Aging and Progression of Beta-Amyloid Pathology in Alzheimer’s Disease Correlates with Microglial Heme-Oxygenase-1 Overexpression



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Tissue Immunofluorescence
2.3. RNAscope in Situ Hybridization
2.4. Human Samples
2.5. Image Analysis
2.6. Statistics
3. Results
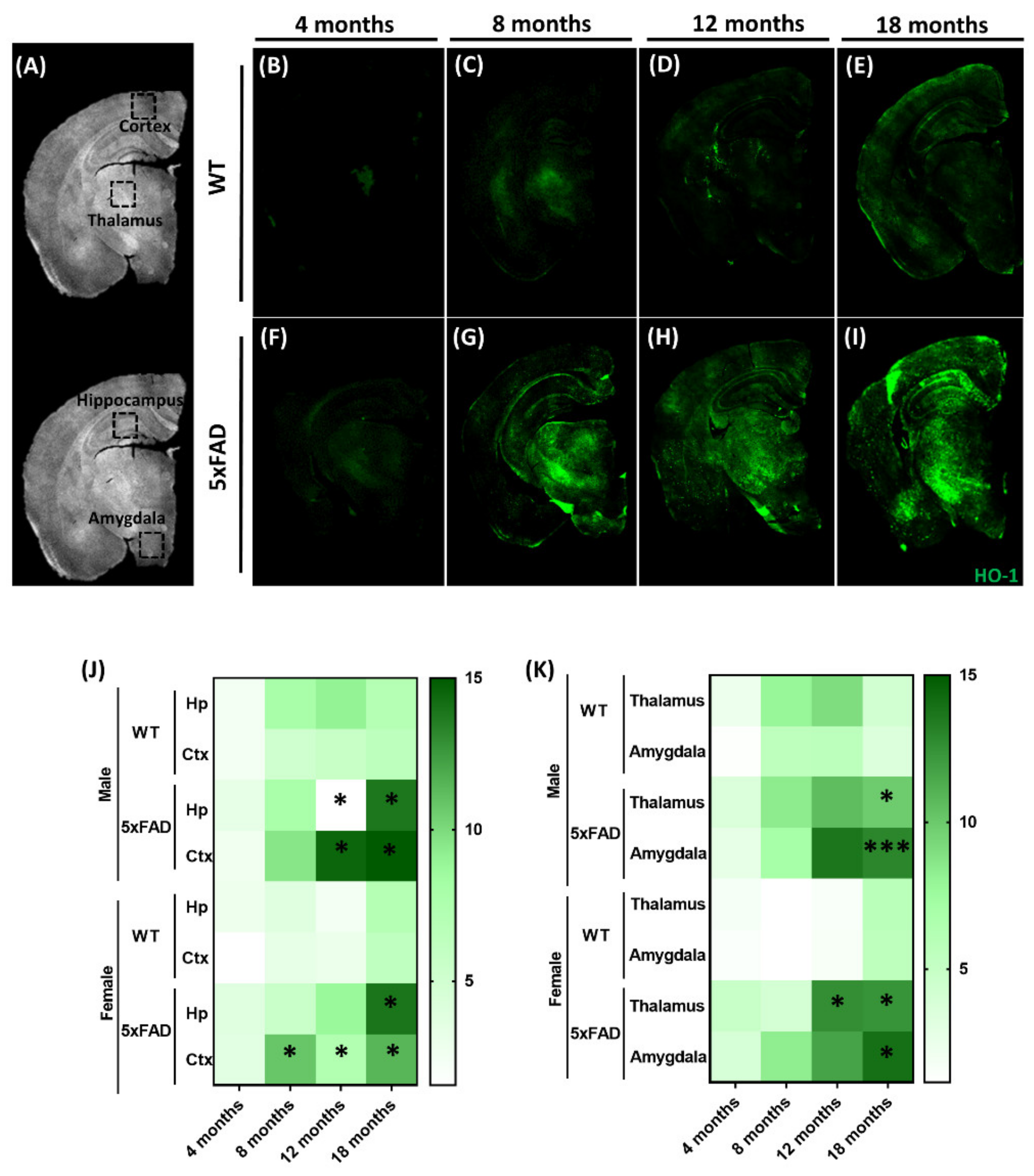
3.1. Expression of Brain HO-1 with Aging in the Alzheimer’s 5xFAD Animal Model
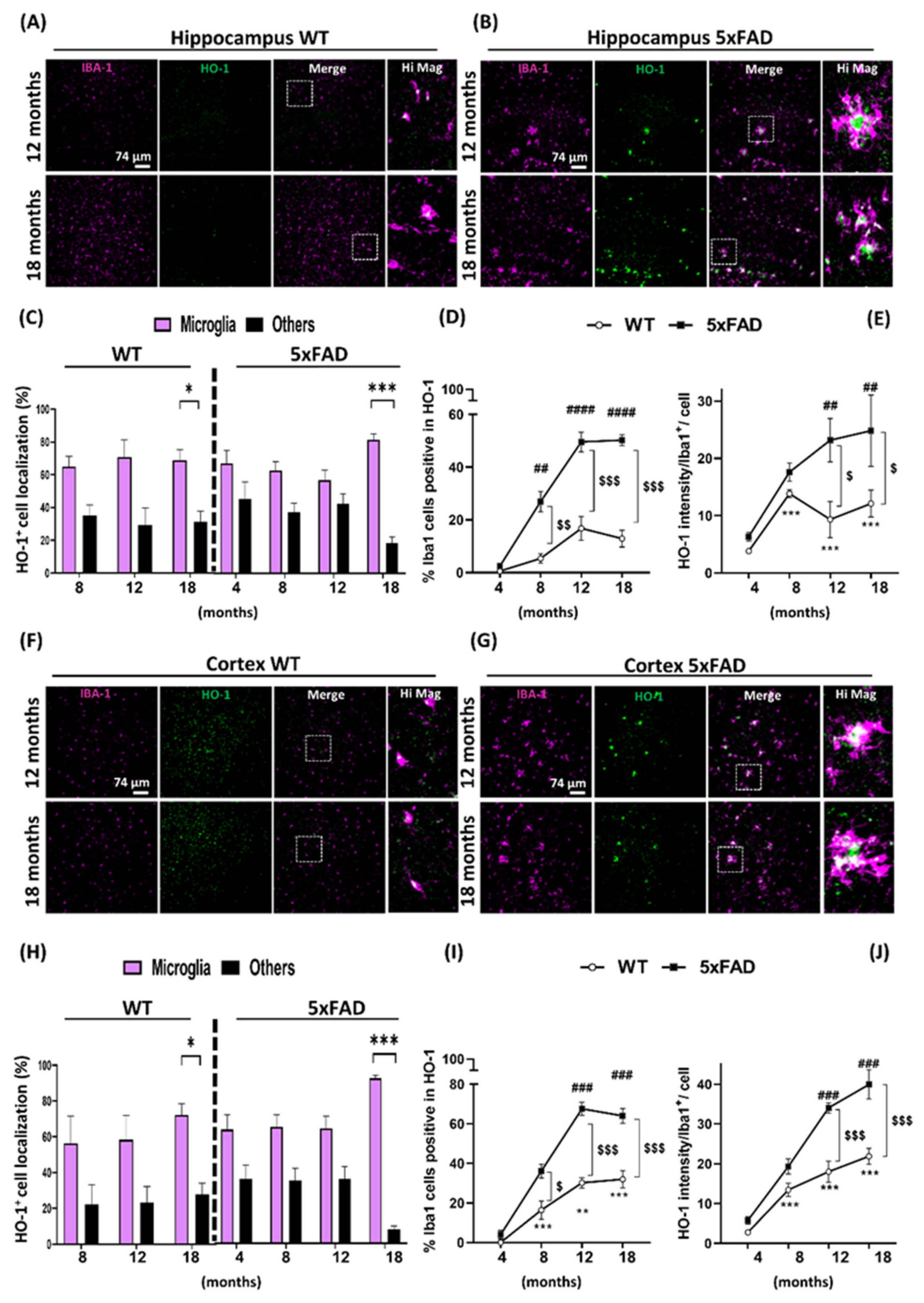
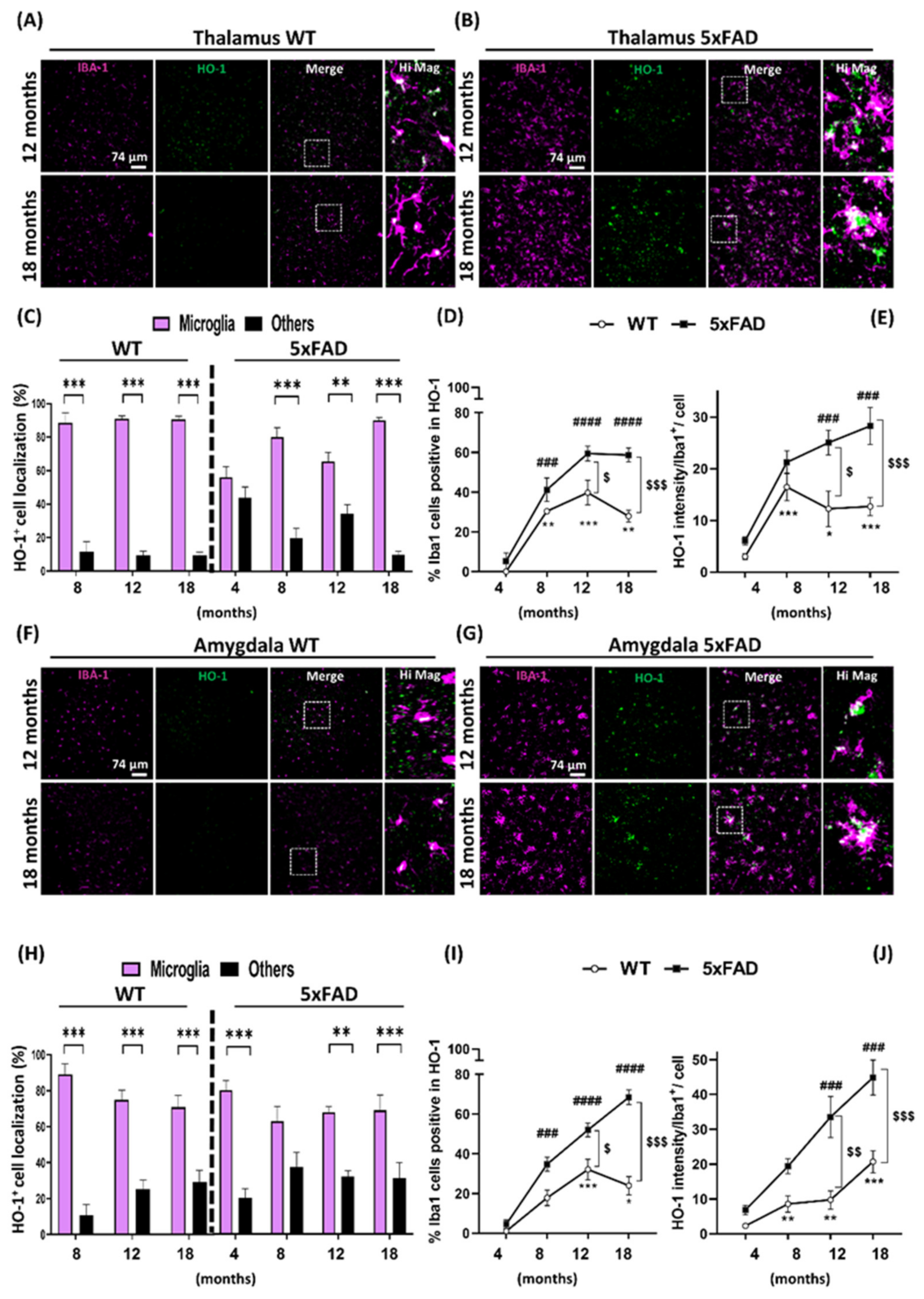
3.2. Microglia HO-1 Expression Profile in WT and 5xFAD Mice with Aging
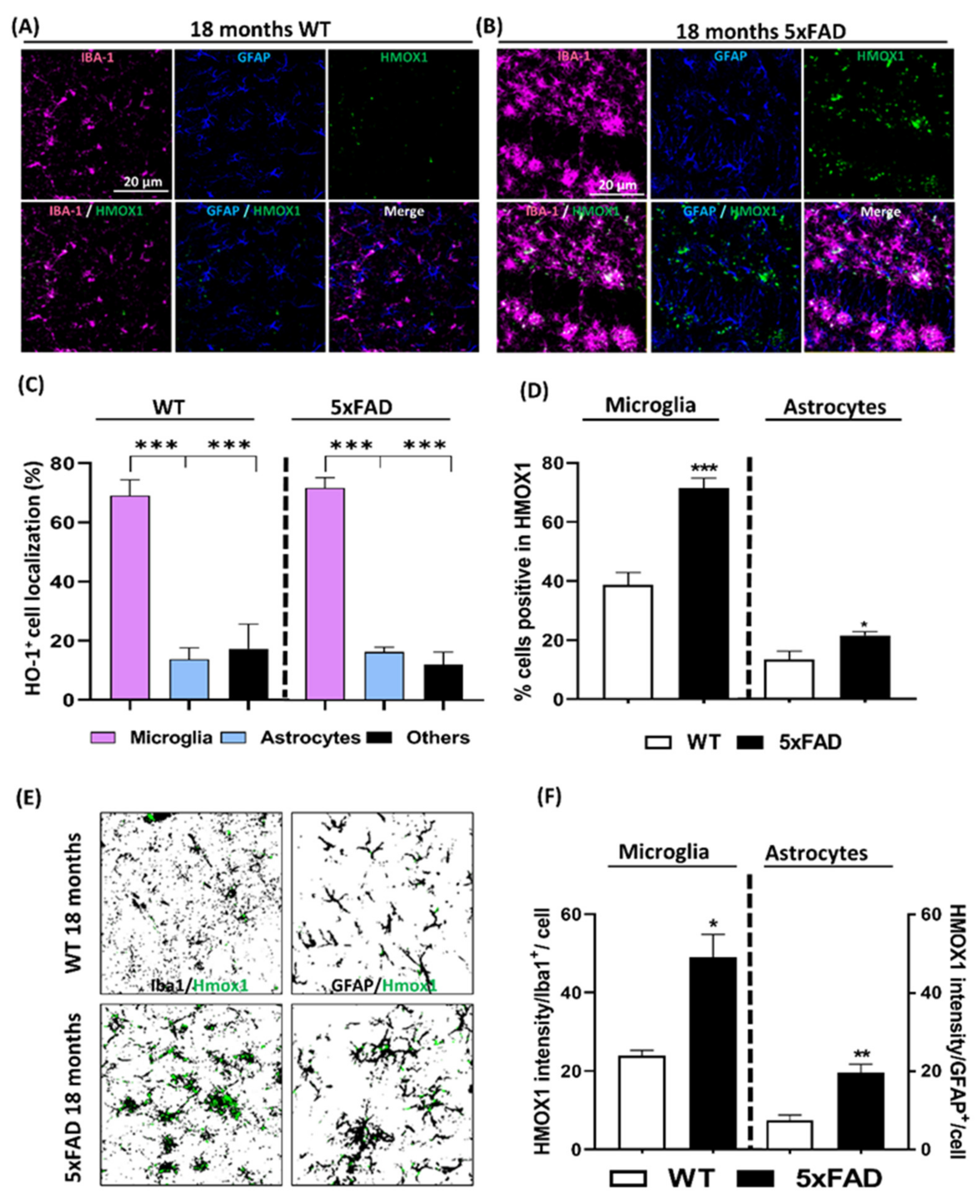
3.3. Localization Analysis of RNA Hmox1 in Glial Cells in Aged 5xFAD Mice
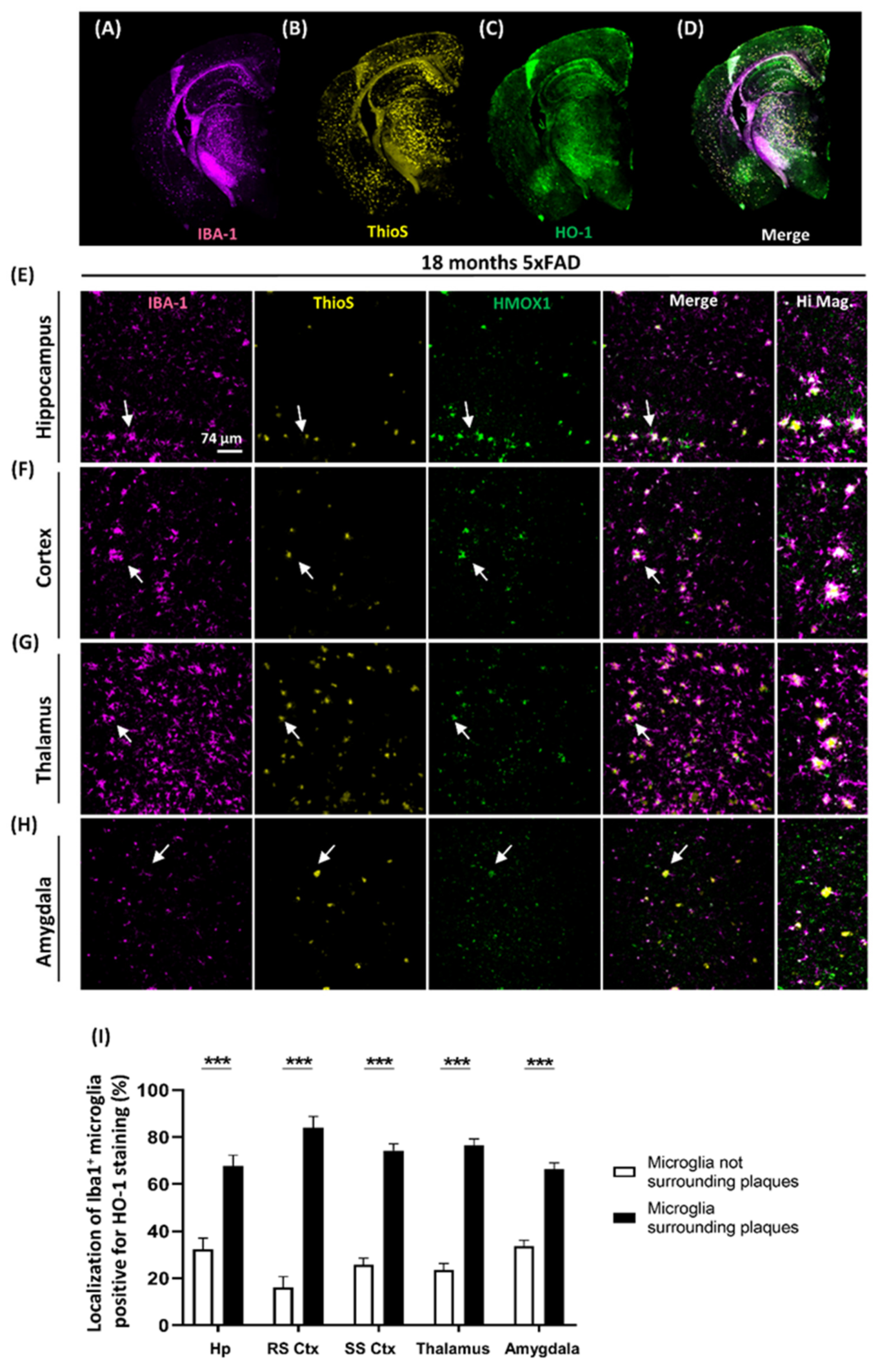
3.4. Localization of Microglia Overexpressing HO-1 Surrounding Aβ Plaques
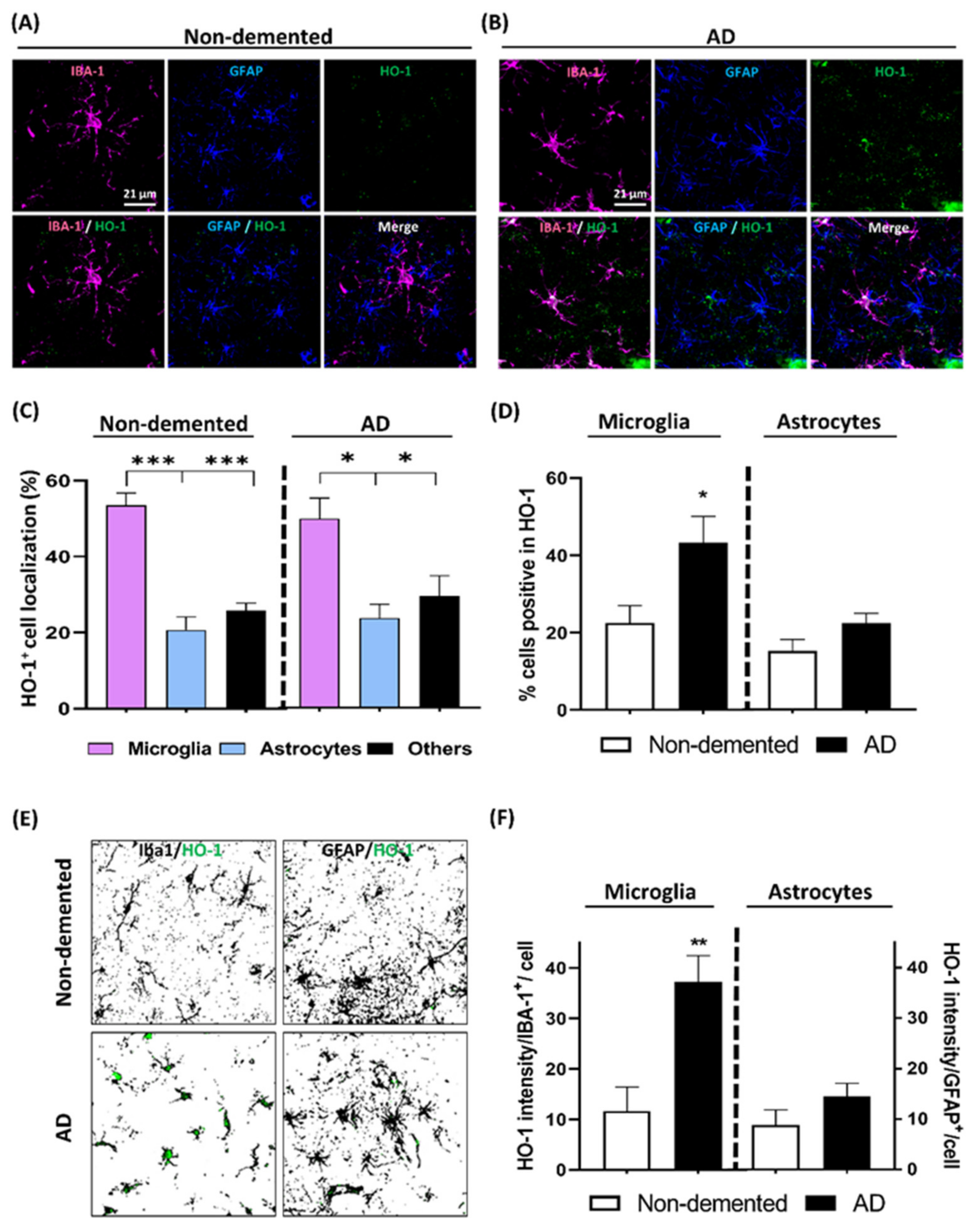
3.5. HO-1 Expression Analysis in the Cortex of AD Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2017, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Shakeri, A.; Rao, P.P.N. Amyloid cascade in Alzheimer’s disease: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2016, 113, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef]
- Richard, B.C.; Kurdakova, A.; Baches, S.; Bayer, T.A.; Weggen, S.; Wirths, O. Gene Dosage Dependent Aggravation of the Neurological Phenotype in the 5XFAD Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 1223–1236. