The Effect of Glutathione Peroxidase-1 Knockout on Anticancer Drug Sensitivities and Reactive Oxygen Species in Haploid HAP-1 Cells

Abstract
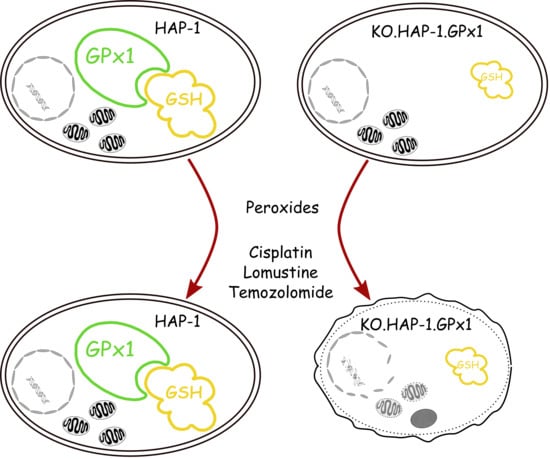
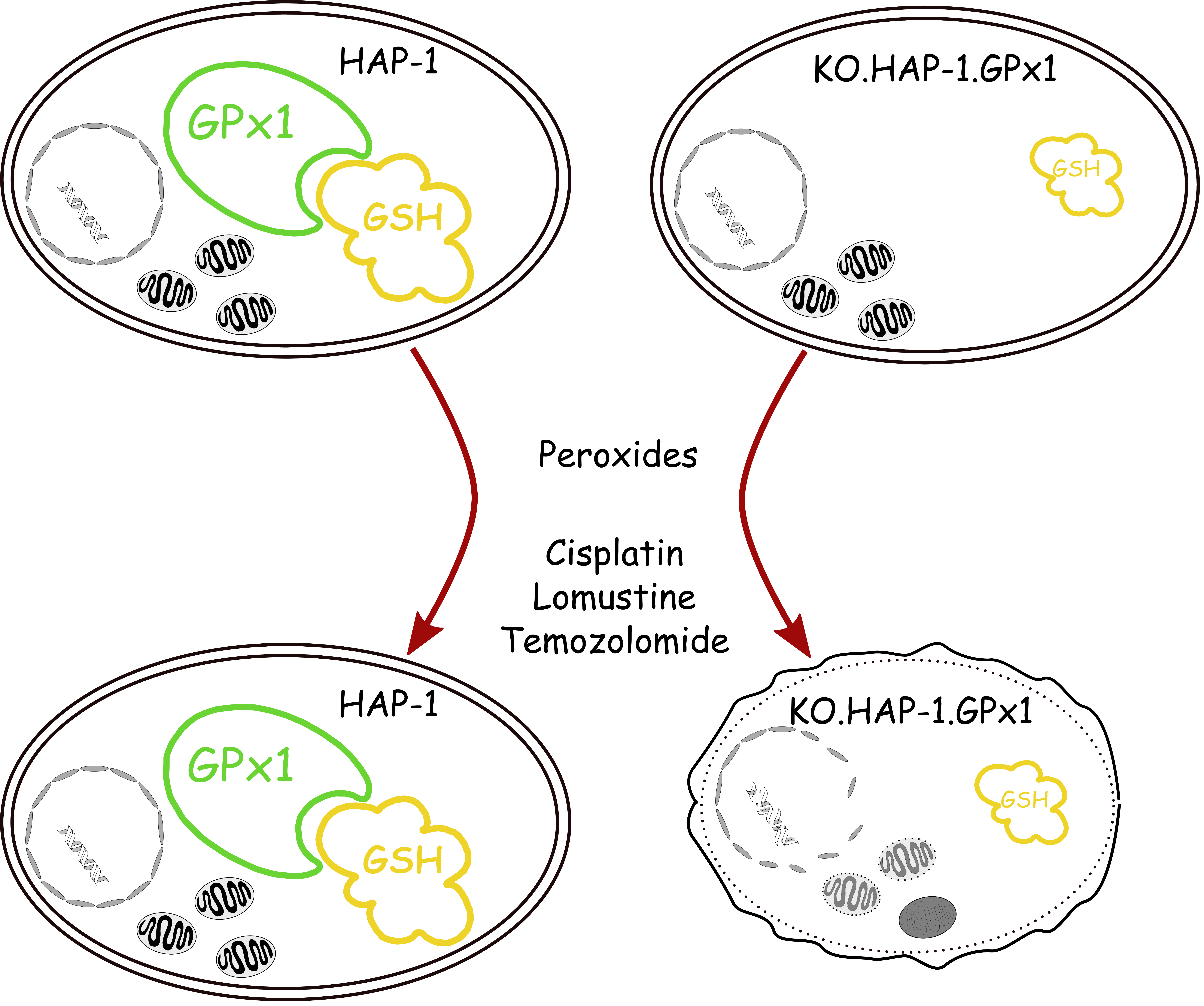
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Determining Cell Sizes by Coulter Counter and EVETM Cell Counter
2.4. Determining Cell Doubling Times via Crystal Violet Assay
2.5. Assessing Rates of Cellular Metabolism with the APH and MTT Assays
2.6. Assessing Cell Viability with the ATP Assay and Annexin-V Assays
2.7. Determination of Total Glutathione Content in Cell Lysates
2.8. Western Blotting of Protein Expression
2.9. Determination of IC50 Values
2.10. Determination of ROS-Levels after Peroxide or Anticancer Drug Treatment
2.11. Statistics
3. Results
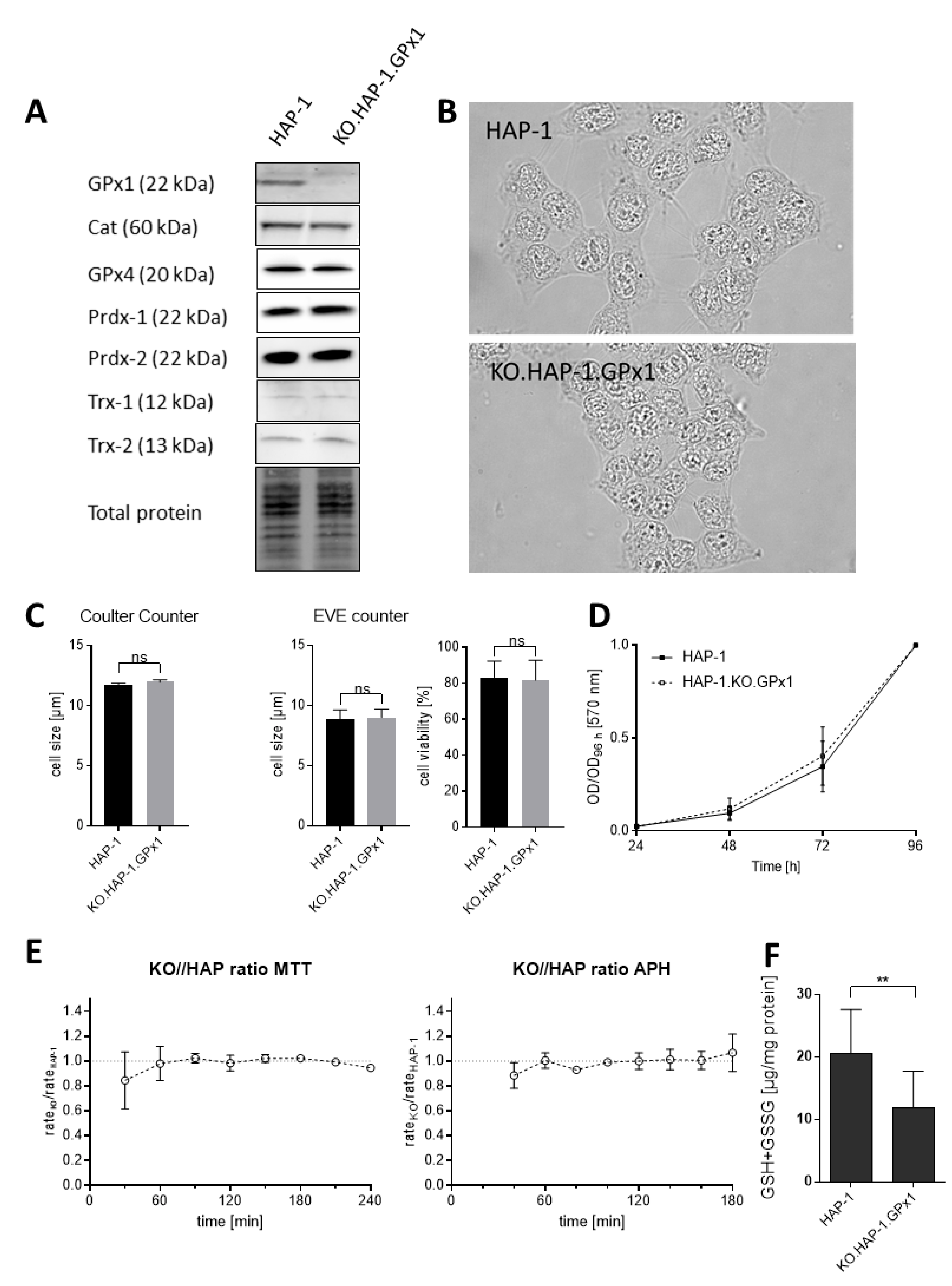
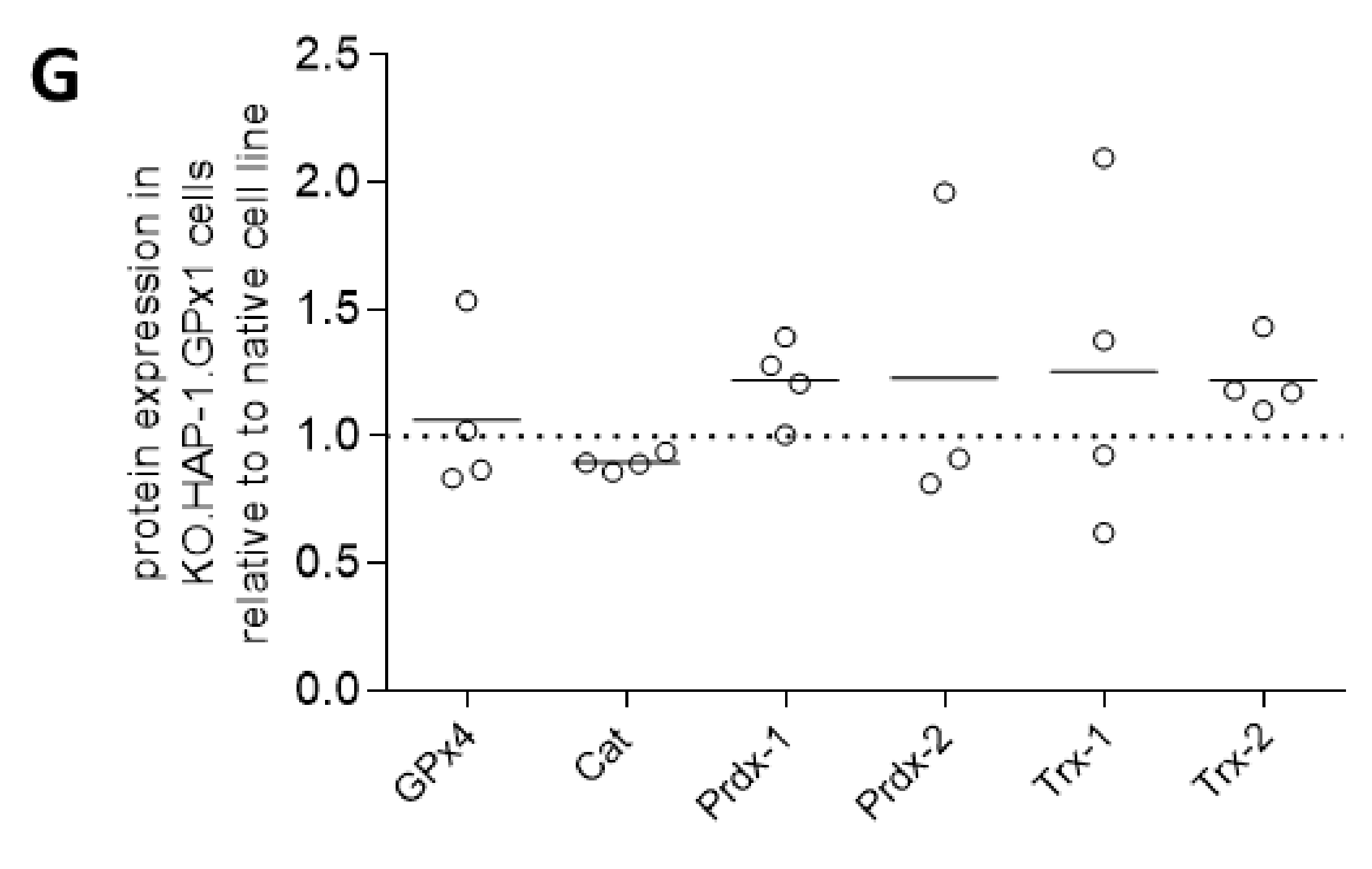
3.1. Characterization of GPx1 Knockout HAP-1 Cells Relative to the Native Cell Line
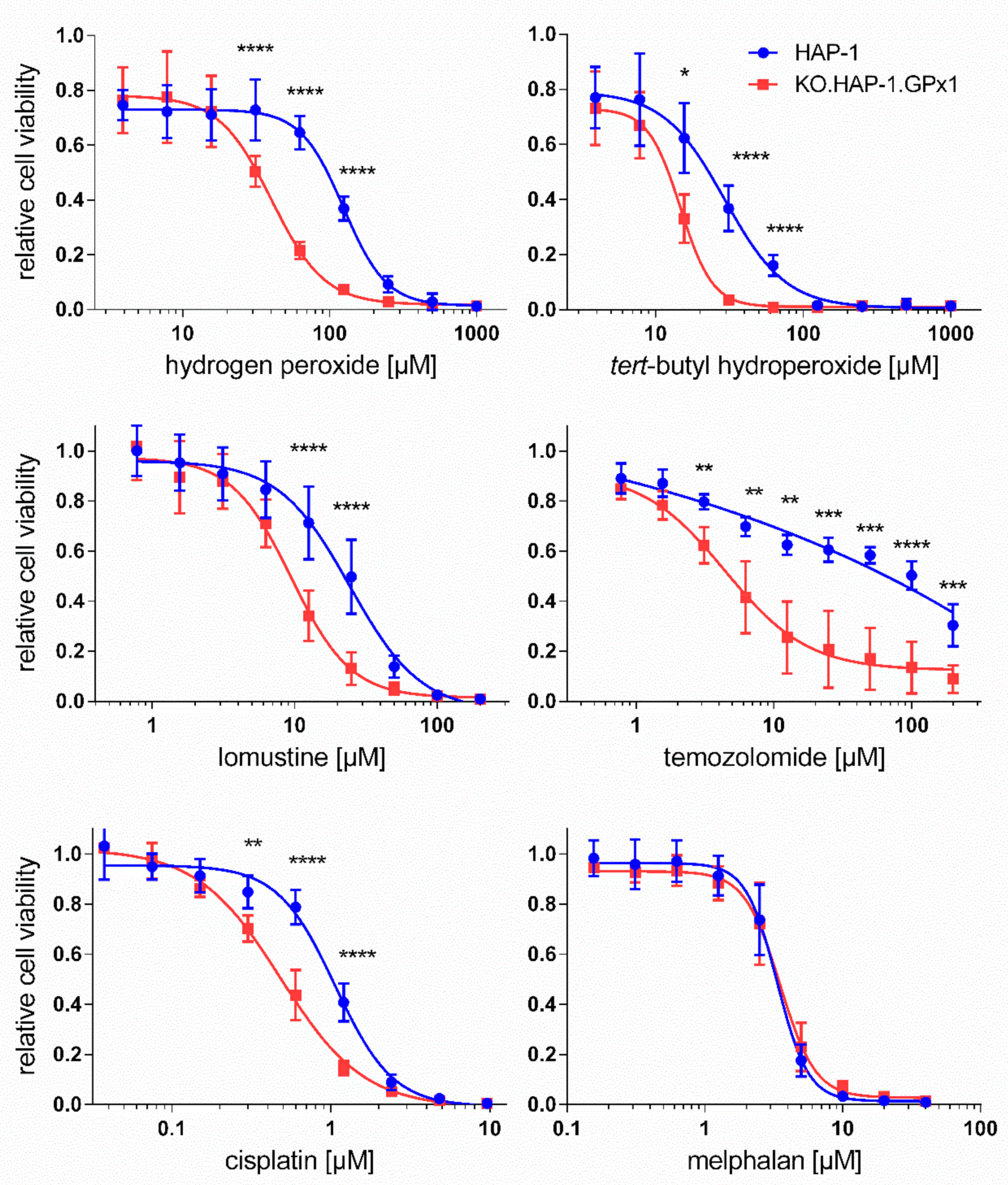
3.2. Effect of GPx1 Knockout in HAP-1 Cells on Toxicity of Peroxides and Anticancer Drugs
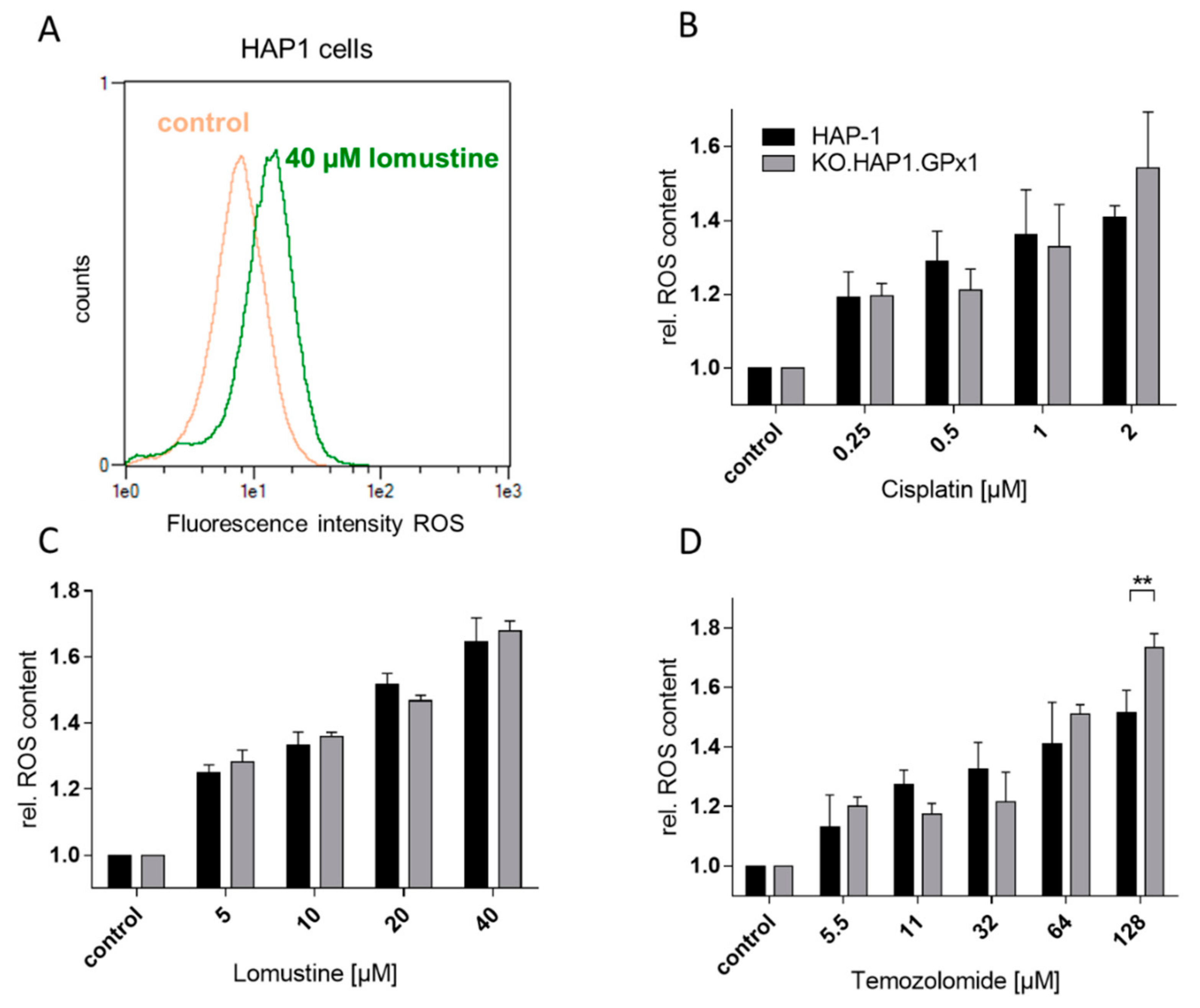
3.3. Determination of ROS Accumulation Induced by Peroxides, Cisplatin, Lomustine and Temozolomide in HAP-1 Cells and Their GPx1 Knockout Strain
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APH | Acidic phosphatase |
| BSA | Bovine serum albumin |
| Cat | Catalase |
| CDF-DA | 2′,7′-dichlorodihydrofluorescein diacetate |
| DTNB | Ellman’s reagent, 5,5-Dithiobis(2-nitrobenzoic acid) |
| FCS | Fetal calf serum |
| GSH | Reduced glutathione |
| GSSG | Glutathione disulfide |
| GSH + GSSG | Total glutathione |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| G6P | Glucose-6-phosphate |
| G6P-DH | Glucose-6-phosphat dehydrogenase |
| KOI | Knockout index |
