Iron Overload, Oxidative Stress and Calcium Mishandling in Cardiomyocytes: Role of the Mitochondrial Permeability Transition Pore


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Chemicals and Reagents
2.3. Cell Isolation
2.4. Measurement of Cytosolic and Mitochondrial Fe Loading
2.5. Measurement of Mitochondrial Reactive Oxygen Species Levels
2.6. Measurement of Mitochondrial Membrane Potential
2.7. Measurements of Intracellular Ca Fluorescence
2.8. Electrocardiograms (ECG) and Arrhythmia Induction Testing
2.9. Statistics
3. Results
3.1. Fe Load in the Cytosol and Mitochondria in WT and CypD KO Ventricular Myocytes
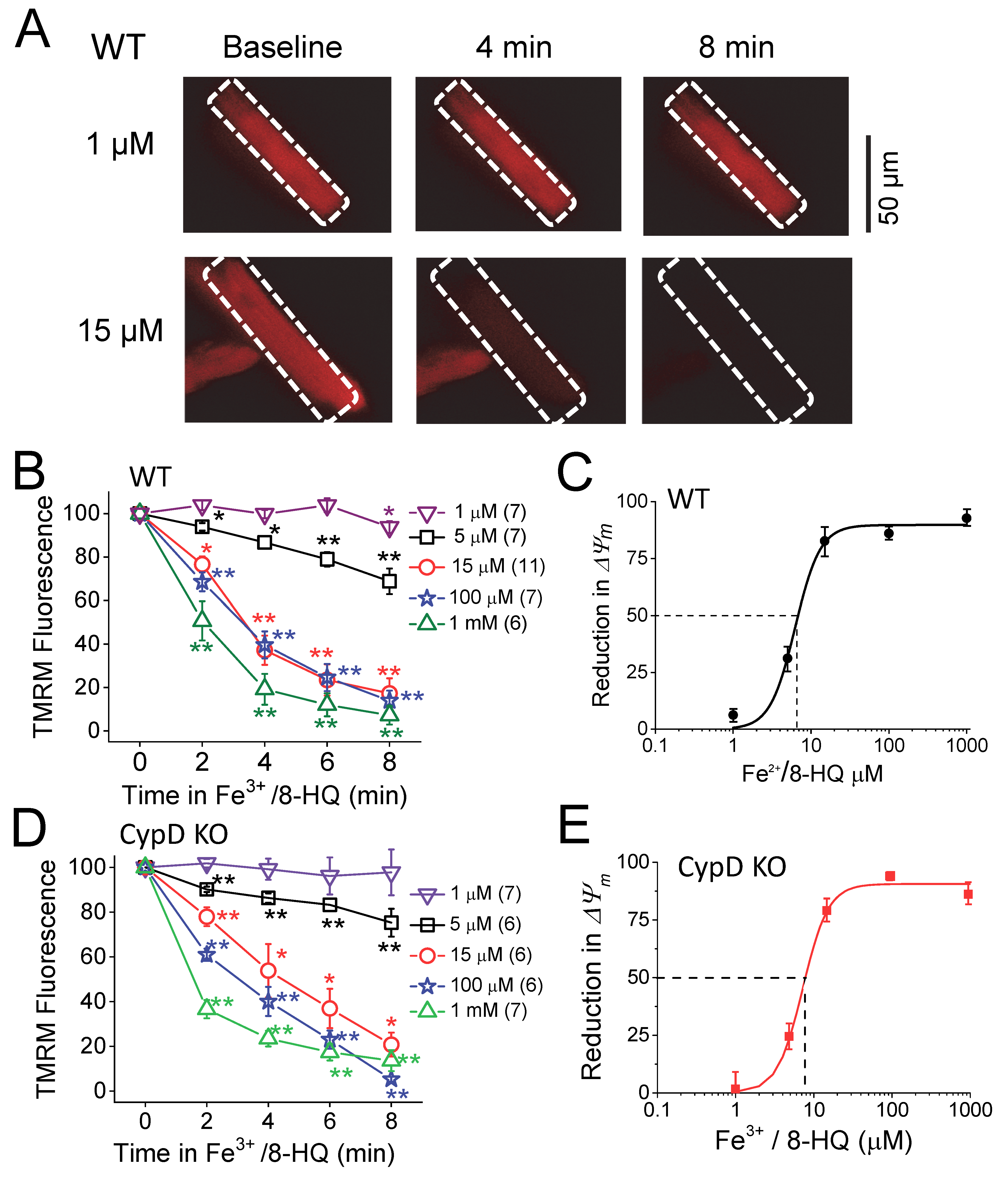
3.2. IO induced Mitochondrial ROS Generation and ΔΨm Depolarization
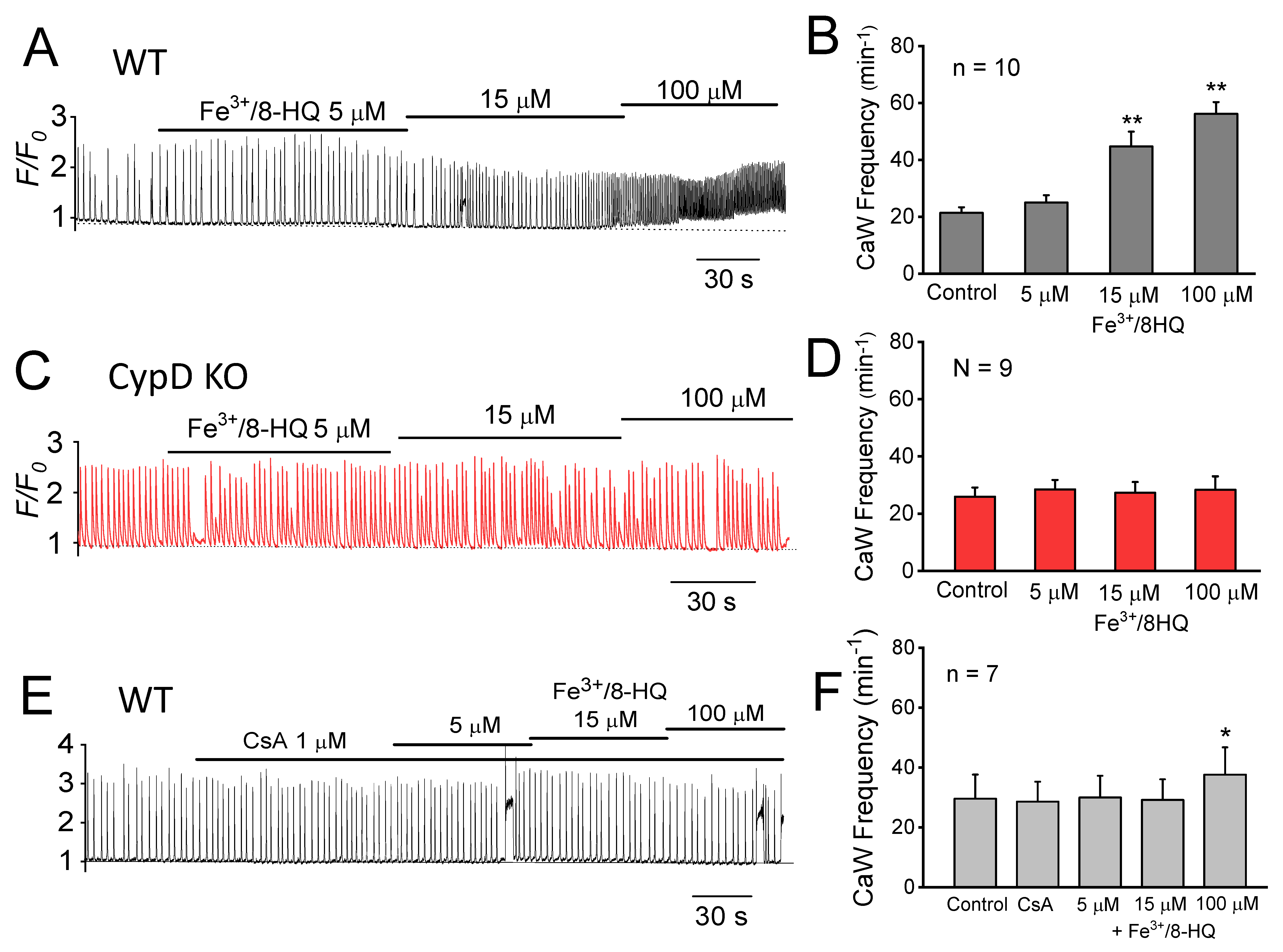
3.3. Promotion of Ca Waves by IO and Protection by mPTP Inhibition
3.4. Preventative Effect of Antioxidants against IO Toxicity
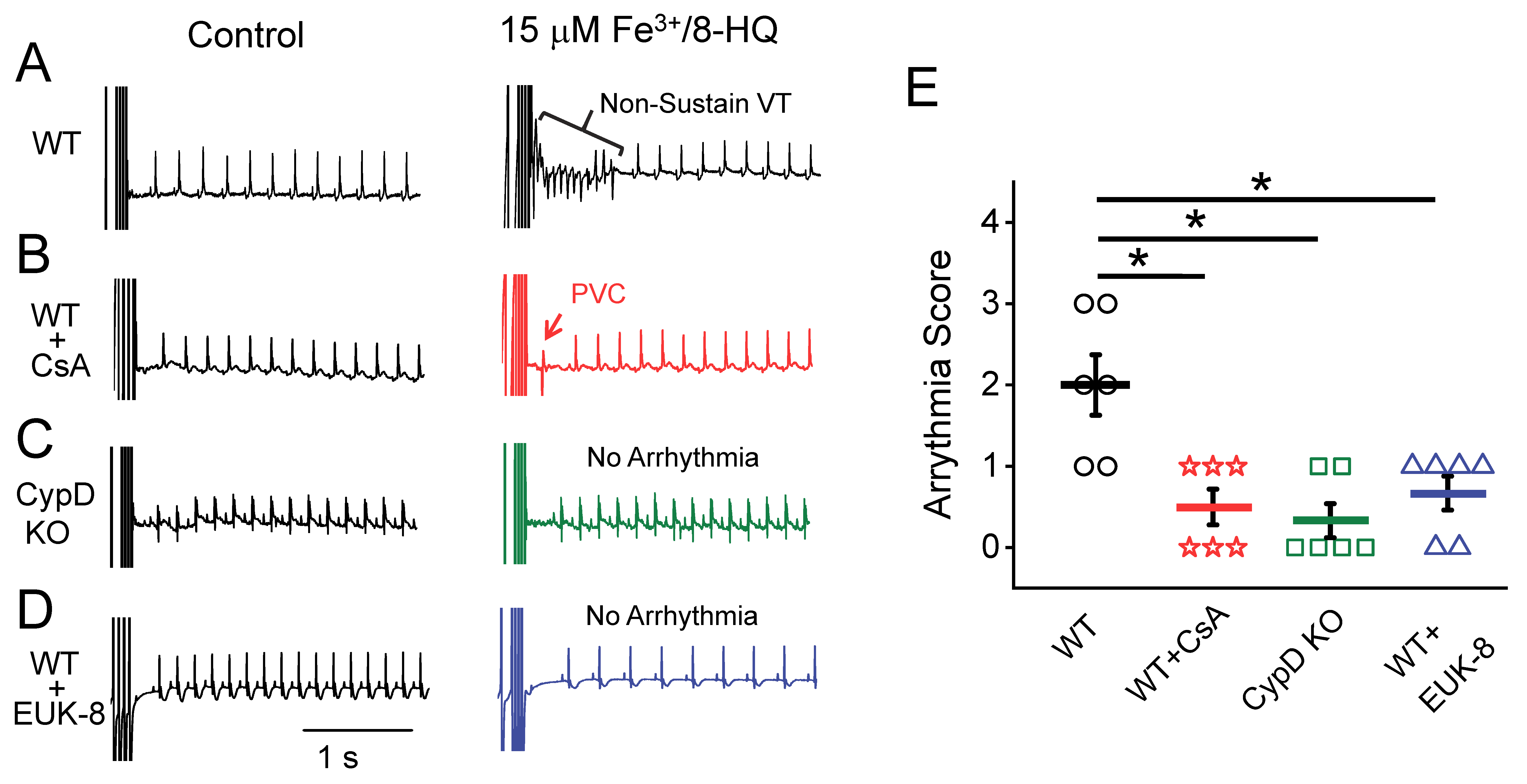
3.5. IO Promoted Arrhythmias and Their Prevention by Antioxidants and mPTP Inhibition in Ex-Vivo Hearts
4. Discussion
4.1. Iron Overload and Arrhythmogenesis—Discrepancies in Clinical and Experimental Settings
4.2. Mechanisms for Ca Mishandling and Arrhythmias under IO: Roles of mPTP and Other Targets
4.3. IO-Induced Oxidative Stress and the Effect of Antioxidants
4.4. Limitations
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Murphy, C.J.; Oudit, G.Y. Iron-overload cardiomyopathy: Pathophysiology, diagnosis, and treatment. J. Card. Fail. 2010, 16, 888–900. [Google Scholar] [CrossRef]
- Das, S.K.; Wang, W.; Zhabyeyev, P.; Basu, R.; McLean, B.; Fan, D.; Parajuli, N.; DesAulniers, J.; Patel, V.B.; Hajjar, R.J.; et al. Iron-overload injury and cardiomyopathy in acquired and genetic models is attenuated by resveratrol therapy. Sci. Rep. 2015, 5, 18132. [Google Scholar] [CrossRef] [Green Version]
