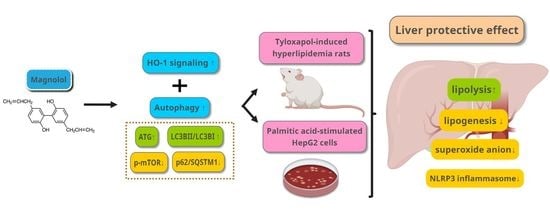
Involvement of HO-1 and Autophagy in the Protective Effect of Magnolol in Hepatic Steatosis-Induced NLRP3 Inflammasome Activation In Vivo and In Vitro



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animal Experimentation
2.2. Cell Culture and Palmitate-Induced Steatosis
2.3. Cell Viability of MG
2.4. Oil Red O Staining
2.5. Measurement of Triglyceride, Total Cholesterol, and IL-1β Levels in Tylo-Injected Rats and PA-Stimulated HepG2 Cells
2.6. Detection of Superoxide Formation in the Liver of Tylo-injected Rats and Reactive Oxygen Species Formation in PA-Stimulated HepG2 Cells
2.7. Western Blot Analysis
2.8. RNA Isolation and Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
2.9. Statistical Analysis
3. Results
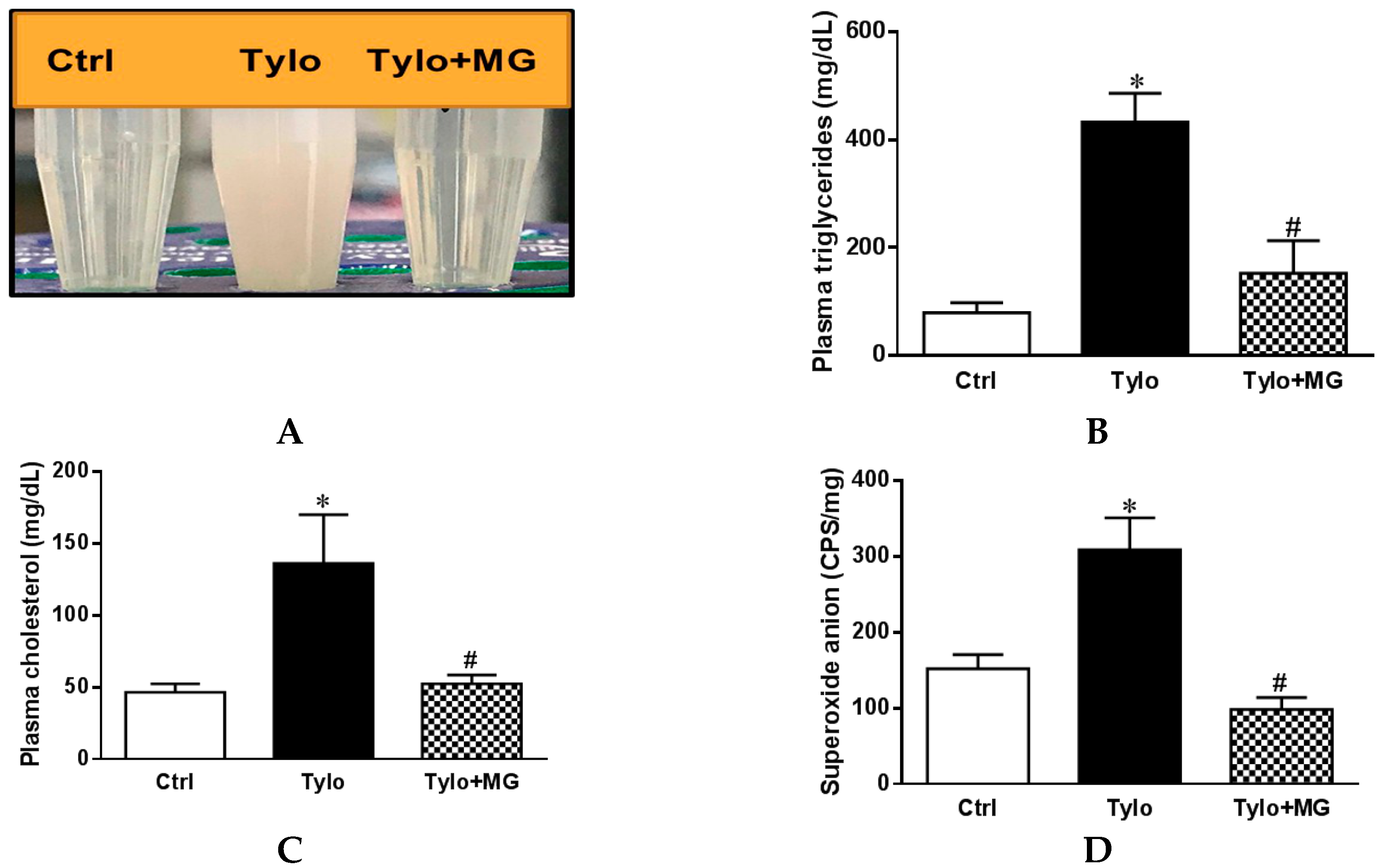
3.1. MG Alleviated Hyperlipidemia and ROS Formation in the Liver of Tylo-Injected Rats
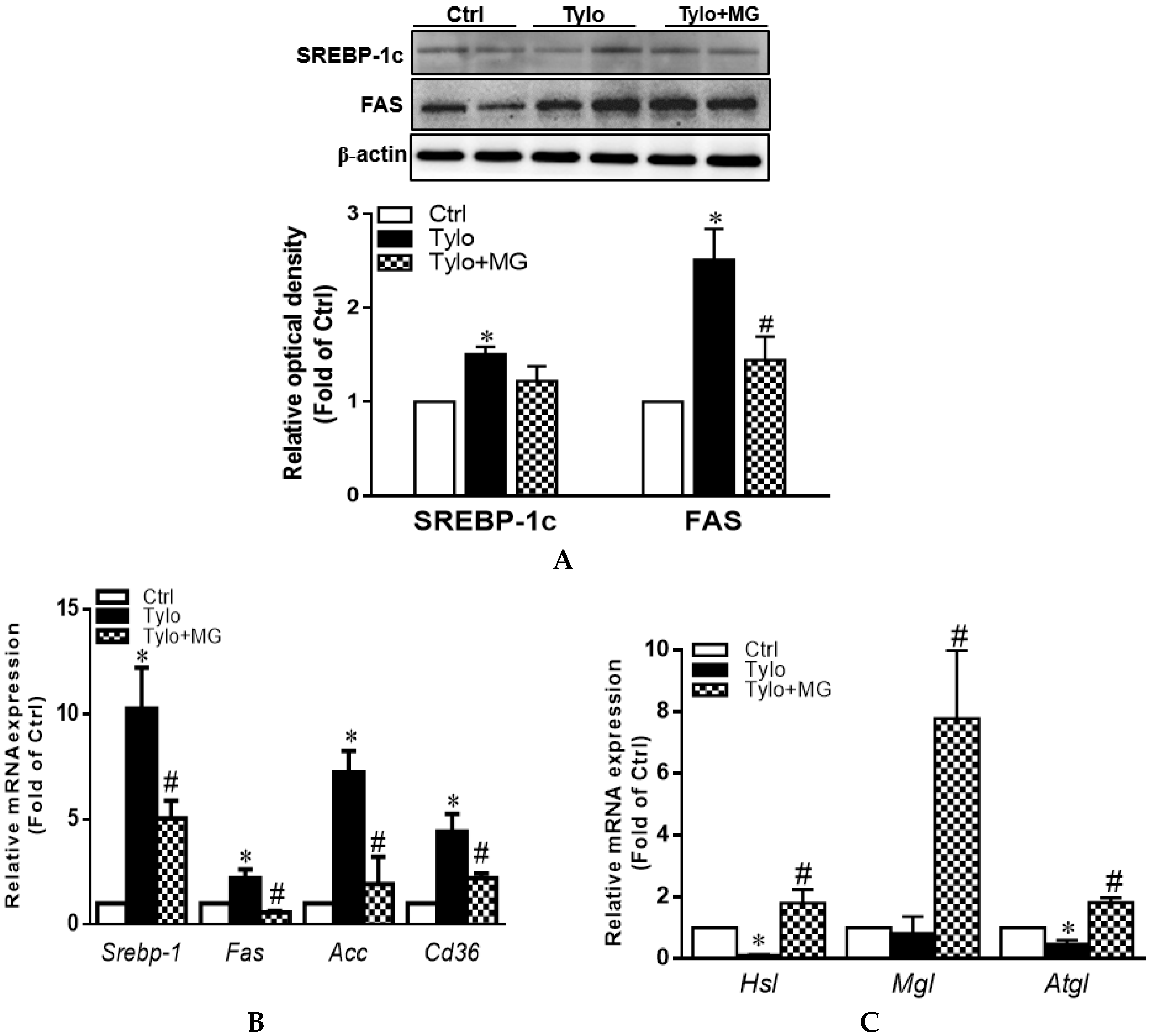
3.2. Effect of MG Treatment on Lipogenesis and Lipolysis Profiles in the Liver of Tylo-induced Hyperlipidemia in Rats
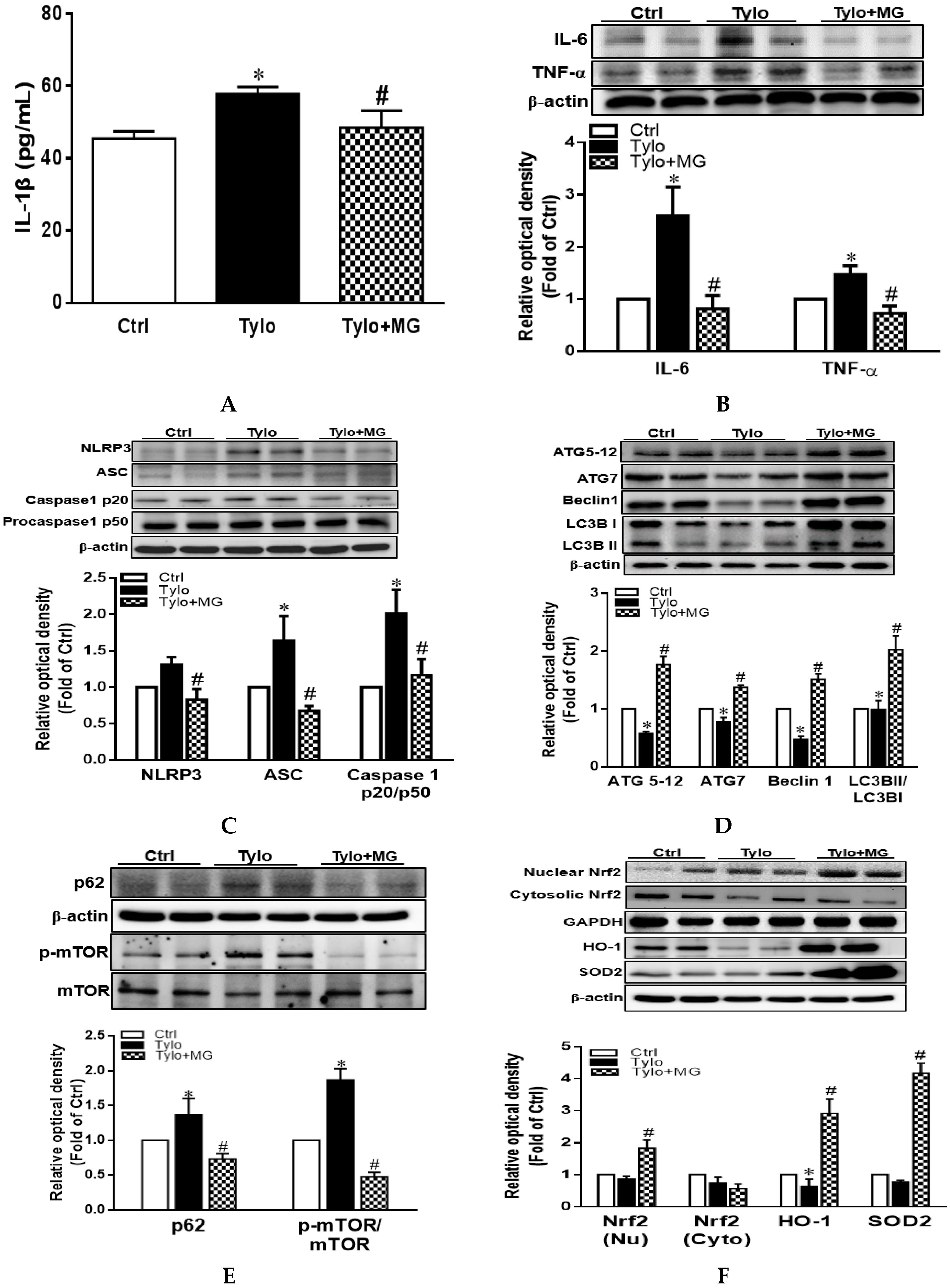
3.3. MG Attenuated Proinflammatory Cytokines and NLRP3 Inflammasome Activation in Tyloxapol-Induced Hyperlipidemia in Rats
3.4. MG Induced Hepatic Autophagy Activation and Nrf2/HO-1 Signaling in Tyloxapol-Induced Hyperlipidemia in Rats
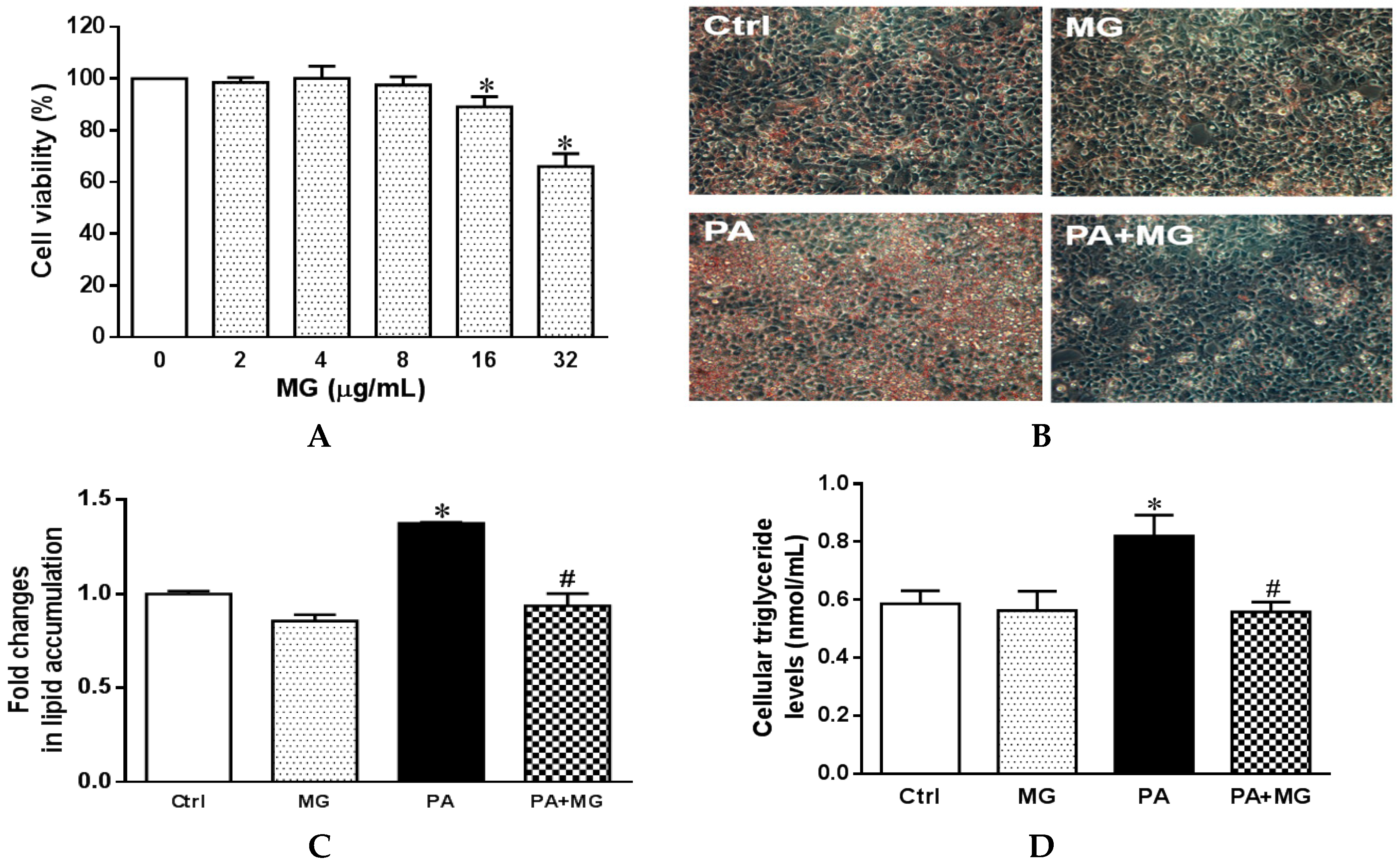
3.5. Cell Viability of HepG2 Cells Treated with MG
3.6. MG Alleviated Steatosis in PA-Stimulated Hepatocytes
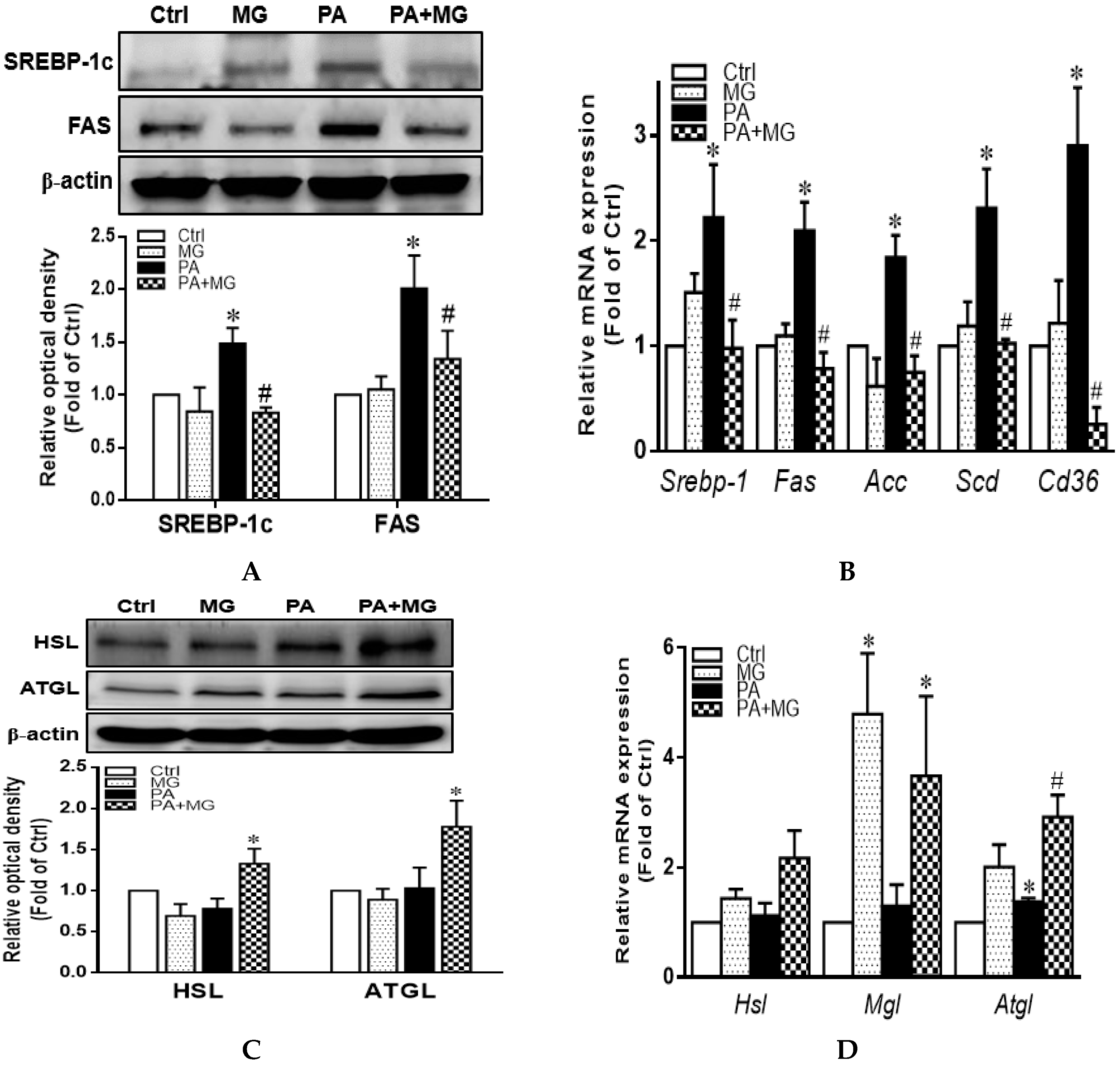