| LPO | Lipid peroxidation |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | Phosphate buffered saline |
| Prdx | Peroxiredoxin |
| ROS | Reactive oxygen species |
| TrxR | Thioredoxin reductase |
References
- Ighodaro, O.M.; Akinloye, O.A. First Line Defence Antioxidants-Superoxide Dismutase (SOD), Catalase (CAT) and Glutathione Peroxidase (GPX): Their Fundamental Role in the Entire Antioxidant Defence Grid. Alex. J. Med. 2019, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Toppo, S.; Flohe, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic Mechanisms and Specificities of Glutathione Peroxidases: Variations of a Basic Scheme. Biochim. Biophys. Acta Gen. Sub. 2009, 1790, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-Dependent Antioxidant Enzymes: Actions and Properties of Selenoproteins. Antioxidants 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esworthy, R.S.; Ho, Y.S.; Chu, F.F. The Gpx1 Gene Encodes Mitochondrial Glutathione Peroxidase in the Mouse Liver. Arch. Biochem. Biophys. 1997, 340, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.S.; Magnenat, J.L.; Bronson, R.T.; Cao, J.; Gargano, M.; Sugawara, M.; Funk, C.D. Mice Deficient in Cellular Glutathione Peroxidase Develop Normally and Show no Increased Sensitivity to Hyperoxia. J. Biol. Chem. 1997, 272, 16644–16651. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.N.; Giblin, F.J.; Lin, L.-R.; Dang, L.; Unakar, N.J.; Musch, D.C.; Boyle, D.L.; Takemoto, L.J.; Ho, Y.-S.; Knoernschild, T.; et al. Glutathione Peroxidase-1 Deficiency Leads to Increased Nuclear Light Scattering, Membrane Damage, and Cataract Formation in Gene-Knockout Mice. Investig. Ophthalmol.Vis. Sci. 2001, 42, 3247–3255. [Google Scholar]