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Ritzel, R.M.; Patel, A.R.; Pan, S.; Crapser, J.; Hammond, M.; Jellison, E.; McCullough, L.D.; Crapser, J. Age- and location-related changes in microglial function. Neurobiol. Aging 2015, 36, 2153–2163. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2017, 217, 459–472. [Google Scholar] [CrossRef]
- Li, J.-W.; Zong, Y.; Cao, X.-P.; Tan, L.; Tan, L. Microglial priming in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 176. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2018, 172, 40–70. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Den, Z. Distal AP-1 binding sites mediate basal level enhancement and TPA induction of the mouse heme oxygenase-1 gene. J. Biol. Chem. 1992, 267, 21894–21900. [Google Scholar]
- Wijayanti, N.; Huber, S.; Samoylenko, A.; Kietzmann, T.; Immenschuh, S. Role of NF-kB and p38 MAP Kinase Signaling Pathways in the Lipopolysaccharide-Dependent Activation of Heme Oxygenase-1 Gene Expression. Antioxid. Redox Signal. 2004, 6, 802–810. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Ling, K.; Men, F.; Wang, W.-C.; Zhou, Y.-Q.; Zhang, H.-W.; Ye, D.-W. Carbon Monoxide and Its Controlled Release: Therapeutic Application, Detection, and Development of Carbon Monoxide Releasing Molecules (CORMs). J. Med. Chem. 2017, 61, 2611–2635. [Google Scholar] [CrossRef]
- Nikam, A.; Ollivier, A.; Rivard, M.; Wilson, J.L.; Mebarki, K.; Martens, T.; Dubois-Randé, J.-L.; Motterlini, R.; Foresti, R. Diverse Nrf2 Activators Coordinated to Cobalt Carbonyls Induce Heme Oxygenase-1 and Release Carbon Monoxide in Vitro and in Vivo. J. Med. Chem. 2016, 59, 756–762. [Google Scholar] [CrossRef]