- Kremastinos, D.T.; Tsetsos, G.A.; Tsiapras, D.P.; Karavolias, G.K.; Ladis, V.A.; Kattamis, C.A. Heart failure in beta thalassemia: A 5-year follow-up study. Am. J. Med. 2001, 111, 349–354. [Google Scholar] [CrossRef]
- Shizukuda, Y.; Rosing, D.R. Iron overload and arrhythmias: Influence of confounding factors. J. Arrhythmia 2019, 35, 575–583. [Google Scholar] [CrossRef]
- Gordan, R.; Wongjaikam, S.; Gwathmey, J.K.; Chattipakorn, N.; Chattipakorn, S.C.; Xie, L.H. Involvement of cytosolic and mitochondrial iron in iron overload cardiomyopathy: An update. Heart Fail. Rev. 2018, 23, 801–816. [Google Scholar] [CrossRef]
- Kumfu, S.; Chattipakorn, S.; Fucharoen, S.; Chattipakorn, N. Mitochondrial calcium uniporter blocker prevents cardiac mitochondrial dysfunction induced by iron overload in thalassemic mice. Biometals 2012, 25, 1167–1175. [Google Scholar] [CrossRef]
- Sripetchwandee, J.; KenKnight, S.B.; Sanit, J.; Chattipakorn, S.; Chattipakorn, N. Blockade of mitochondrial calcium uniporter prevents cardiac mitochondrial dysfunction caused by iron overload. Acta Physiol. 2014, 210, 330–341. [Google Scholar] [CrossRef]
- Fedotcheva, N.I.; Mokhova, E.N. Mitochondrial models of pathologies with oxidative stress. Efficiency of alkalization to reduce mitochondrial damage. Biochemistry 2013, 78, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Berdoukas, V.; Coates, T.D.; Cabantchik, Z.I. Iron and oxidative stress in cardiomyopathy in thalassemia. Free Radic. Biol. Med. 2015, 88, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Gordan, R.; Xie, L.H. Primary effect of reactive oxygen species on electrical remodeling of the heart. Circulation 2014, 78, 1834–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.H.; Chen, F.; Karagueuzian, H.S.; Weiss, J.N. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ. Res. 2009, 104, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Lefer, D.J.; Granger, D.N. Oxidative stress and cardiac disease. Am. J. Med. 2000, 109, 315–323. [Google Scholar] [CrossRef]
- Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 1992, 267, 8834–8839. [Google Scholar] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Mitochondrial Ca2+ and the heart. Cell Calcium 2008, 44, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta 2009, 1787, 1402–1415. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Meana, M.; Fernandez-Sanz, C.; Garcia-Dorado, D. The SR-mitochondria interaction: A new player in cardiac pathophysiology. Cardiovasc. Res. 2010, 88, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Gordan, R.; Wen, H.; Fefelova, N.; Zang, W.J.; Xie, L.H. Modulation of intracellular calcium waves and triggered activities by mitochondrial ca flux in mouse cardiomyocytes. PLoS ONE 2013, 8, e80574. [Google Scholar] [CrossRef] [Green Version]
- Gordan, R.; Fefelova, N.; Gwathmey, J.K.; Xie, L.H. Involvement of mitochondrial permeability transition pore (mPTP) in cardiac arrhythmias: Evidence from cyclophilin D knockout mice. Cell Calcium 2016, 60, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Petrat, F.; Weisheit, D.; Lensen, M.; de Groot, H.; Sustmann, R.; Rauen, U. Selective determination of mitochondrial chelatable iron in viable cells with a new fluorescent sensor. Biochem. J. 2002, 362, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Rauen, U.; Petrat, F.; Sustmann, R.; de Groot, H. Iron-induced mitochondrial permeability transition in cultured hepatocytes. J. Hepatol. 2004, 40, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Kabaeva, Z.; Zhao, M.; Michele, D.E. Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1667–H1674. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Babu, G.J.; Wen, H.; Fefelova, N.; Gordan, R.; Sui, X.; Yan, L.; Vatner, D.E.; Vatner, S.F.; Xie, L.H. Overexpression of adenylyl cyclase type 5 (AC5) confers a proarrhythmic substrate to the heart. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H240–H249. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Zhao, Z.; Fefelova, N.; Xie, L.H. Potential Arrhythmogenic Role of TRPC Channels and Store-Operated Calcium Entry Mechanism in Mouse Ventricular Myocytes. Front. Physiol. 2018, 9, 1785. [Google Scholar] [CrossRef]