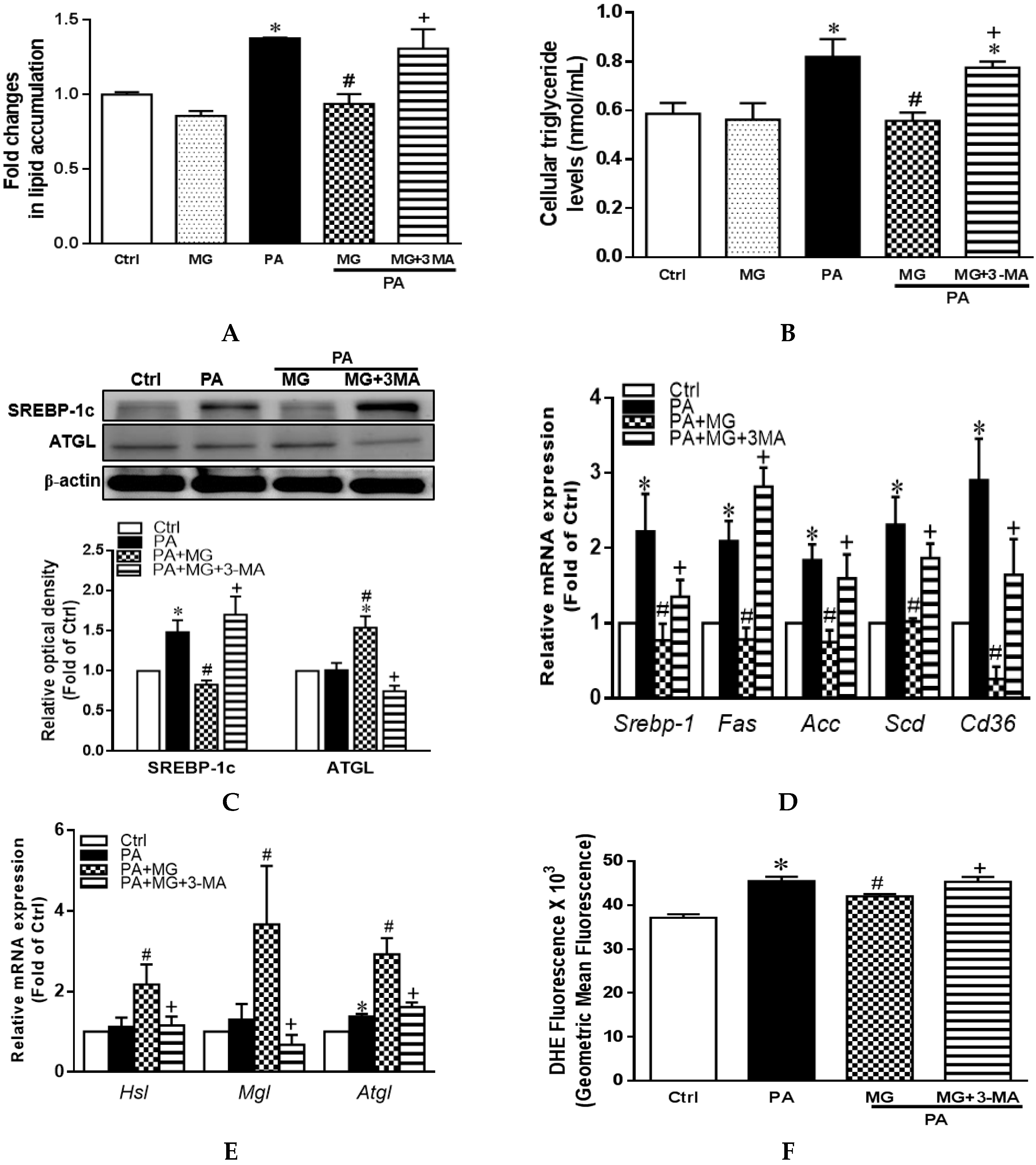
3.7. MG Regulated Lipid Homeostasis in PA-Stimulated HepG2 Cells
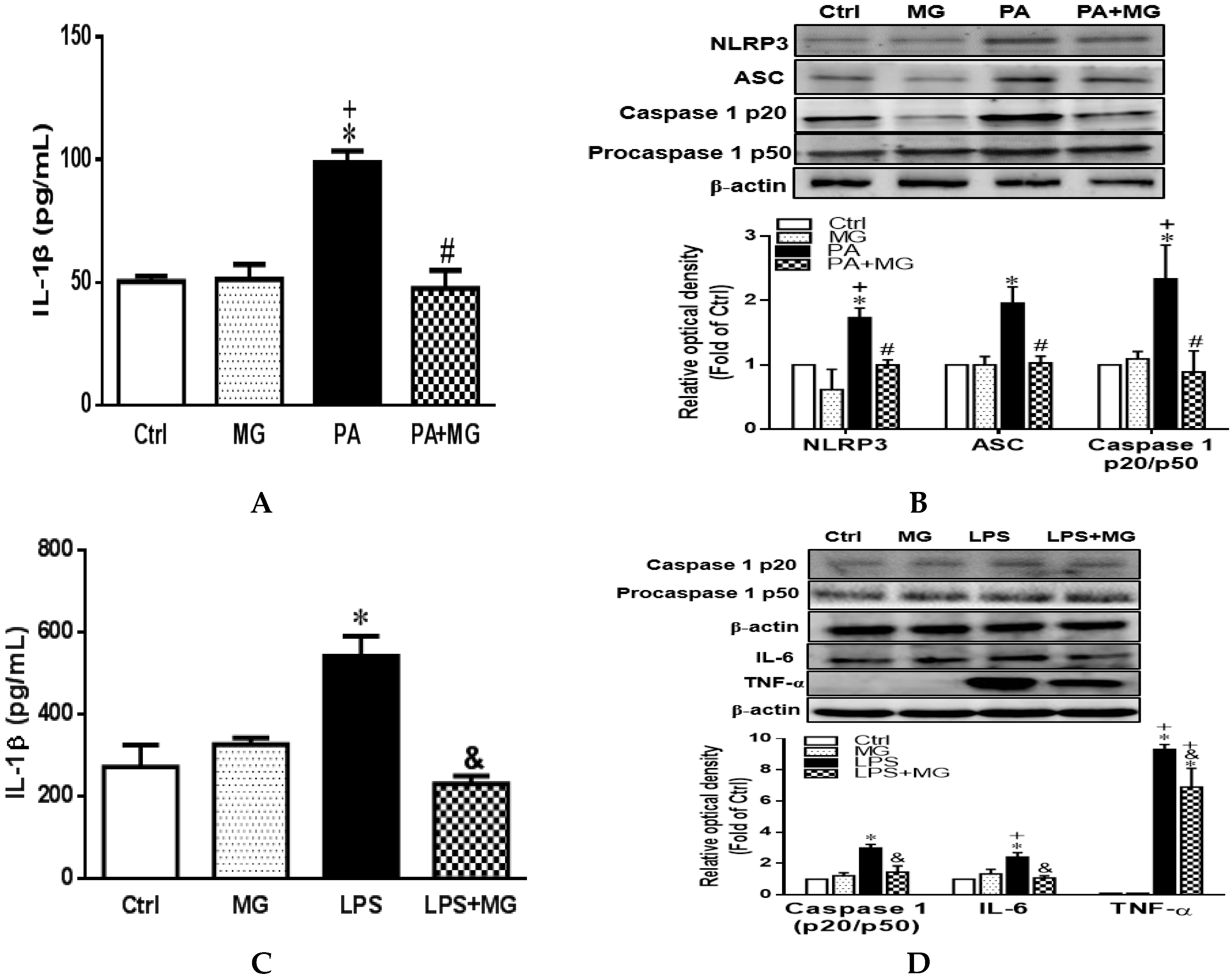
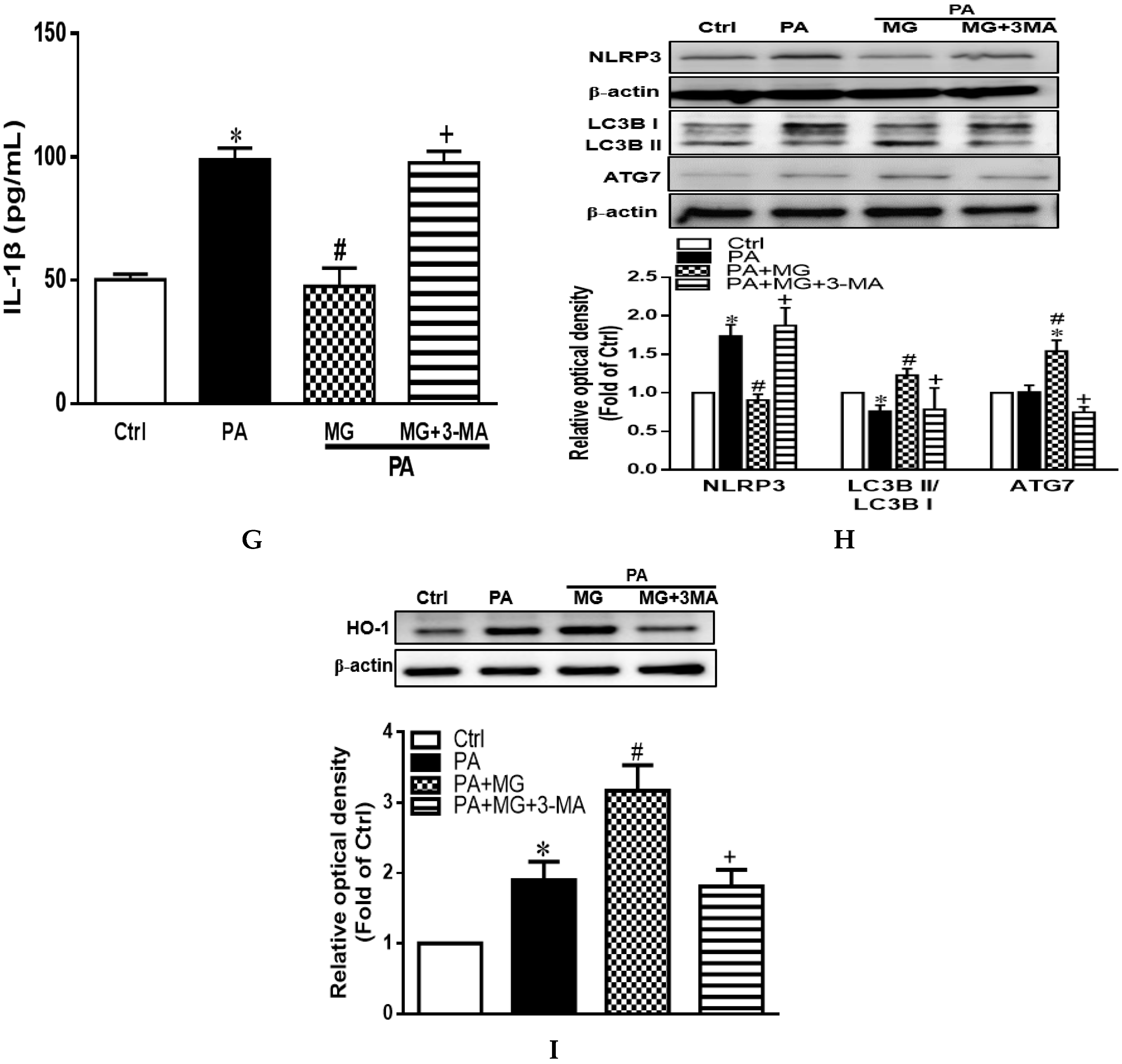
3.8. MG Attenuated IL-1β Secretion and NLRP3 Inflammasome Activation in PA-Induced Steatotic HepG2 Cells
3.9. MG Attenuated NLRP3 Inflammasome and Proinflammatory Cytokine in LPS-Stimulated HepG2 Cells
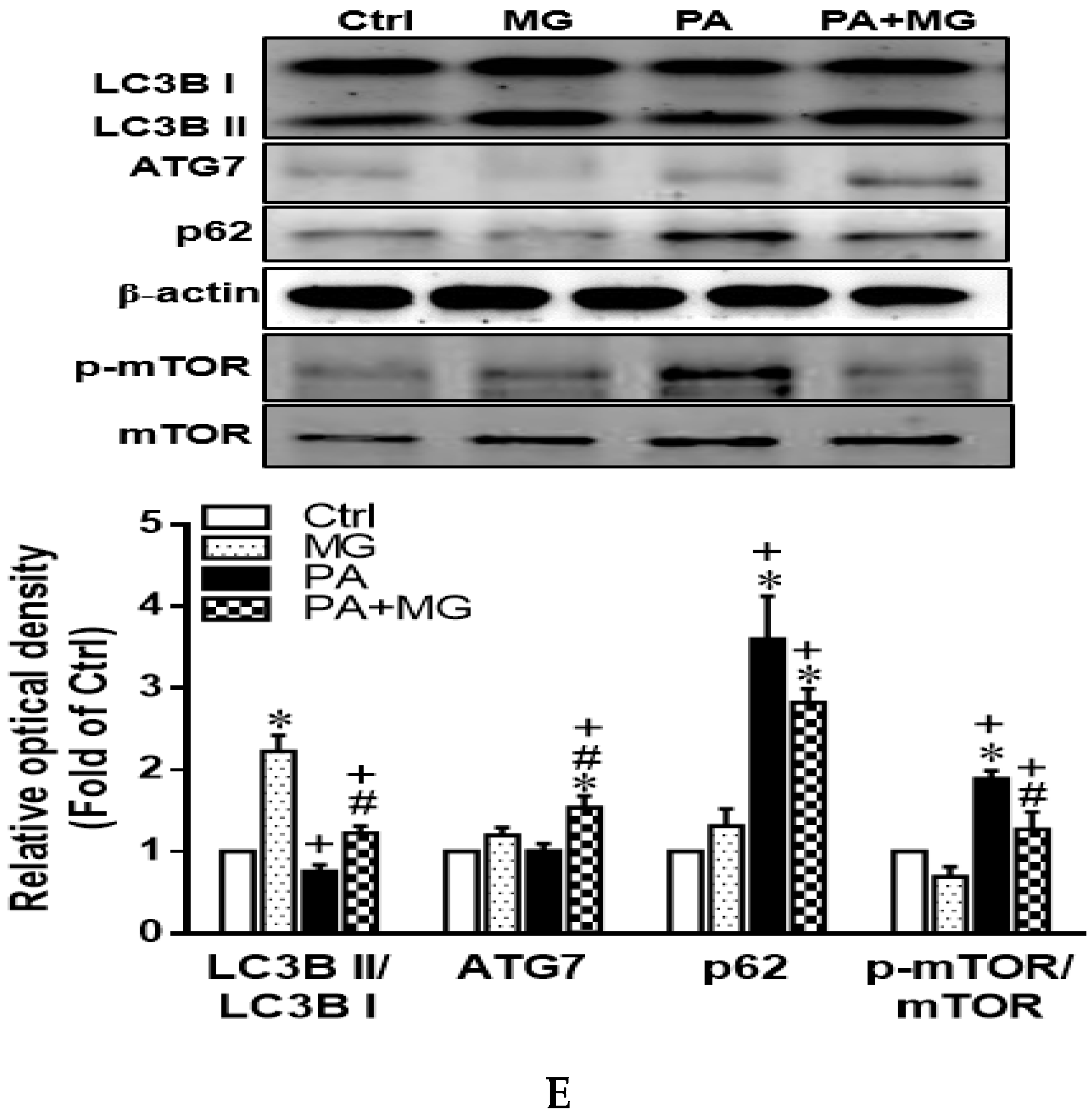
3.10. Activation of Autophagy Contributed to the Protective Effects of MG against PA-Induced Lipotoxicity in Hepatocytes
3.11. MG-Induced Inhibition of Lipid Accumulation, Triglyceride, and NLRP3 Inflammasome and Induction of HO-1 Depend on Autophagy Induction
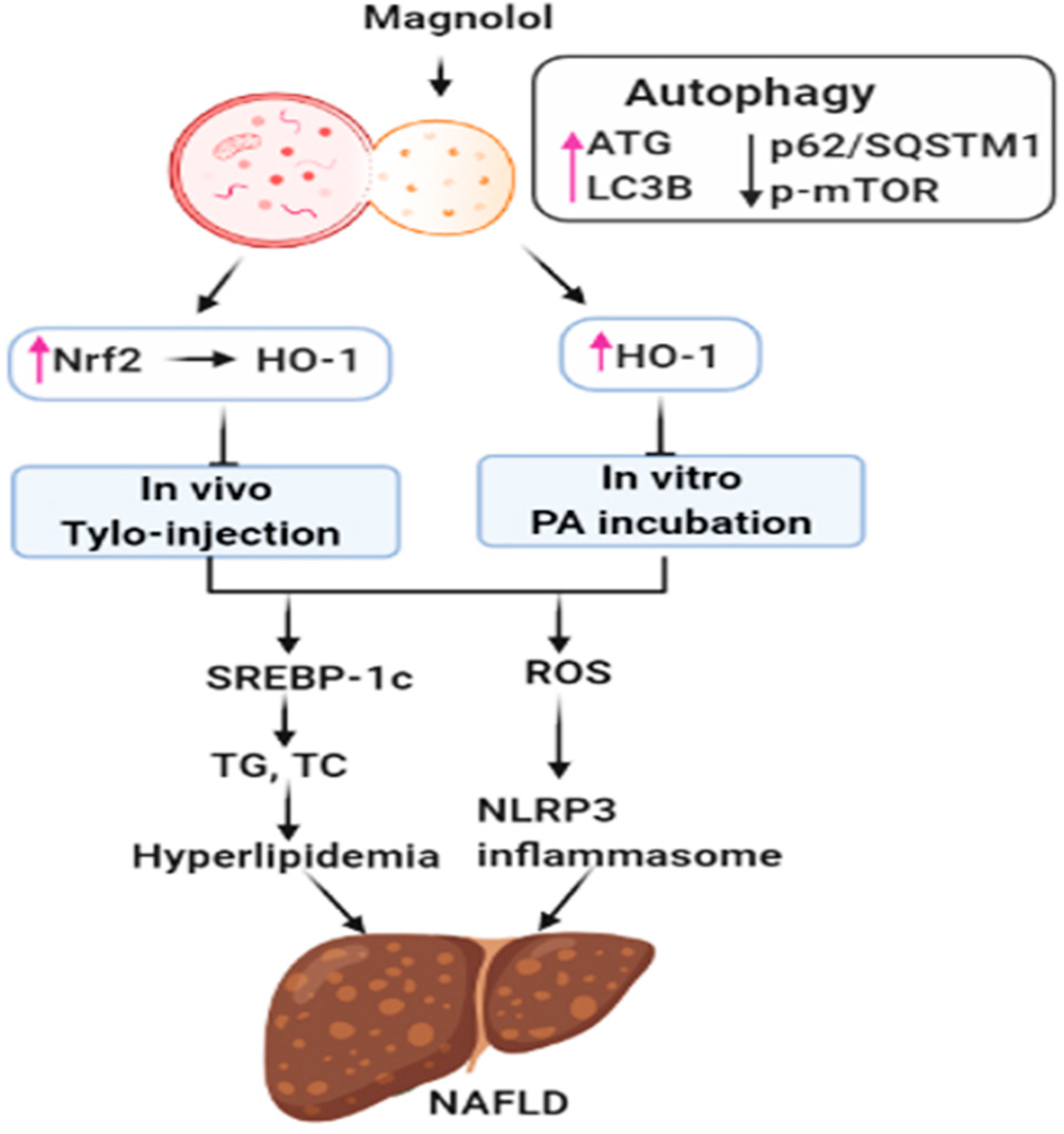
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Greenberg, M.; Glick, Y.; Bellner, L.; Favero, G.; Rezzani, R.; Rodella, L.F.; Agostinucci, K.; Shapiro, J.I.; Abraham, N.G. Adipocyte Specific HO-1 Gene Therapy is Effective in Antioxidant Treatment of Insulin Resistance and Vascular Function in an Obese Mice Model. Antioxidants 2020, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Abraham, N.G.; Junge, J.M.; Drummond, G.S. Translational Significance of Heme Oxygenase in Obesity and Metabolic Syndrome. Trends Pharmacol. Sci. 2016, 37, 17–36. [Google Scholar] [CrossRef] [Green Version]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Wree, A.; McGeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.-Z.; Xu, Z.-Q.; Han, B.-Z.; Su, D.