- Motterlini, R.; Nikam, A.; Manin, S.; Ollivier, A.; Wilson, J.L.; Djouadi, S.; Muchova, L.; Martens, T.; Rivard, M.; Foresti, R. HYCO-3, a dual CO-releaser/Nrf2 activator, reduces tissue inflammation in mice challenged with lipopolysaccharide. Redox Biol. 2018, 20, 334–348. [Google Scholar] [CrossRef]
- Hettiarachchi, N.; Dallas, M.L.; Al-Owais, M.; Jarosz-Griffiths, H.H.; Hooper, N.M.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer’s amyloid-β(1-42)-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, W.; Ikematsu, K.; Tsuda, R. Age-associated increases in heme oxygenase-1 and ferritin immunoreactivity in the autopsied brain. Leg. Med. 2003, 5, S360–S366. [Google Scholar] [CrossRef]
- Schipper, H.M.; Cisse, S.; Stopa, E.G. Expression of heme oxygenase-1 in the senescent and alzheimer-diseased brain. Ann. Neurol. 1995, 37, 758–768. [Google Scholar] [CrossRef]
- Schipper, H.; Liberman, A.; Stopa, E. Neural Heme Oxygenase-1 Expression in Idiopathic Parkinson’s Disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Rojo, A. Heme Oxygenase-1 as a Therapeutic Target in Neurodegenerative Diseases and Brain Infections. Curr. Pharm. Des. 2008, 14, 429–442. [Google Scholar] [CrossRef]
