Investigating the Thioredoxin and Glutathione Systems’ Response in Lymphoma Cells after Treatment with [Au(d2pype)2]Cl

 and
and
Abstract
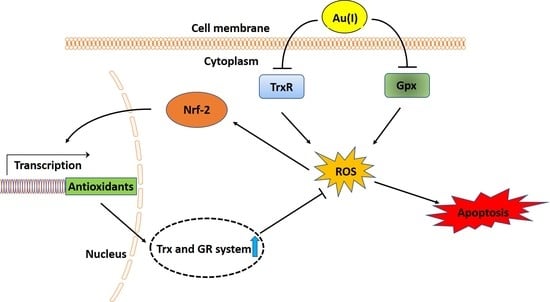
:
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Gold Compound Preparation
2.3. Cell Proliferation Assay
2.4. TrxR Activity Assay
2.5. GR Activity Assay
2.6. Measurement of GSH and GSSG
2.7. Gpx Activity Assay
2.8. Intracellular ROS Measurement Assay
2.9. Caspase-3 Activity Assay
2.10. Reverse Transcriptase-Quantitative PCR (RT-qPCR)
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Results
3.1. Lymphoma Cells Have Up-Regulated Antioxidant Systems
3.2. [Au(d2pype)2]Cl Inhibits Cell Proliferation in Lymphoma Cell Lines
3.3. [Au(d2pype)2]Cl Induces Cell Death via Apoptosis in Lymphoma Cell Lines
3.4. [Au(d2pype)2]Cl Inhibits Selenoproteins Activity
3.5. Cell Death Is Activated via [Au(d2pype)2]Cl-Induced Oxidative Stress
3.6. [Au(d2pype)2]Cl Modulates Expression of Several Antioxidant Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bai, B.; Huang, H.Q.; Cai, Q.Q.; Wang, X.X.; Cai, Q.C.; Lin, Z.X.; Guo, Y. Promising long-term outcome of gemcitabine, vinorelbine, liposomal doxorubicin (GVD) in 14-day schedule as salvage regimen for patients with previously heavily treated Hodgkin’s lymphoma and aggressive non-Hodgkin’s lymphoma. Med. Oncol. 2013, 30, 350. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Herrera, A.F.; Hou, J.; Chen, L.; Wu, J.; Guo, Y.; Newman, E.M. Inhibition of MDR1 overcomes resistance to brentuximab vedotin in hodgkin lymphoma. Clin. Cancer Res. 2020, 26, 1034–1044. [Google Scholar] [CrossRef]
- Melenotte, C.; Mezouar, S.; Mège, J.L.; Gorvel, J.P.; Kroemer, G.; Raoult, D. Bacterial infection and non-Hodgkin’s lymphoma. Crit. Rev. Microbiol. 2020, 46, 270–287. [Google Scholar] [CrossRef] [PubMed]
- Gallamini, A.; Tarella, C.; Viviani, S.; Rossi, A.; Patti, C.; Mulé, A.; Rambaldi, A. Early chemotherapy intensification with escalated BEACOPP in patients with advanced-stage Hodgkin lymphoma with a positive interim positron emission tomography/computed tomography scan after two ABVD cycles: Long-term results of the GITIL/FIL HD 0607 trial. J. Clin. Oncol. 2018, 36, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Lugtenburg, P.J.; Lyon, A.R.; Marks, R.; Luminari, S. Treatment of aggressive non-Hodgkin’s lymphoma in frail patients: Cardiac comorbidities and advanced age. Future Oncol. 2018, 15, 1197–1205. [Google Scholar] [CrossRef]
- Pugazhendhi, A.; Edison, T.N.J.I.; Velmurugan, B.K.; Jacob, J.A.; Karuppusamy, I. Toxicity of Doxorubicin (Dox) to different experimental organ systems. Life Sci. 2018, 200, 26–30. [Google Scholar] [CrossRef]
- Latina, A.; Viticchiè, G.; Lena, A.M.; Piro, M.C.; Annicchiarico-Petruzzelli, M.; Melino, G.; Candi, E. ΔNp63 targets cytoglobin to inhibit oxidative stress-induced apoptosis in keratinocytes and lung cancer. Oncogene 2016, 35, 1493. [Google Scholar] [CrossRef]
