
Immunoinformatics-Based Proteome Mining to Develop a Next-Generation Vaccine Design against Borrelia burgdorferi: The Cause of Lyme Borreliosis




Abstract
:1. Introduction
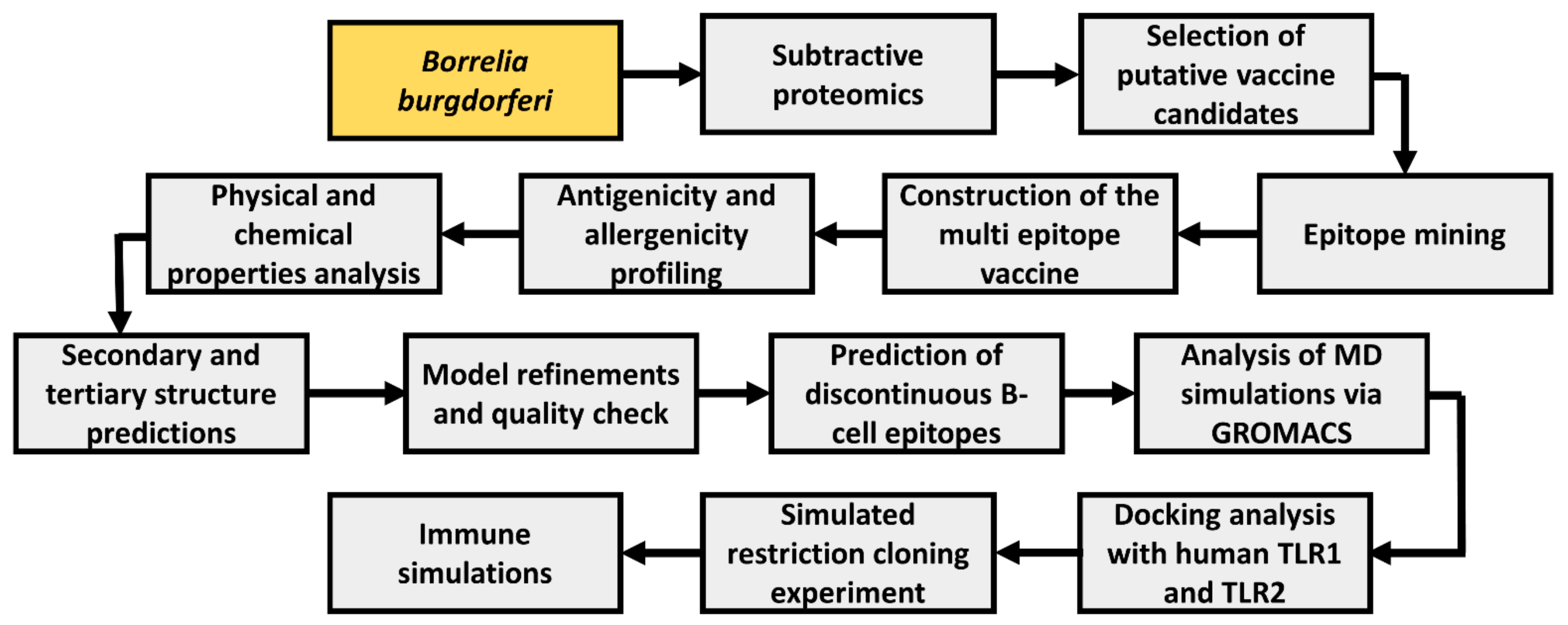
2. Materials and Methods
2.1. Subtractive Proteomics
2.2. Epitope Mining
2.3. Prediction of IFN-γ Epitopes
2.4. Epitope Screening
2.5. Epitope Assemblage
2.6. Conformational B Cell Epitope Analysis