- Brutsch, S.H.; Wang, C.C.; Li, L.; Stender, H.; Neziroglu, N.; Richter, C.; Kuhn, H.; Borchert, A. Expression of Inactive Glutathione Peroxidase 4 Leads to Embryonic Lethality, and Inactivation of the Alox15 Gene does not Rescue Such Knock-in Mice. Antioxid. Redox Signal 2015, 22, 281–293. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Kipp, A.P. Selenium-Dependent Glutathione Peroxidases During Tumor Development. Adv. Cancer Res. 2017, 136, 109–138. [Google Scholar] [CrossRef]
- Jardim, B.V.; Moschetta, M.G.; Leonel, C.; Gelaleti, G.B.; Regiani, V.R.; Ferreira, L.C.; Lopes, J.R.; Zuccari, D.A. Glutathione and Glutathione Peroxidase Expression in Breast Cancer: An Immunohistochemical and Molecular Study. Oncol. Rep. 2013, 30, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Shen, Z.; Wu, D.; Xie, X.; Xu, X.; Lv, L.; Dai, H.; Chen, J.; Gan, X. Glutathione Peroxidase 1 Promotes NSCLC Resistance to Cisplatin via ROS-Induced Activation of PI3K/AKT Pathway. BioMed Res. Int. 2019, 2019, 7640547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, R.; Emmrich, T.; Lemmerhirt, H.; Leffler, U.; Sydow, K.; Hirt, C.; Kiefer, T.; Link, A.; Bednarski, P.J. Identification of a Glutathione Peroxidase Inhibitor that Reverses Resistance to Anticancer Drugs in Human B-Cell Lymphoma Cell Lines. Bioorg. Med. Chem. Lett. 2012, 22, 6712–6715. [Google Scholar] [CrossRef] [PubMed]
- Joncourt, F.; Oberli-Schrammli, A.E.; Stadler, M.; Buser, K.; Franscini, L.; Fey, M.F.; Cerny, T. Patterns of Drug Resistance Parameters in Adult Leukemia. Leuk. Lymphoma 1995, 17, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, S.; Vlantis, A.C.; Liu, S.Y.; Ng, E.K.; Chan, A.B.; Wu, J.; Du, J.; Wei, W.; Liu, X.; et al. Expression of Antioxidant Molecules and Heat Shock Protein 27 in Thyroid Tumors. J. Cell. Biochem. 2016, 117, 2473–2481. [Google Scholar] [CrossRef] [PubMed]