- Deng, W.; Wang, Y.; Gu, L.; Duan, B.; Cui, J.; Zhang, Y.; Du, J. MICAL1 controls cell invasive phenotype via regulating oxidative stress in breast cancer cells. BMC Cancer 2016, 16, 489. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Kamranvar, S.A.; Masucci, M.G. Oxidative stress enables Epstein–Barr virus-induced B-cell transformation by posttranscriptional regulation of viral and cellular growth-promoting factors. Oncogene 2016, 35, 3807. [Google Scholar] [CrossRef]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S. Perspectives of TrxR1-based cancer therapies. In Oxidative Stress; Sies, H., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 639–667. [Google Scholar] [CrossRef]
- Gizi, A.; Papassotiriou, I.; Apostolakou, F.; Lazaropoulou, C.; Papastamataki, M.; Kanavaki, I.; Kanavakis, E. Assessment of oxidative stress in patients with sickle cell disease: The glutathione system and the oxidant–antioxidant status. Blood Cells Mol. Dis. 2011, 46, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A.; Lu, J. Thioredoxin and thioredoxin reductase: Current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010, 396, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, G.D.; Bridge, W.J. The glutathione system and the related thiol network in Caenorhabditis elegans. Redox Biol. 2019, 24, 101171. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A. Thioredoxin and redox signaling: Roles of the thioredoxin system in control of cell fate. Arch. Biochem. Biophys. 2017, 617, 101–105. [Google Scholar] [CrossRef]
- Dóka, É.; Ida, T.; Dagnell, M.; Abiko, Y.; Luong, N.C.; Balog, N.; Nagy, P. Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 2020, 6, eaax8358. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, R.; Holmgren, A. The role of thioredoxin in the regulation of cellular processes by S-nitrosylation. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 689–700. [Google Scholar] [CrossRef]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Lu, J. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Benhar, M.; Shytaj, I.L.; Stamler, J.S.; Savarino, A. Dual targeting of the thioredoxin and glutathione systems in cancer and HIV. J. Clin. Investig. 2016, 126, 1630–1639. [Google Scholar] [CrossRef] [Green Version]
- Raninga, P.V.; di Trapani, G.; Vuckovic, S.; Bhatia, M.; Tonissen, K. Inhibition of thioredoxin 1 leads to apoptosis in drug-resistant multiple myeloma. Oncotarget 2015, 6, 15410. [Google Scholar] [CrossRef] [Green Version]
- Duan, D.; Zhang, J.; Yao, J.; Liu, Y.; Fang, J. Targeting thioredoxin reductase by parthenolide contributes to inducing apoptosis of HeLa cells. J. Biol. Chem. 2016, 291, 10021–10031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berners-Price, S.J.; Filipovska, A. Gold compounds as therapeutic agents for human diseases. Metallomics 2011, 3, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Berners-Price, S.J.; Filipovska, A. The design of gold-based, mitochondria-targeted chemotherapeutics. Aust. J. Chem. 2008, 61, 661–668. [Google Scholar] [CrossRef]
- Karaca, O.; Scalcon, V.; Meier-Menches, S.M.; Bonsignore, R.; Brouwer, J.M.; Tonolo, F.; Casini, A. Characterization of hydrophilic gold (I) N-heterocyclic carbene (NHC) complexes as potent TrxR inhibitors using biochemical and mass spectrometric approaches. Inorg. Chem. 2017, 56, 14237–14250. [Google Scholar] [CrossRef] [Green Version]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef]