- Rahman, M.M.; Sykiotis, G.P.; Nishimura, M.; Bodmer, R.; Bohmann, D. Declining signal dependence of Nrf2-MafS-regulated gene expression correlates with aging phenotypes. Aging Cell 2013, 12, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- Xing, S.; Shen, D.; Chen, C.; Wang, J.; Yu, Z. Early induction of oxidative stress in a mouse model of Alzheimer’s disease with heme oxygenase activity. Mol. Med. Rep. 2014, 10, 599–604. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Kutty, R.K.; Richey, P.L.; Yan, S.D.; Stern, D.; Chader, G.J.; Wiggert, B.; Petersen, R.B.; Perry, G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am. J. Pathol. 1994, 145, 42–47. [Google Scholar] [PubMed]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakimura, J.-I.; Kitamura, Y.; Takata, K.; Umeki, M.; Suzuki, S.; Shibagaki, K.; Taniguchi, T.; Nomura, Y.; Gebicke-Haerter, P.J.; Smith, M.A.; et al. Microglial activation and amyloid-β clearance induced by exogenous heat-shock proteins. FASEB J. 2002, 16, 601–603. [Google Scholar] [CrossRef]
- McIntosh, A.; Mela, V.; Harty, C.; Minogue, A.M.; Costello, D.; Kerskens, C.; Lynch, M.A. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 2019, 29, 606–621. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2017, 14, 450–464. [Google Scholar] [CrossRef]
- Lu, L.-N.; Qian, Z.-M.; Wu, K.-C.; Yung, W.-H.; Ke, Y. Expression of Iron Transporters and Pathological Hallmarks of Parkinson’s and Alzheimer’s Diseases in the Brain of Young, Adult, and Aged Rats. Mol. Neurobiol. 2016, 54, 5213–5224. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Bush, I.A. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Schipper, H.; Gupta, A.; Szarek, W. Suppression of Glial HO-1 Activitiy as a Potential Neurotherapeutic Intervention in AD. Curr. Alzheimer Res. 2009, 6, 424–430. [Google Scholar] [CrossRef]