- Xie, L.H.; Weiss, J.N. Arrhythmogenic consequences of intracellular calcium waves. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H997–H1002. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.C.; Wu, R.; Shang, M.; Sato, T.; Chen, C.; Shapiro, J.S.; Liu, T.; Thakur, A.; Sawicki, K.T.; Prasad, S.V.; et al. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol. Med. 2016, 8, 247–267. [Google Scholar] [CrossRef]
- Bayar, N.; Arslan, S.; Erkal, Z.; Kucukseymen, S. Sustained ventricular tachycardia in a patient with thalassemia major. Ann. Noninvasive Electrocardiol. 2014, 19, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, L.; Mancuso, A.; Bevacqua, E.; Rigano, P. Electrocardiographic abnormalities in thalassemia patients with heart failure. Cardiovasc. Hematol. Disord. Drug Targets 2009, 9, 29–35. [Google Scholar] [CrossRef]
- Cavallaro, L.; Meo, A.; Busa, G.; Coglitore, A.; Sergi, G.; Satullo, G.; Donato, A.; Calabro, M.P.; Miceli, M. Arrhythmia in thalassemia major: Evaluation of iron chelating therapy by dynamic ECG. Minerva Cardioangiol. 1993, 41, 297–301. [Google Scholar] [PubMed]
- Heper, G.; Ozensoy, U.; Korkmaz, M.E. Persistent atrial standstill and idioventricular rhythm in a patient with thalassemia intermedia. Turk Kardiyol. Dern. Ars. Turk Kardiyol. Dern. Yayin Organidir 2009, 37, 256–259. [Google Scholar]
- Nisli, K.; Taner, Y.; Naci, O.; Zafer, S.; Zeynep, K.; Aygun, D.; Umrah, A.; Rukiye, E.; Turkan, E. Electrocardiographic markers for the early detection of cardiac disease in patients with beta-thalassemia major. J. Pediatr. 2010, 86, 159–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, V.C.; Huang, J.W.; Wu, M.S.; Chin, C.Y.; Chiang, F.T.; Liu, Y.B.; Wu, K.D. The effect of iron stores on corrected QT dispersion in patients undergoing peritoneal dialysis. Am. J. Kidney Dis. 2004, 44, 720–728. [Google Scholar] [CrossRef]