- Raninga, P.V.; Lee, A.C.; Sinha, D.; Shih, Y.Y.; Mittal, D.; Makhale, A.; Khanna, K. Therapeutic cooperation between auranofin, a thioredoxin reductase inhibitor and anti-PD-L1 antibody for treatment of triple-negative breast cancer. Int. J. Cancer 2019, 146, 123–136. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, H.; Lu, J.; Holmgren, A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 2012, 287, 38210–38219. [Google Scholar] [CrossRef] [Green Version]
- Rackham, O.; Shearwood, A.M.J.; Thyer, R.; McNamara, E.; Davies, S.M.; Callus, B.A.; Filipovska, A. Substrate and inhibitor specificities differ between human cytosolic and mitochondrial thioredoxin reductases: Implications for development of specific inhibitors. Free Radic. Biol. Med. 2011, 50, 689–699. [Google Scholar] [CrossRef]
- Sze, J.H.; Raninga, P.V.; Nakamura, K.; Casey, M.; Khanna, K.K.; Berners-Price, S.J.; Tonissen, K.F. Anticancer activity of a Gold (I) phosphine thioredoxin reductase inhibitor in multiple myeloma. Redox Biol. 2020, 28, 101310. [Google Scholar] [CrossRef]
- Clapper, E.; Wang, S.; Raninga, P.V.; di Trapani, G.; Tonissen, K.F. Cross-talk between Bcr-abl and the Thioredoxin System in Chronic Myeloid Leukaemia: Implications for CML Treatment. Antioxidants 2020, 9, 207. [Google Scholar] [CrossRef]
- Mannervik, B. Measurement of glutathione reductase activity. Curr. Protoc. Toxicol. 1999, 1, 7.2. [Google Scholar] [CrossRef]
- Anderson, M.E. Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol. 1985, 113, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 1980, 106, 207–212. [Google Scholar] [CrossRef]
- Prohaska, J.R. The glutathione peroxidase activity of glutathione S-transferases. Biochim. Biophys. Acta 1980, 611, 87–98. [Google Scholar] [CrossRef]
- Rushworth, S.A.; MacEwan, D.J.; O’Connell, M.A. Lipopolysaccharide-induced expression of NAD (P) H: Quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J. Immunol. 2008, 181, 6730–6737. [Google Scholar] [CrossRef] [Green Version]
- Nair, H.B.; Sung, B.; Yadav, V.R.; Kannappan, R.; Chaturvedi, M.M.; Aggarwal, B.B. Delivery of antiinflammatory nutraceuticals by nanoparticles for the prevention and treatment of cancer. Biochem. Pharmacol. 2010, 80, 1833–1843. [Google Scholar] [CrossRef] [Green Version]
- Lafleur, M.A.; Drew, A.F.; de Sousa, E.L.; Blick, T.; Bills, M.; Walker, E.C.; Thompson, E.W. Upregulation of matrix metalloproteinases (MMPs) in breast cancer xenografts: A major induction of stromal MMP-13. Int. J. Cancer 2005, 114, 544–554. [Google Scholar] [CrossRef]
- Brune, V.; Tiacci, E.; Pfeil, I.; Döring, C.; Eckerle, S.; van Noesel, C.J.; Küppers, R. Origin and pathogenesis of nodular lymphocyte–predominant Hodgkin lymphoma as revealed by global gene expression analysis. J. Exp. Med. 2008, 205, 2251–2268. [Google Scholar] [CrossRef]
- Oncomine Database. 2008. Available online: https://www.oncomine.org/ (accessed on 31 July 2020).