2.7. Antigenicity and Safety Profiling of the MEV
2.8. Physicochemical Profiling, Structure Projections, Model Refinement, and Quality-Check
2.9. MD Analysis
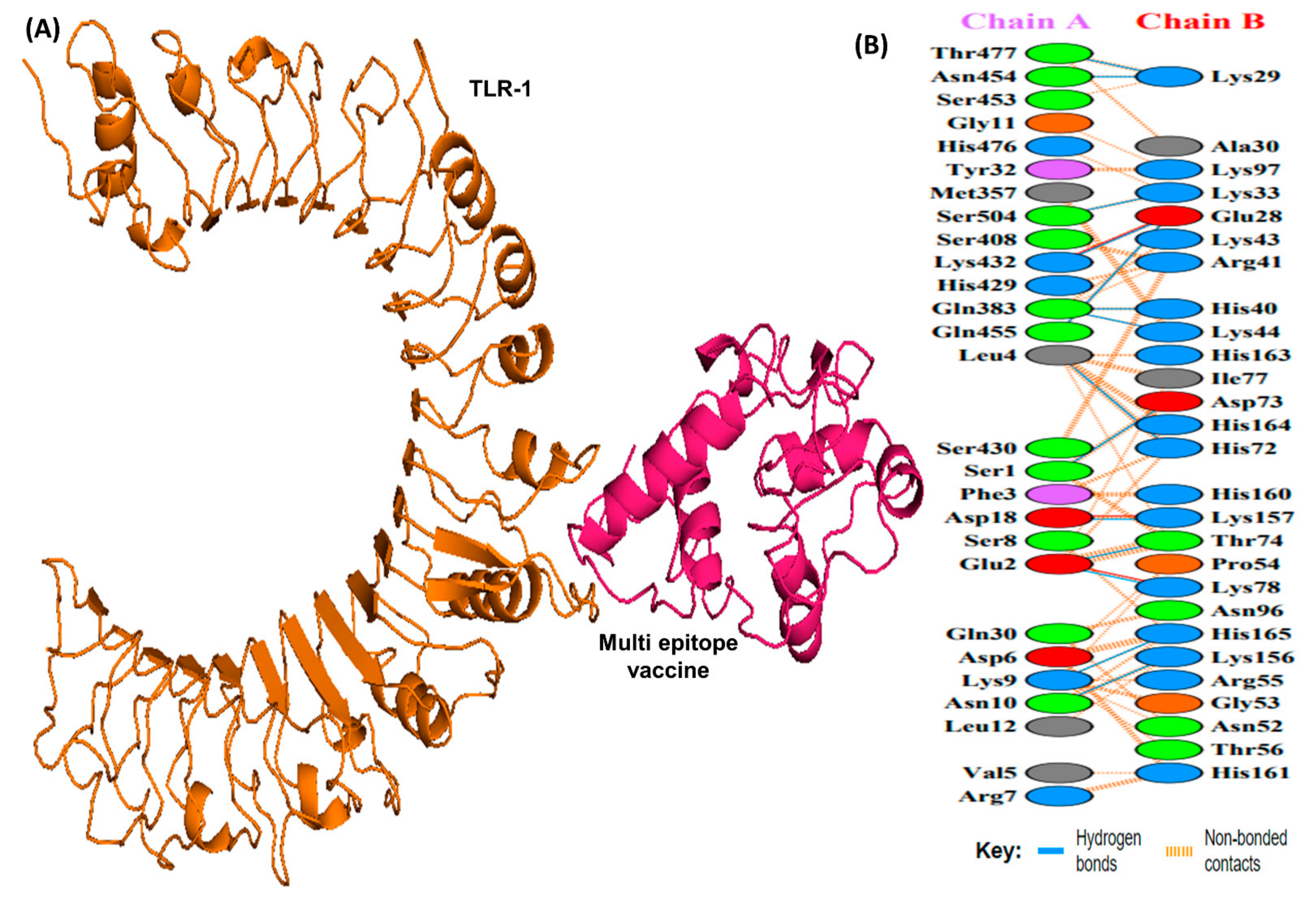
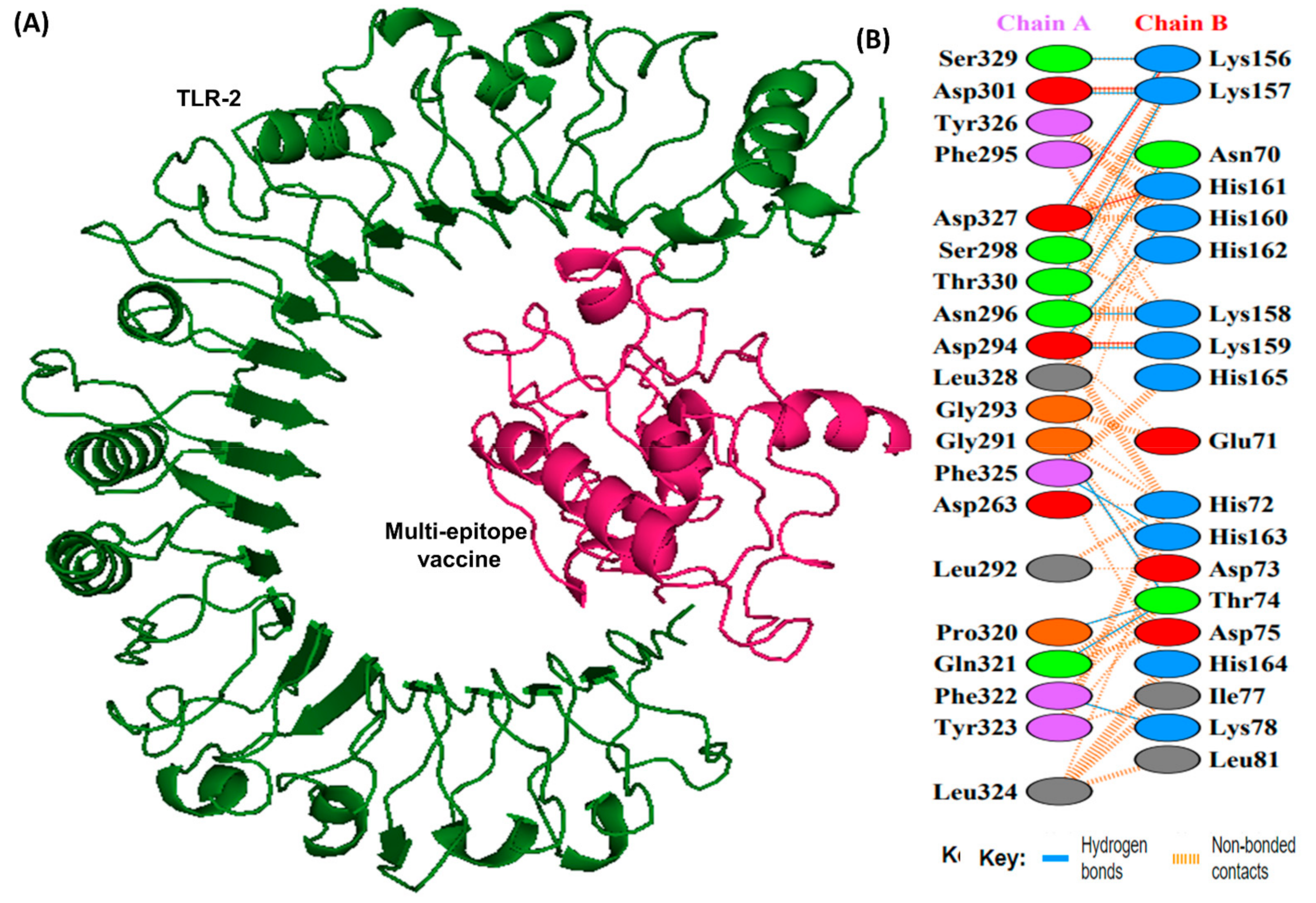
2.10. Molecular Docking with TLR-1 and TLR-2
2.11. In-Silico Cloning Experiment
2.12. Immune Simulations
3. Results
3.1. Data Assemblage and Proteome Subtraction