- Savic-Radojevic, A.; Mimic-Oka, J.; Pljesa-Ercegovac, M.; Opacic, M.; Dragicevic, D.; Kravic, T.; Djokic, M.; Micic, S.; Simic, T. Glutathione S-Transferase-P1 Expression Correlates with Increased Antioxidant Capacity in Transitional Cell Carcinoma of the Urinary Bladder. Eur. Urol. 2007, 52, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Rusolo, F.; Capone, F.; Pasquale, R.; Angiolillo, A.; Colonna, G.; Castello, G.; Costantini, M.; Costantini, S. Comparison of the Seleno-Transcriptome Expression between Human Non-cancerous Mammary Epithelial Cells and Two Human Breast Cancer Cell Lines. Oncol. Lett. 2017, 13, 2411–2417. [Google Scholar] [CrossRef] [PubMed]
- Preuss, M.; Girnun, G.D.; Darby, C.J.; Khoo, N.; Spector, A.A.; Robbins, M.E. Role of Antioxidant Enzyme Expression in the Selective Cytotoxic Response of Glioma Cells to gamma-Linolenic acid Supplementation. Free Radic. Biol. Med. 2000, 28, 1143–1156. [Google Scholar] [CrossRef]
- Pljesa-Ercegovac, M.; Mimic-Oka, J.; Dragicevic, D.; Savic-Radojevic, A.; Opacic, M.; Pljesa, S.; Radosavljevic, R.; Simic, T. Altered Antioxidant Capacity in Human Renal Cell Carcinoma: Role of Glutathione Associated Enzymes. Urol. Oncol. 2008, 26, 175–181. [Google Scholar] [CrossRef]
- Cheng, Y.; Xu, T.; Li, S.; Ruan, H. GPX1, a Biomarker for the Diagnosis and Prognosis of Kidney Cancer, Promotes the Progression of Kidney Cancer. Aging 2019, 11, 12165–12176. [Google Scholar] [CrossRef]
- Da Motta, L.L.; De Bastiani, M.A.; Stapenhorst, F.; Klamt, F. Oxidative Stress Associates with Aggressiveness in Lung Large-Cell Carcinoma. Tumour Biol. 2015, 36, 4681–4688. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Jin, M.; Xiao, L.; Liu, H.; Wei, S. Distinct Prognostic Values of mRNA Expression of Glutathione Peroxidases in Non-Small Cell Lung Cancer. Cancer Manag. Res. 2018, 10, 2997–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.R.; Roh, J.L.; Lee, S.M.; Park, Y.; Cho, K.J.; Choi, S.H.; Nam, S.Y.; Kim, S.Y. Overexpression of Glutathione Peroxidase 1 Predicts Poor Prognosis in Oral Squamous Cell Carcinoma. J.Cancer Res. Clin.Oncol. 2017, 143, 2257–2265. [Google Scholar] [CrossRef] [PubMed]