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Kizek, R. Redox status expressed as GSH: GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Prasai, P.K.; Shrestha, B.; Orr, A.W.; Pattillo, C.B. Decreases in GSH: GSSG activate vascular endothelial growth factor receptor 2 (VEGFR2) in human aortic endothelial cells. Redox Biol. 2018, 19, 22–27. [Google Scholar] [CrossRef]
- Arteel, G.E.; Sies, H. The biochemistry of selenium and the glutathione system. Environ. Toxicol. Pharmacol. 2001, 10, 153–158. [Google Scholar] [CrossRef]
- Grattagliano, I.; Caraceni, P.; Portincasa, P.; Domenicali, M.; Palmieri, V.O.; Trevisani, F.; Palasciano, G. Adaptation of subcellular glutathione detoxification system to stress conditions in choline-deficient diet induced rat fatty liver. Cell Biol. Toxicol. 2003, 19, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2: INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, L.C.; Mishra, S.; Chakraborty, A.; Shekher, A.; Sharma, A.; Gupta, S.C. Oxidative Stress and Cancer Development: Are Noncoding RNAs the Missing Links? Antioxid. Redox Signal. 2020, 33, 1209–1229. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Bao, B.; Mitrea, C.; Wijesinghe, P.; Marchetti, L.; Girsch, E.; Farr, R.L.; Bollig-Fischer, A. Treating triple negative breast cancer cells with erlotinib plus a select antioxidant overcomes drug resistance by targeting cancer cell heterogeneity. Sci. Rep. 2017, 7, 44125. [Google Scholar] [CrossRef] [Green Version]
- Colla, R.; Izzotti, A.; de Ciucis, C.; Fenoglio, D.; Ravera, S.; Speciale, A.; Marengo, B. Glutathione-mediated antioxidant response and aerobic metabolism: Two crucial factors involved in determining the multi-drug resistance of high-risk neuroblastoma. Oncotarget 2016, 7, 70715–70737. [Google Scholar] [CrossRef] [Green Version]
- Gourzones, C.; Bellanger, C.; Lamure, S.; Gadacha, O.K.; de Paco, E.G.; Vincent, L.; Moreaux, J. Antioxidant defenses confer resistance to high dose melphalan in multiple myeloma cells. Cancers 2019, 11, 439. [Google Scholar] [CrossRef] [Green Version]
- Shan, W.; Zhong, W.; Zhao, R.; Oberley, T.D. Thioredoxin 1 as a subcellular biomarker of redox imbalance in human prostate cancer progression. Free Radic. Biol. Med. 2010, 49, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Nipp, M.; Elsner, M.; Balluff, B.; Meding, S.; Sarioglu, H.; Ueffing, M.; Zitzelsberger, H. S100-A10, thioredoxin, and S100-A6 as biomarkers of papillary thyroid carcinoma with lymph node metastasis identified by MALDI imaging. J. Mol. Med. 2012, 90, 163–174. [Google Scholar] [CrossRef]
- Cox, A.G.; Brown, K.K.; Arner, E.S.; Hampton, M.B. The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak-dependent process that involves peroxiredoxin 3 oxidation. Biochem. Pharmacol. 2008, 76, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Du, Y.; Zhang, X.; Lu, J.; Holmgren, A. Glutaredoxin 2 reduces both thioredoxin 2 and thioredoxin 1 and protects cells from apoptosis induced by auranofin and 4-hydroxynonenal. Antioxid. Redox Signal. 2014, 21, 669–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Shi, X.; Zhao, C.; Li, X.; Lan, X.; Liu, S.; Liu, J. Anti-rheumatic agent auranofin induced apoptosis in chronic myeloid leukemia cells resistant to imatinib through both Bcr/Abl-dependent and-independent mechanisms. Oncotarget 2014, 5, 9118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saei, A.A.; Gullberg, H.; Sabatier, P.; Beusch, C.M.; Johansson, K.; Lundgren, B.; Zubarev, R.A. Comprehensive chemical proteomics for target deconvolution of the redox active drug auranofin. Redox Biol. 2020, 32, 101491. [Google Scholar] [CrossRef]