3.2. Immunogenicity and Protein Size Analysis
3.3. Epitope Mining
3.4. Designing of the MEV
3.5. Antigenicity, Allergenicity, and Safety Profiling of the MEV
3.6. Physicochemical Analysis
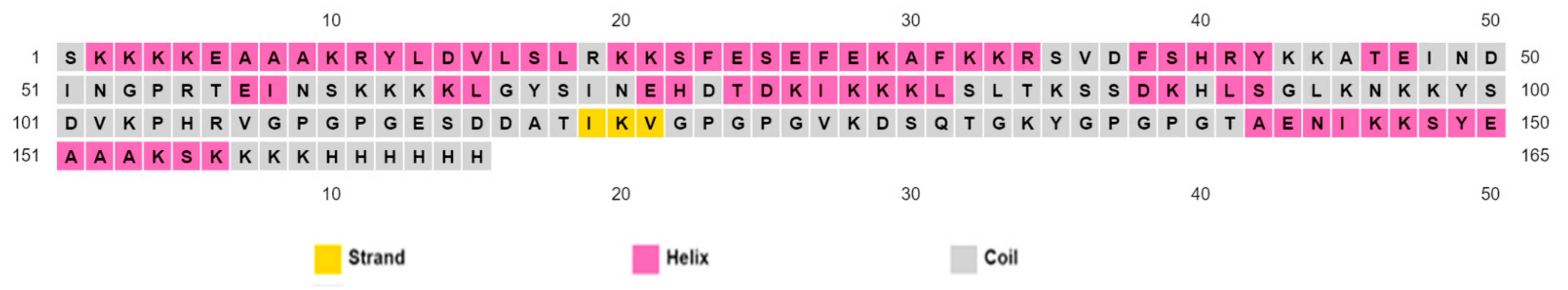
3.7. Projection of Secondary Structure
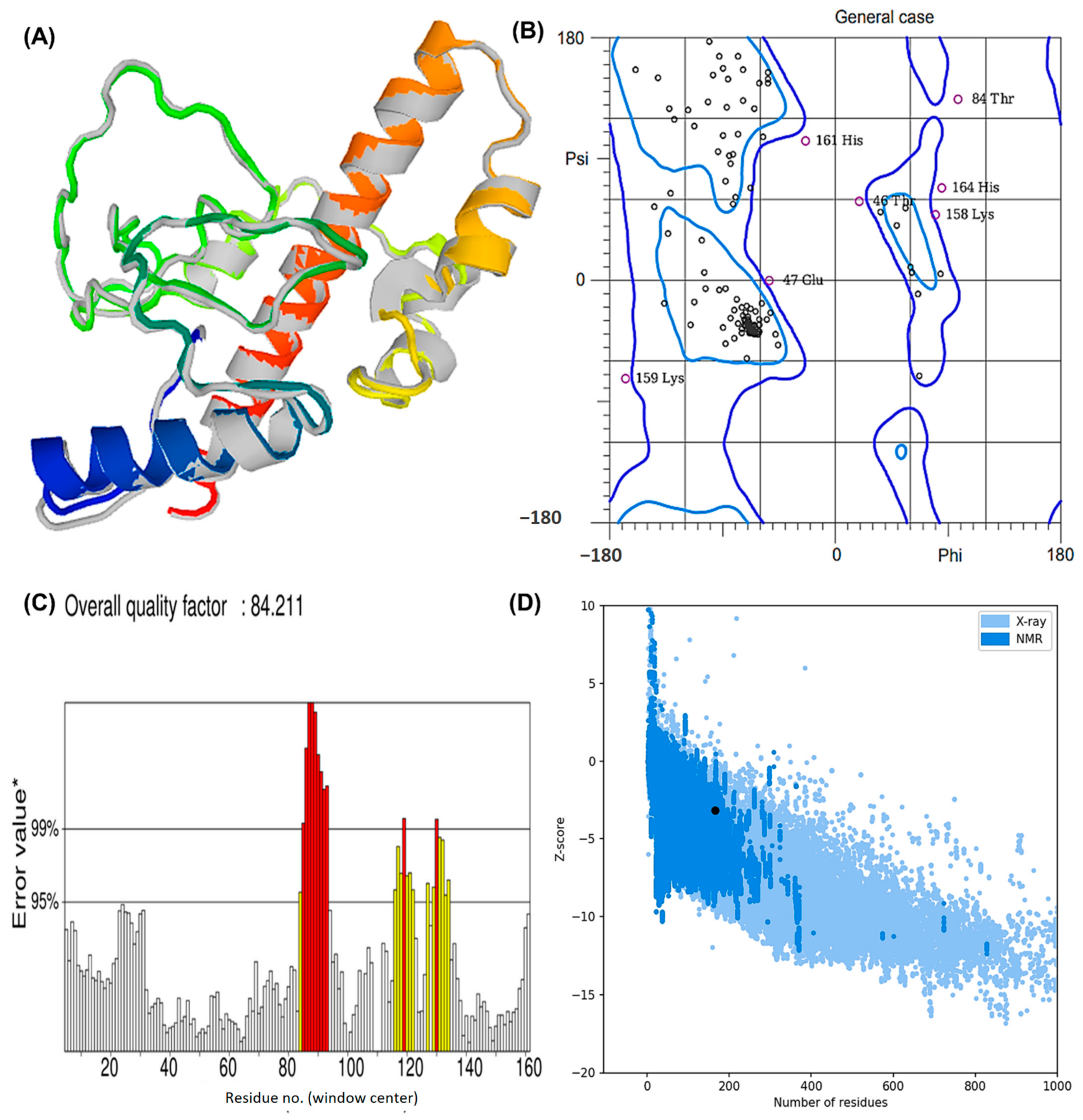
3.8. Projection, Refinement, and Evaluation of 3D Structure
3.9. Discontinuous BCEs