- Adams, R.J.; McKie, V.C.; Brambilla, D.; Carl, E.; Gallagher, D.; Nichols, F.T.; Roach, S.; Abboud, M.; Berman, B.; Driscoll, C.; et al. Stroke prevention trial in sickle cell anemia. Control. Clin. Trials 1998, 19, 110–129. [Google Scholar] [CrossRef]
- Klintschar, M.; Stiller, D. Sudden cardiac death in hereditary hemochromatosis: An underestimated cause of death? Int. J. Leg. Med. 2004, 118, 174–177. [Google Scholar] [CrossRef]
- Aronow, W.S.; Meister, L.; Kent, J.R. Atrioventricular block in familial hemochromatosis treated by permanent synchronous pacemaker. Arch. Intern. Med. 1969, 123, 433–435. [Google Scholar] [CrossRef]
- Rose, R.A.; Sellan, M.; Simpson, J.A.; Izaddoustdar, F.; Cifelli, C.; Panama, B.K.; Davis, M.; Zhao, D.; Markhani, M.; Murphy, G.G.; et al. Iron overload decreases CaV1.3-dependent L-type Ca2+ currents leading to bradycardia, altered electrical conduction, and atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2011, 4, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Obejero-Paz, C.A.; Yang, T.; Dong, W.Q.; Levy, M.N.; Brittenham, G.M.; Kuryshev, Y.A.; Brown, A.M. Deferoxamine promotes survival and prevents electrocardiographic abnormalities in the gerbil model of iron-overload cardiomyopathy. J. Lab. Clin. Med. 2003, 141, 121–130. [Google Scholar] [CrossRef]
- Kaiser, L.; Davis, J.; Patterson, J.; Boyd, R.F.; Olivier, N.B.; Bohart, G.; Schwartz, K.A. Iron does not cause arrhythmias in the guinea pig model of transfusional iron overload. Comp. Med. 2007, 57, 383–389. [Google Scholar] [CrossRef]