- Si, Z.; Wang, X.; Zhang, Z.; Wang, J.; Li, J.; Li, J.; Li, L.; Li, Y.; Peng, Y.; Sun, C.; et al. Heme Oxygenase 1 Induces Tau Oligomer Formation and Synapse Aberrations in Hippocampal Neurons. J. Alzheimer’s Dis. 2018, 65, 409–419. [Google Scholar] [CrossRef]
- Wang, D.; Hui, Y.; Peng, Y.; Tang, L.; Jin, J.; He, R.; Li, Y.; Zhang, S.; Li, L.; Zhou, Y.; et al. Overexpression of Heme Oxygenase 1 Causes Cognitive Decline and Affects Pathways for Tauopathy in Mice. J. Alzheimer’s Dis. 2014, 43, 519–534. [Google Scholar] [CrossRef]
- Li, L.; Peng, Y.; Hui, Y.; Zhang, S.; Zhou, Y.; Li, D.; Li, J.; Si, Z.; Li, J.; Wang, D.; et al. Overexpression of Heme Oxygenase 1 Impairs Cognitive Ability and Changes the Plasticity of the Synapse. J. Alzheimer’s Dis. 2015, 47, 595–608. [Google Scholar] [CrossRef]
- Sung, H.Y.; Choi, B.-O.; Jeong, J.H.; Kong, K.A.; Hwang, J.; Ahn, J.-H. Amyloid Beta-Mediated Hypomethylation of Heme Oxygenase 1 Correlates with Cognitive Impairment in Alzheimer’s Disease. PLoS ONE 2016, 11, e0153156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Lacoste, B.; Pistel, P.J.; Ingram, D.K.; Hamel, E.; Alaoui-Jamali, M.A.; Szarek, W.A.; Vlahakis, J.Z.; Jie, S.; Song, W.; et al. Neurotherapeutic effects of novel HO-1 inhibitorsin vitroand in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2014, 131, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.G.; Caruso, M.; Murchison, C.F.; Zhu, J.Y.; Wright, K.M.; Harris, C.J.; Gray, N.E.; Quinn, J.F.; Soumyanath, A. Centella Asiatica Improves Memory and Promotes Antioxidative Signaling in 5XFAD Mice. Antioxidants 2019, 8, 630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hettiarachchi, N.T.; Boyle, J.P.; Dallas, M.L.; Al-Owais, M.M.; Scragg, J.L.; Peers, C. Heme oxygenase-1 derived carbon monoxide suppresses Aβ1-42 toxicity in astrocytes. Cell Death Dis. 2017, 8, e2884. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective Effect of Caffeic Acid Phenethyl Ester in A Mouse Model of Alzheimer’s Disease Involves Nrf2/HO-1 Pathway. Aging Dis. 2018, 9, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-Q.; Xu, Y.; Zhu, C. Upregulation of Heme Oxygenase-1 by Acteoside Through ERK and PI3 K/Akt Pathway Confer Neuroprotection Against Beta-Amyloid-Induced Neurotoxicity. Neurotox. Res. 2011, 21, 368–378. [Google Scholar] [CrossRef]
- Amin, F.U.; Shah, S.A.; Kim, M.O. Vanillic acid attenuates Aβ1-42-induced oxidative stress and cognitive impairment in mice. Sci. Rep. 2017, 7, 40753. [Google Scholar] [CrossRef]
- Fagone, P.; Patti, F.; Mangano, K.; Mammana, S.; Coco, M.; Touil-Boukoffa, C.; Chikovani, T.; Di Marco, R.; Nicoletti, F. Heme oxygenase-1 expression in peripheral blood mononuclear cells correlates with disease activity in multiple sclerosis. J. Neuroimmunol. 2013, 261, 82–86. [Google Scholar] [CrossRef]