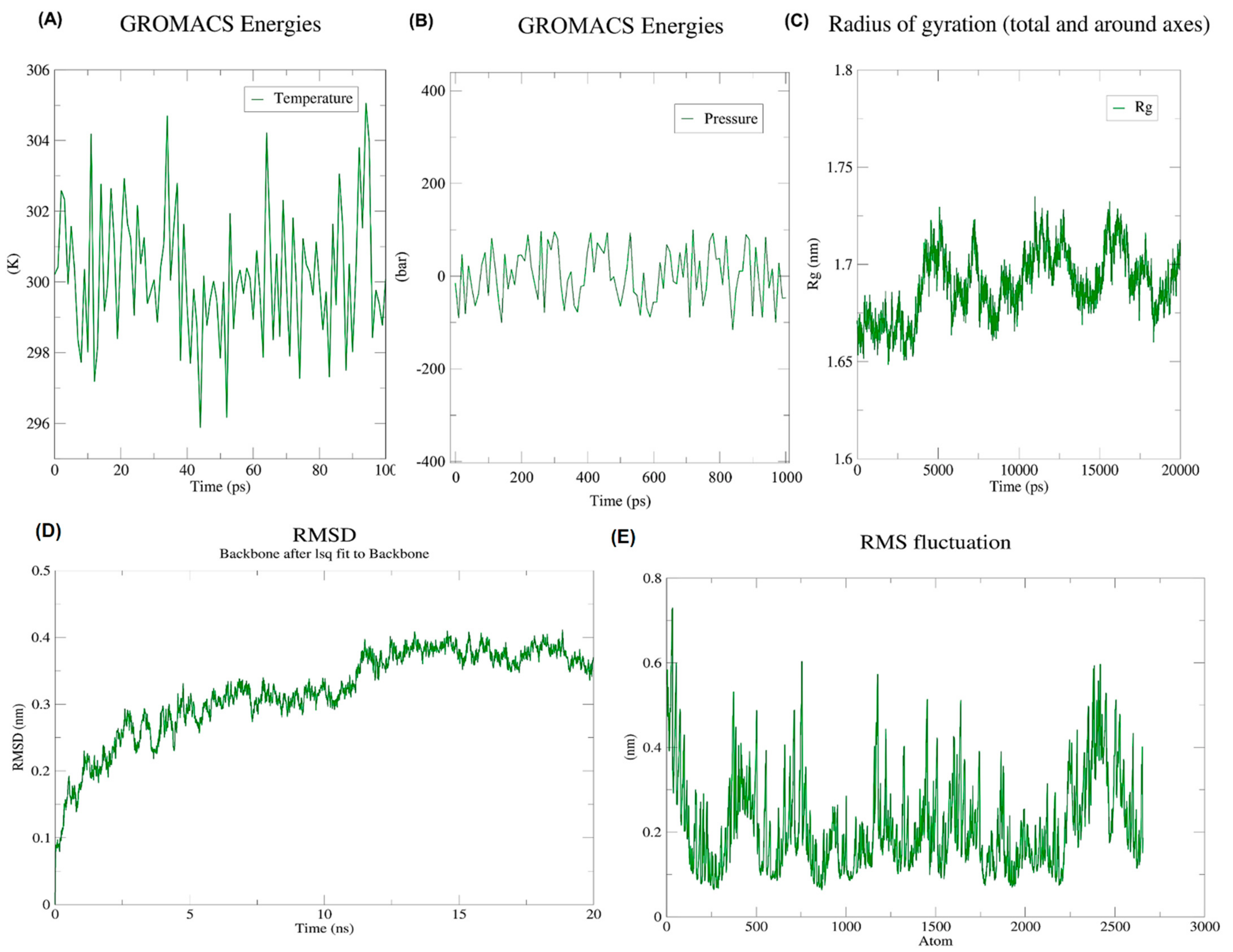
3.10. Analysis of MD Simulations Using GROMACS
3.11. Molecular Docking Analysis
3.12. Restriction Cloning In-Silico Experiment
3.13. Immune Simulations Experiment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Lyme Disease Vaccine | Lyme Disease | CDC. 2017. Available online: https://www.cdc.gov/lyme/prev/vaccine.html (accessed on 17 June 2022).