- Jerome-Morais, A.; Wright, M.E.; Liu, R.; Yang, W.; Jackson, M.I.; Combs, G.F., Jr.; Diamond, A.M. Inverse Association between Glutathione Peroxidase Activity and Both Selenium-Binding Protein 1 Levels and Gleason Score in Human Prostate Tissue. Prostate 2012, 72, 1006–1012. [Google Scholar] [CrossRef] [Green Version]
- Godoy, J.R.; Funke, M.; Ackermann, W.; Haunhorst, P.; Oesteritz, S.; Capani, F.; Elsässer, H.-P.; Lillig, C.H. Redox Atlas of the Mouse: Immunohistochemical Detection of Glutaredoxin-, Peroxiredoxin-, and Thioredoxin-Family Proteins in Various Tissues of the Laboratory Mouse. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 2–92. [Google Scholar] [CrossRef]
- Bracht, K.; Boubakari; Grunert, R.; Bednarski, P.J. Correlations between the Activities of 19 Anti-tumor Agents and the Intracellular Glutathione Concentrations in a Panel of 14 Human Cancer Cell Lines: Comparisons with the National Cancer Institute Data. Anticancer Drugs 2006, 17, 41–51. [Google Scholar] [CrossRef]
- Neumann, C.; Boubakari; Grunert, R.; Bednarski, P.J. Nicotinamide Adenine Dinucleotide Phosphate-Regenerating System Coupled to a Glutathione-Reductase Microtiter Method for Determination of Total Glutathione Concentrations in Adherent Growing Cancer Cell Lines. Anal. Biochem. 2003, 320, 170–178. [Google Scholar] [CrossRef]
- Gürtler, A.; Kunz, N.; Gomolka, M.; Hornhardt, S.; Friedl, A.A.; McDonald, K.; Kohn, J.E.; Posch, A. Stain-Free Technology as a Normalization Tool in Western Blot Analysis. Anal.l Biochem. 2013, 433, 105–111. [Google Scholar] [CrossRef]