-F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Ralston, J.C.; Lyons, C.L.; Kennedy, E.B.; Kirwan, A.M.; Roche, H.M. Fatty Acids and NLRP3 Inflammasome-Mediated Inflammation in Metabolic Tissues. Annu. Rev. Nutr. 2017, 37, 77–102. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, S.M. Medicine, NLRP3 inflammasome activation in D-galactosamine and lipopolysaccharide-induced acute liver failure: Role of heme oxygenase-1. Free Radic. Biol. Med. 2013, 65, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J.J.N. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.J.; Xie, G.; Zhang, N.; Li, Y. Inhibiting autophagy promotes endoplasmic reticulum stress and the ROSinduced nodlike receptor 3dependent proinflammatory response in HepG2 cells. Mol. Med. Rep. 2016, 14, 3999–4007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licari, M.; Raffaele, M.; Rosman, Z.F.; Schragenheim, J.; Bellner, L.; Vanella, L.; Arad, M. Beneficial Effects of Thymoquinone on Metabolic Function and Fatty Liver in a Murine Model of Obesity. J. Nutr. Food Sci. 2019, 9, 751. [Google Scholar]
- Shen, H.H.; Peterson, S.J.; Bellner, L.; Choudhary, A.; Levy, L.; Gancz, L.; Sasson, A.; Trainer, J.; Rezzani, R.; Resnick, A.; et al. Cold-Pressed Nigella Sativa Oil Standardized to 3% Thymoquinone Potentiates Omega-3 Protection against Obesity-Induced Oxidative Stress, Inflammation, and Markers of Insulin Resistance Accompanied with Conversion of White to Beige Fat in Mice. Antioxidants 2020, 9, 489. [Google Scholar] [CrossRef]