- Steere, A.C.; Strle, F.; Wormser, G.P.; Hu, L.T.; Branda, J.A.; Hovius, J.W.R.; Li, X.; Mead, P.S. Lyme Borreliosis. Nat. Rev. Dis. Prim. 2016, 2, 16090. [Google Scholar] [CrossRef] [PubMed]
- How Many People Get Lyme Disease? | Lyme Disease | CDC. Available online: https://www.cdc.gov/lyme/stats/humancases.html (accessed on 5 April 2022).
- Baranton, G.; Assous, M.; Postic, D. Three Bacterial Species Associated with Lyme Borreliosis. CLinical and Diagnostic Implications. Bull. Acad. Natl. Med. 1992, 176, 1075–1085. [Google Scholar] [PubMed]
- Kingry, L.C.; Batra, D.; Replogle, A.; Rowe, L.A.; Pritt, B.S.; Petersen, J.M. Whole Genome Sequence and Comparative Genomics of the Novel Lyme Borreliosis Causing Pathogen, Borrelia Mayonii. PLoS ONE 2016, 11, e0168994. [Google Scholar] [CrossRef] [PubMed]
- Rebman, A.W.; Aucott, J.N. Post-Treatment Lyme Disease as a Model for Persistent Symptoms in Lyme Disease. Front. Med. 2020, 7, 57. [Google Scholar] [CrossRef]
- Kamp, H.D.; Swanson, K.A.; Wei, R.R.; Dhal, P.K.; Dharanipragada, R.; Kern, A.; Sharma, B.; Sima, R.; Hajdusek, O.; Hu, L.T.; et al. Design of a Broadly Reactive Lyme Disease Vaccine. NPJ Vaccines 2020, 5, 33. [Google Scholar] [CrossRef]
- Schrestha, K.; Kadkhoda, K. Early Lyme Disease-Associated Guillain Barre Syndrome: A Case Report. IDCases 2022, 27, e01432. [Google Scholar] [CrossRef] [PubMed]
- Lyme Disease Vaccines | NIH: National Institute of Allergy and Infectious Diseases. Available online: https://www.niaid.nih.gov/diseases-conditions/lyme-disease-vaccines (accessed on 11 July 2022).
- Wormser, G.P. A Brief History of OspA Vaccines Including Their Impact on Diagnostic Testing for Lyme Disease. Diagn. Microbiol. Infect. Dis. 2022, 102, 115572. [Google Scholar] [CrossRef]
- Kitsou, C.; Pal, U. Vaccines Against Vector-Borne Diseases. Methods Mol. Biol. 2022, 2411, 269–286. [Google Scholar] [CrossRef]
- Badawi, A.; Shering, M.; Rahman, S.; Lindsay, L.R. A Systematic Review and Meta-Analysis for the Adverse Effects, Immunogenicity and Efficacy of Lyme Disease Vaccines: Guiding Novel Vaccine Development. Can. J. Public Health 2017, 108, e62–e70. [Google Scholar] [CrossRef]
- Fatima, I.; Ahmad, S.; Abbasi, S.W.; Ashfaq, U.A.; Shahid, F.; Tahir ul Qamar, M.; Rehman, A.; Allemailem, K.S. Designing of a Multi-Epitopes-Based Peptide Vaccine against Rift Valley Fever Virus and Its Validation through Integrated Computational Approaches. Comput. Biol. Med. 2022, 141, 105151. [Google Scholar] [CrossRef] [PubMed]
- Tahir Ul Qamar, M.; Ismail, S.; Ahmad, S.; Mirza, M.U.; Abbasi, S.W.; Ashfaq, U.A.; Chen, L.L. Development of a Novel Multi-Epitope Vaccine Against Crimean-Congo Hemorrhagic Fever Virus: An Integrated Reverse Vaccinology, Vaccine Informatics and Biophysics Approach. Front. Immunol. 2021, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Doolan, B.J.; Christie, M.; Dolianitis, C. A Ticking Time Bomb: A Case of Lyme Disease. Australas. J. Dermatol. 2019, 60, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Bidmos, F.A.; Siris, S.; Gladstone, C.A.; Langford, P.R. Bacterial Vaccine Antigen Discovery in the Reverse Vaccinology 2.0 Era: Progress and Challenges. Front. Immunol. 2018, 9, 2315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Multi-Epitope Vaccines: A Promising Strategy against Tumors and Viral Infections. Cell. Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef]
- Bobe, J.R.; Jutras, B.L.; Horn, E.J.; Embers, M.E.; Bailey, A.; Moritz, R.L.; Zhang, Y.; Soloski, M.J.; Ostfeld, R.S.; Marconi, R.T.; et al. Recent Progress in Lyme Disease and Remaining Challenges. Front. Med. 2021, 8, 1276. [Google Scholar] [CrossRef]
- Cassiani-Ingoni, R.; Cabral, E.S.; Lünemann, J.D.; Garza, Z.; Magnus, T.; Gelderblom, H.; Munson, P.J.; Marques, A.; Martin, R. Borrelia Burgdorferi Induces TLR1 and TLR2 in Human Microglia and Peripheral Blood Monocytes but Differentially Regulates HLA-Class II Expression. J. Neuropathol. Exp. Neurol. 2006, 65, 540–548. [Google Scholar] [CrossRef]
- Cabral, E.S.; Gelderblom, H.; Hornung, R.L.; Munson, P.J.; Martin, R.; Marques, A.R. Borrelia Burgdorferi Lipoprotein-Mediated TLR2 Stimulation Causes the down-Regulation of TLR5 in Human Monocytes. J. Infect. Dis. 2006, 193, 849–859. [Google Scholar] [CrossRef]