- Rackham, O.; Nichols, S.J.; Leedman, P.J.; Berners-Price, S.J.; Filipovska, A. A gold (I) phosphine complex selectively induces apoptosis in breast cancer cells: Implications for anticancer therapeutics targeted to mitochondria. Biochem. Pharmacol. 2007, 74, 992–1002. [Google Scholar] [CrossRef]
- Hondal, R.J.; Marino, S.M.; Gladyshev, V.N. Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid. Redox Signal. 2013, 18, 1675–1689. [Google Scholar] [CrossRef] [Green Version]
- Berners-Price, S.J.; Bowen, R.J.; Fernandes, M.A.; Layh, M.; Lesueur, W.J.; Mahepal, S.; van Rensburg, C.E. Gold(I) and silver(I) complexes of 2,3-bis(diphenylphosphino)maleic acid: Structural studies and antitumour activity. Inorg. Chim. Acta 2005, 358, 4237–4246. [Google Scholar] [CrossRef]
- Arnér, E.S. Selenoproteins—What unique properties can arise with selenocysteine in place of cysteine? Exp. Cell Res. 2010, 316, 1296–1303. [Google Scholar] [CrossRef]
- Jia, J.J.; Geng, W.S.; Wang, Z.Q.; Chen, L.; Zeng, X.S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Peroja, P.; Pasanen, A.K.; Haapasaari, K.M.; Jantunen, E.; Soini, Y.; Turpeenniemi-Hujanen, T.; Karihtala, P. Oxidative stress and redox state-regulating enzymes have prognostic relevance in diffuse large B-cell lymphoma. Exp. Hematol. Oncol. 2012, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Pasanen, A.K.; Kuitunen, H.; Haapasaari, K.M.; Karihtala, P.; Kyllönen, H.; Soini, Y.; Kuittinen, O. Expression and prognostic evaluation of oxidative stress markers in an immunohistochemical study of B-cell derived lymphomas. Leuk. Lymphoma 2012, 53, 624–631. [Google Scholar] [CrossRef]
- Gustafson, H.L.; Yao, S.; Goldman, B.H.; Lee, K.; Spier, C.M.; le Blanc, M.L.; Briehl, M.M. Genetic polymorphisms in oxidative stress-related genes are associated with outcomes following treatment for aggressive B-cell non-Hodgkin lymphoma. Am. J. Hematol. 2014, 89, 639–645. [Google Scholar] [CrossRef] [Green Version]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Mak, T.W. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.K.; Schneider, M.; Kölle, P.; Kuhlencordt, P.; Förster, H.; Beck, H.; Conrad, M. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 2010, 70, 9505–9514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledano, M.B.; Delaunay-Moisan, A.; Outten, C.E.; Igbaria, A. Functions and cellular compartmentation of the thioredoxin and glutathione pathways in yeast. Antioxid. Redox Signal. 2013, 18, 1699–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Zhang, H.; Zhang, X.; Lu, J.; Holmgren, A. Thioredoxin 1 is inactivated due to oxidation induced by peroxiredoxin under oxidative stress and reactivated by the glutaredoxin system. J. Biol. Chem. 2013, 288, 32241–32247. [Google Scholar] [CrossRef] [Green Version]