- Colella, A.D.; Chegenii, N.; Tea, M.N.; Gibbins, I.L.; Williams, K.A.; Chataway, T.K. Comparison of Stain-Free Cels with Traditional Immunoblot Loading Control Methodology. Anal. Biochem. 2012, 430, 108–110. [Google Scholar] [CrossRef]
- Behnisch-Cornwell, S.; Bandaru, S.S.M.; Napierkowski, M.; Wolff, L.; Zubair, M.; Urbainsky, C.; Lillig, C.; Schulzke, C.; Bednarski, P.J. Pentathiepins: A Novel Class of Glutathione Peroxidase 1 Inhibitors that Induce Oxidative Stress, Loss of Mitochondrial Membrane Potential and Apoptosis in Human Cancer Cells. ChemMedChem 2020, 15, 1515–1528. [Google Scholar] [CrossRef]
- Essletzbichler, P.; Konopka, T.; Santoro, F.; Chen, D.; Gapp, B.V.; Kralovics, R.; Brummelkamp, T.R.; Nijman, S.M.; Bürckstümmer, T. Megabase-scale deletion using CRISPR/Cas9 to Generate a Fully Haploid Human Cell Line. Genome Res. 2014, 24, 2059–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Cheng, W.H.; Porres, J.M.; Ross, D.A.; Lei, X.G. Knockout of Cellular Glutathione Peroxidase Gene Renders Mice Susceptible to Diquat-Induced Oxidative Stress. Free Radic. Biol. Med. 1999, 27, 605–611. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Dai, M.; Yuan, Z. Methods for the Detection of Reactive Oxygen Species. Anal. Methods 2018, 10, 4625–4638. [Google Scholar] [CrossRef]
- De Haan, J.B.; Bladier, C.; Griffiths, P.; Kelner, M.; O’Shea, R.D.; Cheung, N.S.; Bronson, R.T.; Silvestro, M.J.; Wild, S.; Zheng, S.S.; et al. Mice with a Homozygous Null Mutation for the Most Abundant Glutathione Peroxidase, Gpx1, Show Increased Susceptibility to the Oxidative Stress-Inducing Agents Paraquat and Hydrogen Peroxide. J. Biol. Chem. 1998, 273, 22528–22536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oancea-Castillo, L.R.; Klein, C.; Abdollahi, A.; Weber, K.J.; Regnier-Vigouroux, A.; Dokic, I. Comparative Analysis of the Effects of a Sphingosine Kinase Inhibitor to Temozolomide and Radiation Treatment on Glioblastoma Cell Lines. Cancer Biol. Ther. 2017, 18, 400–406. [Google Scholar] [CrossRef]