- Cheng, A.S.; Cheng, Y.H.; Chiou, C.H.; Chang, T.L. Resveratrol upregulates Nrf2 expression to attenuate methylglyoxal-induced insulin resistance in Hep G2 cells. J. Agric. Food Chem. 2012, 60, 9180–9187. [Google Scholar] [CrossRef]
- Raffaele, M.; Licari, M.; Amin, S.; Alex, R.; Shen, H.H.; Singh, S.P.; Vanella, L.; Rezzani, R.; Bonomini, F.; Peterson, S.J.; et al. Cold Press Pomegranate Seed Oil Attenuates Dietary-Obesity Induced Hepatic Steatosis and Fibrosis through Antioxidant and Mitochondrial Pathways in Obese Mice. Int. J. Mol. Sci. 2020, 21, 5469. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, Z.; Huang, X.; Shi, W.; Zhang, R.; Chen, M.; Huang, H.; Wu, L. Insights on the Multifunctional Activities of Magnolol. BioMed Res. Int. 2019, 2019, 1847130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Feng, H.; Han, L.; Wu, L.; Lv, H.; Shen, B.; Li, Z.; Zhang, Q.; Liu, G. Magnolol Alleviates Inflammatory Responses and Lipid Accumulation by AMP-Activated Protein Kinase-Dependent Peroxisome Proliferator-Activated Receptor α Activation. Front. Immunol. 2018, 9, 147. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.J.; Lee, C.W.; Sung, H.C.; Chen, Y.H.; Wang, S.H.; Wu, P.J.; Chiang, Y.C.; Tsai, J.S.; Wu, C.C.; Li, C.Y.; et al. Magnolol reduced TNF-α-induced vascular cell adhesion molecule-1 expression in endothelial cells via JNK/p38 and NF-κB signaling pathways. Am. J. Chin. Med. 2014, 42, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Kim, C.S.; Kim, Y.S.; Jung, D.H.; Jang, D.S.; Lee, Y.M.; Kim, J.S. Effects of magnolol (5,5′-diallyl-2,2′-dihydroxybiphenyl) on diabetic nephropathy