- Muri, J.; Thut, H.; Heer, S.; Krueger, C.C.; Bornkamm, G.W.; Bachmann, M.F.; Kopf, M. The thioredoxin-1 and glutathione/glutaredoxin-1 systems redundantly fuel murine B-cell development and responses. Eur. J. Immunol. 2019, 49, 709–723. [Google Scholar] [CrossRef]
- Berkholz, D.S.; Faber, H.R.; Savvides, S.N.; Karplus, P.A. Catalytic cycle of human glutathione reductase near 1 Å resolution. J. Mol. Biol. 2008, 382, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Benhar, M. Roles of mammalian glutathione peroxidase and thioredoxin reductase enzymes in the cellular response to nitrosative stress. Free Radic. Biol. Med. 2018, 127, 160–164. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Wrzaczek, M.; Brosché, M.; Kangasjärvi, J. ROS signaling loops—production, perception, regulation. Curr. Opin. Plant Biol. 2013, 16, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, H.; Tada-Oikawa, S.; Hiraku, Y.; Kojima, M.; Kawanishi, S. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci. 2005, 76, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Jenner, A.; Bruchelt, G.; Niethammer, D.; Halliwell, B. Effect of concentration on the cytotoxic mechanism of doxorubicin—apoptosis and oxidative DNA damage. Biochem. Biophys. Res. Commun. 1997, 230, 254–257. [Google Scholar] [CrossRef]
- Cappetta, D.; de Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Urbanek, K. Oxidative stress and cellular response to doxorubicin: A common factor in the complex milieu of anthracycline cardiotoxicity. Oxidative Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Zou, X.; Feng, Z.; Li, Y.; Wang, Y.; Wertz, K.; Weber, P.; Liu, J. Stimulation of GSH synthesis to prevent oxidative stress-induced apoptosis by hydroxytyrosol in human retinal pigment epithelial cells: Activation of Nrf2 and JNK–p62/SQSTM1 pathways. J. Nutr. Biochem. 2012, 23, 994–1006. [Google Scholar] [CrossRef]
- Lu, J.; Chew, E.H.; Holmgren, A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. USA 2007, 104, 12288–12293. [Google Scholar] [CrossRef] [Green Version]
- Habermann, K.J.; Grünewald, L.; van Wijk, S.; Fulda, S. Targeting redox homeostasis in rhabdomyosarcoma cells: GSH–depleting agents enhance auranofin–induced cell death. Cell Death Dis. 2017, 8, e3067. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Qiu, H.; Xu, J.; Mo, J.; Liu, Y.; Gui, Y.; Gan, Z. Expression and prognostic potential of GPX1 in human cancers based on data mining. Ann. Transl. Med. 2020, 8, 124. [Google Scholar] [CrossRef]
- Kinowaki, Y.; Kurata, M.; Ishibashi, S.; Ikeda, M.; Tatsuzawa, A.; Yamamoto, M.; Yamamoto, K. Glutathione peroxidase 4 overexpression inhibits ROS–induced cell death in diffuse large B–cell lymphoma. Lab. Investig. 2018, 98, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Reinke, E.N.; Ekoue, D.N.; Bera, S.; Mahmud, N.; Diamond, A.M. Translational regulation of GPx–1 and GPx–4 by the mTOR pathway. PLoS ONE 2014, 9, e93472. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase–1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Hu, J.; Wu, S.; Wang, L.; Cao, X.; Zhang, X.; Fang, B. Auranofin–mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non–small cell lung cancer cells. Oncotarget 2016, 7, 3548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014, 4, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.W.; Lee, S.H.; Woo, G.H.; Kwon, H.J.; Kim, D.Y. Downregulation of TXNIP leads to high proliferative activity and estrogen–dependent cell growth in breast cancer. Biochem. Biophys. Res. Commun. 