- UniProt Consortium, T. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Cenk Sahinalp, S.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved Protein Subcellular Localization Prediction with Refined Localization Subcategories and Predictive Capabilities for All Prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Gupta, D. VirulentPred: A SVM Based Prediction Method for Virulent Proteins in Bacterial Pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ali, S.S.; Zaheer, I.; Saleem, S.; Ziaullah; Zaman, N.; Iqbal, A.; Suleman, M.; Wadood, A.; Rehman, A.U.; et al. Proteome-Wide Mapping and Reverse Vaccinology-Based B and T Cell Multi-Epitope Subunit Vaccine Designing for Immune Response Reinforcement against Porphyromonas Gingivalis. J. Biomol. Struct. Dyn. 2022, 40, 833–847. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-Scale Validation of Methods for Cytotoxic T-Lymphocyte Epitope Prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of Continuous B-Cell Epitopes in an Antigen Using Recurrent Neural Network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, P.; Kim, Y.; Haste-Andersen, P.; Beaver, J.; Bourne, P.E.; Bui, H.-H.; Buus, S.; Frankild, S.; Greenbaum, J.; et al. Immune Epitope Database Analysis Resource (IEDB-AR). Nucleic Acids Res. 2008, 36, W513–W518. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved Methods for Predicting Peptide Binding Affinity to MHC Class II Molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Validi, M.; Karkhah, A.; Prajapati, V.K.; Nouri, H.R. Immuno-Informatics Based Approaches to Design a Novel Multi Epitope-Based Vaccine for Immune Response Reinforcement against Leptospirosis. Mol. Immunol. 2018, 104, 128–138. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. Peptide Toxicity Prediction. Methods Mol. Biol. 2015, 1268, 143–157. [Google Scholar] [CrossRef]
- Peptide Solubility Calculator. Available online: https://pepcalc.com/peptide-solubility-calculator.php?msclkid=f30d6654d10711ec8686f820ae9d4dad (accessed on 11 May 2022).
- BLAST. Available online: https://www.uniprot.org/blast/ (accessed on 11 May 2022).
- Sanami, S.; Azadegan-Dehkordi, F.; Rafieian-Kopaei, M.; Salehi, M.; Ghasemi-Dehnoo, M.; Mahooti, M.; Alizadeh, M.; Bagheri, N. Design of a Multi-Epitope Vaccine against Cervical Cancer Using Immunoinformatics Approaches. Sci. Rep. 2021, 11, 12397. [Google Scholar] [CrossRef]
- Khan, M.; Khan, S.; Ali, A.; Akbar, H.; Sayaf, A.M.; Khan, A.; Wei, D.Q. Immunoinformatics Approaches to Explore Helicobacter Pylori Proteome (Virulence Factors) to Design B and T Cell Multi-Epitope Subunit Vaccine. Sci. Rep. 2019, 9, 13321. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.; Bui, H.H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A New Structure-Based Tool for the Prediction of Antibody Epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A Server for In Silico Prediction of Allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P.S. AlgPred: Prediction of Allergenic Proteins and Mapping of IgE Epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef]
- McGinnis, S.; Madden, T.L. BLAST: At the Core of a Powerful and Diverse Set of Sequence Analysis Tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef]
- Shende, G.; Haldankar, H.; Barai, R.S.; Bharmal, M.H.; Shetty, V.; Idicula-Thomas, S.; Hancock, J. PBIT: Pipeline Builder for Identification of Drug Targets for Infectious Diseases. Bioinformatics 2017, 33, 929–931. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB Bioinformatics Resource Portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Omoniyi, A.A.; Adebisi, S.S.; Musa, S.A.; Nzalak, J.O.; Bauchi, Z.M.; Bako, K.W.; Olatomide, O.D.; Zachariah, R.; Nyengaard, J.R. In Silico Design and Analyses of a Multi-Epitope Vaccine against Crimean-Congo Hemorrhagic Fever Virus through Reverse Vaccinology and Immunoinformatics Approaches. Sci. Rep. 2022, 12, 8736. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED Protein Structure Prediction Server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Tahir Ul Qamar, M.; Shokat, Z.; Muneer, I.; Ashfaq, U.A.; Javed, H.; Anwar, F.; Bari, A.; Zahid, B.; Saari, N. Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach. Vaccines 2020, 8, 288. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Pikkemaat, M.G.; Linssen, A.B.M.; Berendsen, H.J.C.; Janssen, D.B. Molecular Dynamics Simulations as a Tool for Improving Protein Stability. Protein Eng. Des. Sel. 2002, 15, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to Its Potential Expression Host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Cronan, J.E. Escherichia Coli as an Experimental Organism. In eLS; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014. [Google Scholar]
- SnapGene | Software for Everyday Molecular Biology. Available online: https://www.snapgene.com/ (accessed on 27 July 2022).