in type 2 diabetic Goto-Kakizaki rats. Life Sci. 2007, 80, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Cha, B.Y.; Lee, Y.S.; Yonezawa, T.; Teruya, T.; Nagai, K.; Woo, J.T. Magnolol enhances adipocyte differentiation and glucose uptake in 3T3-L1 cells. Life Sci. 2009, 84, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Variya, B.C.; Bakrania, A.K.; Chen, Y.; Han, J.; Patel, S.S. Suppression of abdominal fat and anti-hyperlipidemic potential of Emblica officinalis: Upregulation of PPARs and identification of active moiety. Biomed. Pharmacother. 2018, 108, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Kawanabe, Y.; Shinozaki, F.; Sato, S.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Sueishi, M. Triglyceride is strongly associated with nonalcoholic fatty liver disease among markers of hyperlipidemia and diabetes. Biomed. Rep. 2014, 2, 633–636. [Google Scholar] [CrossRef]
- Bahirat, U.A.; Shenoy, R.R.; Goel, R.N.; Nemmani, K.V. APD668, a G protein-coupled receptor 119 agonist improves fat tolerance and attenuates fatty liver in high-trans fat diet induced steatohepatitis model in C57BL/6 mice. Eur. J. Pharmacol. 2017, 801, 35–45. [Google Scholar] [CrossRef]
- Shen, B.; Zhao, C.; Wang, Y.; Peng, Y.; Cheng, J.; Li, Z.; Wu, L.; Jin, M.; Feng, H. Aucubin inhibited lipid accumulation and oxidative stress via Nrf2/HO-1 and AMPK signalling pathways. J. Cell. Mol. Med. 2019, 23, 4063–4075. [Google Scholar] [CrossRef] [Green Version]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Yang, G.; Lee, H.E.; Lee, J.Y. A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a