2018, 498, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, M.J.; Kim, D.O.; Kim, W.S.; Yoon, S.J.; Park, Y.J.; Choi, I. TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metab. 2013, 18, 75–85. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | * Accession Number | Forward | Reverse |
|---|---|---|---|
| L32 | NC_000003.12 | 5′CAGGGTTCGTAGAAGATTCAA GGG 3′ | 5′CTTGGAGGAAAACATTGTGAGCGATC 3′ |
| Trx | NC_000009.12 | 5′GC AGTTTATAAAGGGAGAGAGCA 3′ | 5′TGATCATTTTGCAAGGCCCA 3′ |
| TrxR | NC_000012.12 | 5′GGAATCCACCCTGTCTCTGC 3′ | 5′ACGAGCCAGTGG TTTGCAGT 3′ |
| TXNIP | NC_000001.11 | 5′GGCACCTGTGTCTGCTAAAA 3′ | 5′CGGGAACATGTATTCTCAAA 3′ |
| GR | NC_000008.11 | 5′GCTGCTGGCCGAAAACTTG 3′ | 5′GAATGGCTTCATCTTCCGTGA 3′ |
| Grx | NC_000005.10 | 5′AACCACACTAACGAGATTCAAGAT 3′ | 5′AGAGACTAGATCACTGCATCCGC 3′ |
| Gpx1 | NC_000003.12 | 5′CAGTTTGGGCATCAGGAGAAC 3′ | 5′TCATAAGCGCGGTGGCGT 3′ |
| Gpx4 | NC_000019.10 | 5′AACGTGGCCTCCCAGTGAG 3′ | 5′GCTTCCCGAACTGGTTACACG 3′ |
| Nrf-2 | NC_000002.12 | 5′GCTCAGTTACACTAGATGAAGAGACA 3′ | 5′CAGTCATCAAAGTACAAAGCATCT 3′ |
| HL | DLBCL | ||||
|---|---|---|---|---|---|
| Gene Name | Fold Change | p-Value | Gene Name | Fold Change | p-Value |
| TXN * | 4.871 | 3.31 × 10−10 | TXN * | 3.108 | 8.72 × 10−13 |
| TXN2 | 1.127 | 0.003 | TXN2 | 1.126 | 8.88 × 10−4 |
| TXNRD1 | 1.178 | 0.004 | TXNRD1 | 1.44 | 0.02 |
| TXNRD2 | 1.076 | 0.127 | TXNRD2 | 1.086 | 0.01 |
| TXNRD3 | 1.085 | 0.059 | TXNRD3 | 1.232 | 0.011 |
| TXNIP | −1.038 | 0.659 | TXNIP | −1.069 | 0.75 |
| GPX1 * | 5.107 | 1.12 × 10−7 | GPX1 * | 5.757 | 4.88 × 10−8 |
| GPX2 | 1.123 | 0.012 | GPX2 | 1.059 | 0.11 |
| GPX3 | 1.423 | 0.039 | GPX3 | 1.483 | 0.016 |
| GPX4 * | 2.961 | 8.16 × 10−6 | GPX4 * | 3.642 | 2.24 × 10−7 |
| GPX5 | 1.027 | 0.239 | GPX5 | −1.055 | 0.898 |
| GPX7 | −1.389 | 1 | GPX7 | 1.481 | 0.021 |
| GLRX | −1.107 | 0.726 | GLRX | 1.388 | 0.107 |
| GLRX2 * | 1.926 | 7.52 × 10−6 | GLRX2 * | 1.899 | 6.03 × 10−6 |
| GLRX3 | 1.339 | 0.0012 | GLRX3 * | 1.616 | 1.35 × 10−6 |
| GLRX5 | 1.212 | 0.121 | GLRX5 | 2.164 | 9.98 × 10−6 |
| GSR | −1.074 | 0.862 | GSR | 1.247 | 0.005 |
| NRE2L2 ^ | 1.188 | 0.003 | NRE2L2 | 1.203 | 1.90 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Lu, Y.; Woods, K.; Di Trapani, G.; Tonissen, K.F. Investigating the Thioredoxin and Glutathione Systems’ Response in Lymphoma Cells after Treatment with [Au(d2pype)2]Cl. Antioxidants 2021, 10, 104. https://doi.org/10.3390/antiox10010104
Wang S, Lu Y, Woods K, Di Trapani G, Tonissen KF. Investigating the Thioredoxin and Glutathione Systems’ Response in Lymphoma Cells after Treatment with [Au(d2pype)2]Cl. Antioxidants. 2021; 10(1):104. https://doi.org/10.3390/antiox10010104
Chicago/Turabian StyleWang, Sicong, Yaoying Lu, Kyra Woods, Giovanna Di Trapani, and Kathryn F. Tonissen. 2021. "Investigating the Thioredoxin and Glutathione Systems’ Response in Lymphoma Cells after Treatment with [Au(d2pype)2]Cl" Antioxidants 10, no. 1: 104. https://doi.org/10.3390/antiox10010104