- Gan, X.; Chen, B.; Shen, Z.; Liu, Y.; Li, H.; Xie, X.; Xu, X.; Li, H.; Huang, Z.; Chen, J. High GPX1 Expression Promotes Esophageal Squamous Cell Carcinoma Invasion, Migration, Proliferation and Cisplatin-Resistance but can be Reduced by Vitamin D. Int. J. Clin. Exp. Med. 2014, 7, 2530–2540. [Google Scholar] [CrossRef] [Green Version]
- Stojkovic, S.; Podolski-Renic, A.; Dinic, J.; Stankovic, T.; Bankovic, J.; Hadzic, S.; Paunovic, V.; Isakovic, A.; Tanic, N.; Pesic, M. Development of Resistance to Antiglioma Agents in Rat C6 Cells Caused Collateral Sensitivity to Doxorubicin. Exp. Cell Res. 2015, 335, 248–257. [Google Scholar] [CrossRef]
- Salatino, A.; Aversa, I.; Battaglia, A.M.; Sacco, A.; Di Vito, A.; Santamaria, G.; Chirillo, R.; Veltri, P.; Tradigo, G.; Di Cello, A.; et al. H-Ferritin Affects Cisplatin-Induced Cytotoxicity in Ovarian Cancer Cells through the Modulation of ROS. Oxid. Med.Cell. Longev. 2019, 2019, 3461251. [Google Scholar] [CrossRef]
- Tse, A.K.; Chen, Y.J.; Fu, X.Q.; Su, T.; Li, T.; Guo, H.; Zhu, P.L.; Kwan, H.Y.; Cheng, B.C.; Cao, H.H.; et al. Sensitization of Melanoma Cells to Alkylating Agent-Induced DNA Damage and Cell Death via Orchestrating Oxidative Stress and IKKβ Inhibition. Redox Biol. 2017, 11, 562–576. [Google Scholar] [CrossRef]
- McConnell, D.D.; McGreevy, J.W.; Williams, M.N.; Litofsky, N.S. Do Anti-Oxidants Vitamin D(3,) Melatonin, and Alpha-Lipoic Acid Have Synergistic Effects with Temozolomide on Cultured Glioblastoma Cells? Medicines 2018, 5, 58. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HAP-1 | KO.HAP-1.GPx1 | KOI | p | |
|---|---|---|---|---|
| Hydrogen peroxide | 92.4 ± 7.3 µM | 29.6 ± 7.0 µM | 0.32 | 0.0001 |
| tert-Butyl hydroperoxide | 22.1 ± 7.7 µM | 10.7 ± 3.2 µM | 0.48 | 0.05 |