mouse model induced by high fat diet. Sci. Rep. 2016, 6, 24399. [Google Scholar] [PubMed]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [PubMed] [Green Version]
- Gong, P.; Cederbaum, A.I. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology 2006, 43, 144–153. [Google Scholar]
- Yao, H.R.; Liu, J.; Plumeri, D.; Cao, Y.B.; He, T.; Lin, L.; Li, Y.; Jiang, Y.Y.; Li, J.; Shang, J. Lipotoxicity in HepG2 cells triggered by free fatty acids. Am. J. Transl. Res. 2011, 3, 284–291. [Google Scholar]
- Wesolowski, S.R.; Kasmi, K.C.; Jonscher, K.R.; Friedman, J.E. Developmental origins of NAFLD: A womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 81–96. [Google Scholar]
- Rempel, V.; Fuchs, A.; Hinz, S.; Karcz, T.; Lehr, M.; Koetter, U.; Müller, C.E. Magnolia Extract, Magnolol, and Metabolites: Activation of Cannabinoid CB2 Receptors and Blockade of the Related GPR55. ACS Med. Chem. Lett. 2013, 4, 41–45. [Google Scholar]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar]
- Ogawa, Y.; Imajo, K.; Honda, Y.; Kessoku, T.; Tomeno, W.; Kato, S.; Fujita, K.; Yoneda, M.; Saito, S.; Saigusa, Y.; et al. Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci. Rep. 2018, 8, 11365. [Google Scholar]
- Singh, S.P.; Grant, I.; Meissner, A.; Kappas, A.; Abraham, N.G. Ablation of adipose-HO-1 expression increases white fat over beige fat through inhibition of mitochondrial fusion and of PGC1α in female mice. Horm. Mol. Biol. Clin. Investig. 2017, 31. [Google Scholar] [CrossRef]