- Li, Y.; Pan, K.; Chen, L.; Ning, J.; Li, X.; Yang, T.; Terrando, N.; Gu, J.; Tao, G. Deferoxamine regulates neuroinflammation and iron homeostasis in a mouse model of postoperative cognitive dysfunction. J. Neuroinflamm. 2016, 13, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblanc, R.H.; Chen, R.; Selim, M.H.; Hanafy, K.A. Heme oxygenase-1-mediated neuroprotection in subarachnoid hemorrhage via intracerebroventricular deferoxamine. J. Neuroinflamm. 2016, 13, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-Y.; Cao, J.; Zhang, L.-M.; Li, Y.-F.; Mi, W.-D. Deferoxamine attenuates lipopolysaccharide-induced neuroinflammation and memory impairment in mice. J. Neuroinflamm. 2015, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappalá, A.; Volti, G.L. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murohashi, K.; Hara, Y.; Shinada, K.; Nagai, K.; Shinkai, M.; Kawana, A.; Kaneko, T. Clinical Significance of Serum Hemeoxygenase-1 as a New Biomarker for the Patients with Interstitial Pneumonia. Can. Respir. J. 2018, 2018, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K. Heme oxygenase-1 and acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Hatano, T.; Saiki, S.; Okuzumi, A.; Mohney, R.P.; Hattori, N. Identification of novel biomarkers for Parkinson’s disease by metabolomic technologies. J. Neurol. Neurosurg. Psychiatry 2015, 87, 295–301. [Google Scholar] [CrossRef]
- Song, W.; Kothari, V.; Velly, A.M.; Cressatti, M.; Liberman, A.; Gornitsky, M.; Schipper, H.M. Evaluation of salivary heme oxygenase-1 as a potential biomarker of early Parkinson’s disease. Mov. Disord. 2018, 33, 583–591. [Google Scholar] [CrossRef]