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, 9862. [Google Scholar] [CrossRef]
- Khalid, K.; Irum, S.; Ullah, S.R.; Andleeb, S. In-Silico Vaccine Design Based on a Novel Vaccine Candidate Against Infections Caused by Acinetobacter Baumannii. Int. J. Pept. Res. Ther. 2022, 28, 16. [Google Scholar] [CrossRef] [PubMed]
- ul Ain, Q.; Ahmad, S.; Azam, S.S. Subtractive Proteomics and Immunoinformatics Revealed Novel B-Cell Derived T-Cell Epitopes against Yersinia Enterocolitica: An Etiological Agent of Yersiniosis. Microb. Pathog. 2018, 125, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Grandi, G. Bacterial Surface Proteins and Vaccines. F1000 Biol. Rep. 2010, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Barve, N.; Gupta, K.; Chandra, S.; Jain, N.; Tiwari, S.; Leon-Sicairos, N.; Canizalez-Roman, A.; Rodrigues dos Santos, A.; Hassan, S.S.; et al. Exoproteome and Secretome Derived Broad Spectrum Novel Drug and Vaccine Candidates in Vibrio Cholerae Targeted by Piper Betel Derived Compounds. PLoS ONE 2013, 8, e52773. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y. How Significant Is a Protein Structure Similarity with TM-Score = 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef]
- De Vries, S.J.; Van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK Web Server for Data-Driven Biomolecular Docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef]
- Eisen, R.J.; Eisen, L.; Beard, C.B. County-Scale Distribution of Ixodes Scapularis and Ixodes Pacificus (Acari: Ixodidae) in the Continental United States. J. Med. Entomol. 2016, 53, 349–386. [Google Scholar] [CrossRef]
- Lyme BorreLiosis in Europe. Available online: https://caudwelllymedotnet.files.wordpress.com/2016/01/who-factsheet-lyme-borreliosis-epidemiology.pdf (accessed on 5 April 2022).
- Schoen, R.T. Challenges in the Diagnosis and Treatment of Lyme Disease. Curr. Rheumatol. Rep. 2020, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sigal, L.H.; Curran, A.S. LYME DISEASE: A MULTIFOCAL WORLDWIDE EPIDEMIC. Annu. Rev. Public Health 1991, 12, 85–109. [Google Scholar] [CrossRef]
- Stricker, R.B.; Johnson, L. Lyme Disease: The next Decade. Infect. Drug Resist. 2011, 4, 1. [Google Scholar] [CrossRef]
- Berende, A.; Ter Hofstede, H.J.M.; Vos, F.J.; Vogelaar, M.L.; Van Middendorp, H.; Evers, A.W.M.; Kessels, R.P.C.; Kullberg, B.J. Effect of Prolonged Antibiotic Treatment on Cognition in Patients with Lyme Borreliosis. Neurology 2019, 92, e1447–e1455. [Google Scholar] [CrossRef]
- Beaujean, D.; Crutzen, R.; Kengen, C.; Van Steenbergen, J.; Ruwaard, D. Increase in Ticks and Lyme Borreliosis, Yet Research into Its Prevention on the Wane. Vector-Borne Zoonotic Dis. 2016, 16, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Caoili, S.E.C.; Dangott, L.J.; Gerasimov, E.; Ionov, Y.; Piontkivska, H.; Zelikovsky, A.; Waghela, S.D.; Rogovskyy, A.S. Identification of Surface Epitopes Associated with Protection against Highly Immune-Evasive VlsE-Expressing Lyme Disease Spirochetes. Infect. Immun. 2018, 86, e00182-18. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania Secretory Proteins to Design B and T Cell Multi-Epitope Subunit Vaccine Using Immunoinformatics Approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef] [PubMed]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-Silico Design of a Multi-Epitope Vaccine Candidate against Onchocerciasis and Related Filarial Diseases. Sci. Rep. 2019, 9, 4409. [Google Scholar] [CrossRef]
- Pizza, M.; Grandi, G.; Telford, J.L.; Rappuoli, R. Reverse Vaccinology: A Genome-Based Approach to Vaccine Development. Chim. Oggi 2002, 20, 32–36. [Google Scholar]
- Bacchetta, R.; Gregori, S.; Roncarolo, M.G. CD4+ Regulatory T Cells: Mechanisms of Induction and Effector Function. Autoimmun. Rev. 2005, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Shamriz, S.; Ofoghi, H.; Moazami, N. Effect of Linker Length and Residues on the Structure and Stability of a Fusion Protein with Malaria Vaccine Application. Comput. Biol. Med. 2016, 76, 24–29. [Google Scholar] [CrossRef]