- Kaiser, L.; Davis, J.M.; Patterson, J.; Johnson, A.L.; Bohart, G.; Olivier, N.B.; Schwartz, K.A. Iron sufficient to cause hepatic fibrosis and ascites does not cause cardiac arrhythmias in the gerbil. Transl. Res. 2009, 154, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Lane, D.J.; Becker, E.M.; Huang, M.L.; Whitnall, M.; Suryo Rahmanto, Y.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Ginsburg, K.S.; Kettlewell, S.; Bossuyt, J.; Smith, G.L.; Bers, D.M. Measuring local gradients of intramitochondrial [Ca2+] in cardiac myocytes during sarcoplasmic reticulum Ca2+ release. Circ. Res. 2013, 112, 424–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieminen, A.L.; Schwartz, J.; Hung, H.I.; Blocker, E.R.; Gooz, M.; Lemasters, J.J. Mitoferrin-2 (MFRN2) Regulates the Electrogenic Mitochondrial Calcium Uniporter and Interacts Physically with MCU. Biophys. J. 2014, 106, 581. [Google Scholar] [CrossRef] [Green Version]
- Siri-Angkul, N.; Xie, L.H.; Chattipakorn, S.C.; Chattipakorn, N. Cellular Electrophysiology of Iron-Overloaded Cardiomyocytes. Front. Physiol. 2018, 9, 1615. [Google Scholar] [CrossRef]
- Siri-Angkul, N.; Gordan, R.; Wongjaikam, S.; Fefelova, N.; Gwathmey, J.K.; Chattipakorn, S.; Chattipakorn, N.; Xie, L.H. Activation of transient receptor potential canonical channel currents in iron-overloaded cardiac myocytes. Circ. Res. 2019, 125, A507. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [Green Version]
- Tomaselli, G.F.; Barth, A.S. Sudden cardio arrest: Oxidative stress irritates the heart. Nat. Med. 2010, 16, 648–649. [Google Scholar] [CrossRef]
- Zhao, Z.; Fefelova, N.; Shanmugam, M.; Bishara, P.; Babu, G.J.; Xie, L.H. Angiotensin II induces afterdepolarizations via reactive oxygen species and calmodulin kinase II signaling. J. Mol. Cell. Cardiol. 2011, 50, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.H.; Shi, C.X.; Dong, F.; Sheng, J.W.; Xu, Y.F. Inhibition of the rapid component of the delayed rectifier potassium current in ventricular myocytes by angiotensin II via the AT1 receptor. Br. J. Pharmacol. 