- Bauer, M.; Bauer, I. Heme oxygenase-1: Redox regulation and role in the hepatic response to oxidative stress. Antioxid. Redox Signal. 2002, 4, 749–758. [Google Scholar] [CrossRef]
- Peterson, S.J.; Rubinstein, R.; Faroqui, M.; Raza, A.; Boumaza, I.; Zhang, Y.; Stec, D.; Abraham, N.G. Positive Effects of Heme Oxygenase Upregulation on Adiposity and Vascular Dysfunction: Gene Targeting vs. Pharmacologic Therapy. Int. J. Mol. Sci. 2019, 20, 2514. [Google Scholar]
- Drummond, G.S.; Baum, J.; Greenberg, M.; Lewis, D.; Abraham, N.G. HO-1 overexpression and underexpression: Clinical implications. Arch. Biochem. Biophys. 2019, 673, 108073. [Google Scholar] [CrossRef]
- Lu, S.H.; Hsu, W.L.; Chen, T.H.; Chou, T.C. Activation of Nrf2/HO-1signaling pathway involves the anti-inflammatory activity of magnolol in Porphyromonas gingivalis lipopolysaccharide-stimulated mouse RAW 264.7 macrophages. Int. Immunopharmacol. 2015, 29, 770–778. [Google Scholar]
- Brunt, E.M.; Wong, V.W.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers 2015, 1, 15080. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.J.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Dong, L.; Lin, X.; Li, J. Relevance of the NLRP3 Inflammasome in the Pathogenesis of Chronic Liver Disease. Front. Immunol. 2017, 8, 1728. [Google Scholar] [CrossRef] [Green Version]