| Melphalan | 3.44 ± 0.56 µM | 3.55 ± 0.73 µM | 1.03 | - |
| Chlorambucil | 6.96 ± 1.78 µM | 7.01 ± 1.82 µM | 1.01 | - |
| Thiotepa | 6.21 ± 1.06 µM | 6.34 ± 0.56 µM | 1.02 | - |
| Lomustine | 23.0 ± 6.9 µM | 9.84 ± 1.94 µM | 0.43 | 0.005 |
| Temozolomide | 63.9 ± 21.7 µM | 5.53 ± 2.42 µM | 0.09 | 0.005 |
| Vinblastine | 1.58 ± 0.33 nM | 1.74 ± 0.50 nM | 1.10 | - |
| Paclitaxel | 2.82 ± 0.42 nM | 2.52 ± 0.53 nM | 0.90 | - |
| Camptothecin | 3.88 ± 0.09 nM | 3.83 ± 0.43 nM | 0.99 | - |
| Colchicine | 13.0 ± 2.6 nM | 14.0 ± 4.4 nM | 1.08 | - |
| Podophyllotoxin | 19.5 ± 1.0 nM | 20.1 ± 1.4 nM | 1.03 | - |
| Bortezomib | 13.3 ± 4.8 nM | 11.4 ± 2.3 nM | 0.85 | - |
| Doxorubicin | 24.4 ± 7.4 nM | 21.5 ± 4.4 nM | 0.82 | - |
| Methotrexate | 64.7 ± 5.9 nM | 64.8 ± 4.4 nM | 1.00 | - |
| Cisplatin | 1.06 ± 0.13 µM | 0.52 ± 0.07 µM | 0.49 | 0.0005 |
| Carboplatin | 29.6 ± 18.6 µM | 24.4 ± 16.0 µM | 0.83 | 0.05 |
| Oxaliplatin | 0.94 ± 0.29 µM | 0.67 ± 0.17 µM | 0.71 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behnisch-Cornwell, S.; Wolff, L.; Bednarski, P.J. The Effect of Glutathione Peroxidase-1 Knockout on Anticancer Drug Sensitivities and Reactive Oxygen Species in Haploid HAP-1 Cells. Antioxidants 2020, 9, 1300. https://doi.org/10.3390/antiox9121300
Behnisch-Cornwell S, Wolff L, Bednarski PJ. The Effect of Glutathione Peroxidase-1 Knockout on Anticancer Drug Sensitivities and Reactive Oxygen Species in Haploid HAP-1 Cells. Antioxidants. 2020; 9(12):1300. https://doi.org/10.3390/antiox9121300
Chicago/Turabian StyleBehnisch-Cornwell, Steven, Lisa Wolff, and Patrick J. Bednarski. 2020. "The Effect of Glutathione Peroxidase-1 Knockout on Anticancer Drug Sensitivities and Reactive Oxygen Species in Haploid HAP-1 Cells" Antioxidants 9, no. 12: 1300. https://doi.org/10.3390/antiox9121300