2008, 154, 429–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saotome, M.; Katoh, H.; Yaguchi, Y.; Tanaka, T.; Urushida, T.; Satoh, H.; Hayashi, H. Transient opening of mitochondrial permeability transition pore by reactive oxygen species protects myocardium from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1125–H1132. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. The Mitochondrial Permeability Transition—A Pore Way for the Heart to Die. J. Clin. Basic Cardiol. 2002, 5, 29–41. [Google Scholar]
- Fraysse, B.; Nagi, S.M.; Boher, B.; Ragot, H.; Laine, J.; Salmon, A.; Fiszman, M.Y.; Toussaint, M.; Fromes, Y. Ca2+ overload and mitochondrial permeability transition pore activation in living delta-sarcoglycan-deficient cardiomyocytes. Am. J. Physiol. Cell Physiol. 2010, 299, C706–C713. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Davidson, A.M. Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 1990, 268, 153–160. [Google Scholar]
- Gordan, R.; Wongjaikam, S.; Fefelova, N.; Siri-Angkul, N.; Gwathmey, J.K.; Chattipakorn, N.; Chattipakorn, S.; Xie, L.H. Mitochondrial permeability transition pore, calcium uniporter, and iron overload in the heart. Circ. Res. 2018, 123, A254. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordan, R.; Fefelova, N.; Gwathmey, J.K.; Xie, L.-H. Iron Overload, Oxidative Stress and Calcium Mishandling in Cardiomyocytes: Role of the Mitochondrial Permeability Transition Pore. Antioxidants 2020, 9, 758. https://doi.org/10.3390/antiox9080758
Gordan R, Fefelova N, Gwathmey JK, Xie L-H. Iron Overload, Oxidative Stress and Calcium Mishandling in Cardiomyocytes: Role of the Mitochondrial Permeability Transition Pore. Antioxidants. 2020; 9(8):758. https://doi.org/10.3390/antiox9080758
Chicago/Turabian StyleGordan, Richard, Nadezhda Fefelova, Judith K. Gwathmey, and Lai-Hua Xie. 2020. "Iron Overload, Oxidative Stress and Calcium Mishandling in Cardiomyocytes: Role of the Mitochondrial Permeability Transition Pore" Antioxidants 9, no. 8: 758. https://doi.org/10.3390/antiox9080758