
| MODEL 3 | 1.288 | 1.011 | 1 | 0 | 96.6 | −5711.32 |
| MODEL 4 | 1.243 | 0.797 | 0.7 | 0 | 97.7 | −5704.9 |
| MODEL 5 | 1.125 | 0.789 | 1 | 0 | 98.1 | −5701.56 |
| MODEL 6 | 1.249 | 0.917 | 1 | 0 | 97.3 | −5699.96 |
| MODEL 7 | 1.053 | 0.862 | 0.7 | 0 | 97.3 | −5698.35 |
| MODEL 8 | 1.15 | 0.733 | 0.7 | 0.5 | 98.1 | −5698.25 |
| MODEL 9 | 1.247 | 1.051 | 1.7 | 0 | 97.3 | −5695.52 |
| MODEL 10 | 1.174 | 0.903 | 1.2 | 0 | 97.7 | −5690.57 |
| MEV–MHC-I Protein–Protein Docking Results | ||
| Docked Complex | Cluster Members | Binding Energy (kcal/mol) |
| 1 | 97 | −855.4 |
| 2 | 97 | −876.2 |
| 3 | 89 | −766.8 |
| 4 | 87 | −938.1 |
| 5 | 74 | −889.4 |
| MEV–MHC-II Protein–Protein Docking Results | ||
| Docked Complex | Cluster Members | Binding Energy (kcal/mol) |
| 1 | 99 | −773.7 |
| 2 | 84 | −843.8 |
| 3 | 70 | −777.4 |
| 4 | 68 | −770.0 |
| 5 | 57 | −763.5 |
| MM/GBSA | ||
|---|---|---|
| ENERGY PARAMETER | MHC-I–Vaccine Complex | MHC-II–Vaccine Complex |
| VDWAALS | −188.00 | −192.00 |
| EEL | −68.00 | −56.00 |
| DELTA G GAS | −256.00 | −248.00 |
| DELTA G SOLV | 39.00 | 34.00 |
| DELTA TOTAL | −217 | −214 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousaf, M.; Ullah, A.; Sarosh, N.; Abbasi, S.W.; Ismail, S.; Bibi, S.; Hasan, M.M.; Albadrani, G.M.; Talaat Nouh, N.A.; Abdulhakim, J.A.; et al. Design of Multi-Epitope Vaccine for Staphylococcus saprophyticus: Pan-Genome and Reverse Vaccinology Approach. Vaccines 2022, 10, 1192. https://doi.org/10.3390/vaccines10081192
Yousaf M, Ullah A, Sarosh N, Abbasi SW, Ismail S, Bibi S, Hasan MM, Albadrani GM, Talaat Nouh NA, Abdulhakim JA, et al. Design of Multi-Epitope Vaccine for Staphylococcus saprophyticus: Pan-Genome and Reverse Vaccinology Approach. Vaccines. 2022; 10(8):1192. https://doi.org/10.3390/vaccines10081192
Chicago/Turabian StyleYousaf, Maha, Asad Ullah, Nida Sarosh, Sumra Wajid Abbasi, Saba Ismail, Shabana Bibi, Mohammad Mehedi Hasan, Ghadeer M. Albadrani, Nehal Ahmed Talaat Nouh, Jawaher A. Abdulhakim, and et al. 2022. "Design of Multi-Epitope Vaccine for Staphylococcus saprophyticus: Pan-Genome and Reverse Vaccinology Approach" Vaccines 10, no. 8: 1192. https://doi.org/10.3390/vaccines10081192