- Mateo, I.; Infante, J.; Sánchez-Juan, P.; García-Gorostiaga, I.; Rodríguez-Rodríguez, E.; Vázquez-Higuera, J.L.; Berciano, J.; Combarros, O. Serum heme oxygenase-1 levels are increased in Parkinson’s disease but not in Alzheimer’s disease. Acta Neurol. Scand. 2010, 121, 136–138. [Google Scholar] [CrossRef]
- Schipper, H.M. Biomarker potential of heme oxygenase-1 in Alzheimer’s disease and mild cognitive impairment. Biomarkers Med. 2007, 1, 375–385. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Mendívil, C.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N.; Lopez, M.G. Aging and Progression of Beta-Amyloid Pathology in Alzheimer’s Disease Correlates with Microglial Heme-Oxygenase-1 Overexpression. Antioxidants 2020, 9, 644. https://doi.org/10.3390/antiox9070644
Fernández-Mendívil C, Arreola MA, Hohsfield LA, Green KN, Lopez MG. Aging and Progression of Beta-Amyloid Pathology in Alzheimer’s Disease Correlates with Microglial Heme-Oxygenase-1 Overexpression. Antioxidants. 2020; 9(7):644. https://doi.org/10.3390/antiox9070644
Chicago/Turabian StyleFernández-Mendívil, Cristina, Miguel A. Arreola, Lindsay A. Hohsfield, Kim N. Green, and Manuela G. Lopez. 2020. "Aging and Progression of Beta-Amyloid Pathology in Alzheimer’s Disease Correlates with Microglial Heme-Oxygenase-1 Overexpression" Antioxidants 9, no. 7: 644. https://doi.org/10.3390/antiox9070644