- Meza, B.; Ascencio, F.; Sierra-Beltrán, A.P.; Torres, J.; Angulo, C. A Novel Design of a Multi-Antigenic, Multistage and Multi-Epitope Vaccine against Helicobacter Pylori: An In Silico Approach. Infect. Genet. Evol. 2017, 49, 309–317. [Google Scholar] [CrossRef]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the Linkers Which Effectively Separate Domains of a Bifunctional Fusion Protein. Protein Eng. Des. Sel. 2001, 14, 529–532. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Protein ID | Antigenicity (>0.4) | Virulence | Allergenicity | Homology to Humans | Size (kDa) |
|---|---|---|---|---|---|---|
| TsaE | WP_002556784.1 | 0.68 | ✓ | ✗ | Not found | 15.4 |
| FliP | WP_002556874.1 | 0.71 | ✓ | ✗ | Not found | 29.0 |
| ABC transporter permease | WP_002557338.1 | 0.53 | ✓ | ✗ | Not found | 27.6 |
| MreD | WP_002656052.1 | 0.71 | ✓ | ✗ | Not found | 20.9 |
| YggT family protein | WP_002656831.1 | 0.63 | ✓ | ✗ | Not found | 22.2 |
| MurJ | WP_002657239.1 | 0.64 | ✓ | ✗ | Not found | 58 |
| CTL Epitope (9-mer) | Protein ID | Combined Score | VaxiJen | Toxin | Conservancy in B. burgdorfei sp. | Solubility |
|---|---|---|---|---|---|---|
| KSEKKMINF | WP_002556784 | 0.9186 | 0.5478 | No | 100% | Good |
| TTNGLNFPF | WP_002556874 | 0.8427 | 1.3018 | No | 100% | Good |
| DLGIILLQY | WP_002557338 | 0.8397 | 0.8354 | No | 100% | Good |
| IIFAKPIMY | WP_002657239 | 0.8012 | 0.5076 | No | 100% | Good |
| Epitope (9-mer) | Protein ID | Percentile Rank | Antigenicity | Toxin | IFN Epitope | Conservancy | Solubility |
|---|---|---|---|---|---|---|---|
| IILLQYLGI | WP_002557338 | 0.01 | 0.6290 | No | Positive | 100% | Good |
| FQWDVGFSF | WP_002657239 | 0.01 | 1.82 | No | positive | 100% | Good |
| ILILIRILL | WP_002656831 | 0.01 | 1.2411 | No | positive | 100% | Good |
| Linear B Cell Epitope (15-mer) | Protein | Probability Score | Antigenicity | Toxin | Conservancy (%) | Solubility |
|---|---|---|---|---|---|---|
| IALSIVPKDRLFSLTF | WP_002556784.1 | 0.85 | 0.7432 | No | 100 | Good |
| MGMIMLPPVMISLPFK | WP_002556874.1 | 0.92 | 1.2510 | No | 100 | Good |
| YFTGLPLGFFVFGYTI | WP_002656052.1 | 0.75 | 0.9086 | No | 100 | Good |
| Docking Analysis | TLR-1 | TLR-2 |
|---|---|---|
| HADDOCK Parameters | ||
| HADDOCK score | −79.2 ± 14.1 | −103.0 ± 2.5 |
| Z-Score | −1.3 | −0.8 |
| RMSD | 1.2 ± 1.5 | 0.3 ± 0.1 |
| Van der Waals energy | −81.4 ± 2.4 | −82.7 ± 4.1 |
| Electrostatic energy | −332.3 ± 50.7 | −335.2 ± 49.5 |
| Desolvation energy | −14.0 ± 10.2 | −51.2 ± 10.6 |
| Buried Surface Area | 2567.8 ± 210.5 | 1938.2 ± 63.0 |
| Binding affinity and kD prediction | ||
| ΔG (kcal mol−1) | −12.1 | −12.0 |
| Kd (M) at 25.0 °C | 1.3 × 10−9 | 1.5 × 10−9 |
| Number of Interfacial Contacts (ICs) per property | ||
| Charged-charged | 22 | 14 |
| Charged-polar | 42 | 15 |
| Charged-apolar | 23 | 36 |
| Polar-polar | 3 | 2 |
| Polar-apolar | 9 | 8 |
| Apolar-apolar | 3 | 5 |
| Protein-protein interface interaction statistics | ||
| Salt bridges | 3 | 4 |
| Hydrogen bonds | 14 | 13 |
| No. of non-bonded contacts | 149 | 204 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalid, K.; Ahsan, O.; Khaliq, T.; Muhammad, K.; Waheed, Y. Immunoinformatics-Based Proteome Mining to Develop a Next-Generation Vaccine Design against Borrelia burgdorferi: The Cause of Lyme Borreliosis. Vaccines 2022, 10, 1239. https://doi.org/10.3390/vaccines10081239
Khalid K, Ahsan O, Khaliq T, Muhammad K, Waheed Y. Immunoinformatics-Based Proteome Mining to Develop a Next-Generation Vaccine Design against Borrelia burgdorferi: The Cause of Lyme Borreliosis. Vaccines. 2022; 10(8):1239. https://doi.org/10.3390/vaccines10081239
Chicago/Turabian StyleKhalid, Kashaf, Omar Ahsan, Tanwir Khaliq, Khalid Muhammad, and Yasir Waheed. 2022. "Immunoinformatics-Based Proteome Mining to Develop a Next-Generation Vaccine Design against Borrelia burgdorferi: The Cause of Lyme Borreliosis" Vaccines 10, no. 8: 1239. https://doi.org/10.3390/vaccines10081239