- Kagan, J.C.; Horng, T. NLRP3 inflammasome activation: CD36 serves double duty. Nat. Immunol. 2013, 14, 772–774. [Google Scholar] [CrossRef] [Green Version]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Yu, F.; Wang, J.; Guo, C.; Fan, X. Autophagy: A new target for nonalcoholic fatty liver disease therapy. Hepat. Med. 2016, 8, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, D.; Dallaglio, K.; Torquato, P.; Piroddi, M.; Galli, F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. J. Lab. Clin. Med. 2018, 193, 54–71. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, J.; Yan, S.; Lei, G.; Lee, C.H.; Yin, X.M. Ethanol-triggered Lipophagy Requires SQSTM1 in AML12 Hepatic Cells. Sci. Rep. 2017, 7, 12307. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, X.; Chu, E.S.H.; Chen, X.; Kang, W.; Wu, F.; To, K.F.; Wong, V.W.S.; Chan, H.L.Y.; Chan, M.T.V.; et al. Defective lysosomal clearance of autophagosomes and its clinical implications in nonalcoholic steatohepatitis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 37–51. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′→3′) | Reverse (3′→5′) | Accession No. |
|---|---|---|---|
| Fas | AAGGACCTGTCTAGGTTTGATGC | TGGCTTCATAGGTGACTTCCA | NM_017332.1 |
| Srebp-1 | GCCCCTGTAACGACCACTG | CAGCGAGTCTGCCTTGATG | NM_001276707.1 |
| Acc | ATGTCTGGCTTGCACCTAGTA | CCCCAAAGCGAGTAACAAATTCT | NM_022193.1 |
| Scd | TCTAGCTCCTATACCACCACCA | TCGTCTCCAACTTATCTCCTCC | NM_139192.2 |
| Cd36 | GGCTGTGACCGGAACTGTG | AGGTCTCCAACTGGCATTAGAA | NM_031561.2 |
| Hsl | TCAGTGTCTAGGTCAGACTGG | AGGCTTCTGTTGGGTATTGGA | NM_012859.1 |
| Mgl | ATGCCAGAGGAAAGTTCCCC | CGTCTGCATTGACCAGGTG | NM_138502.2 |
| Atgl | GGCTTCCTCGGCGTCTACTA | TTTACCAGGTTGAAGGAGGGG | NM_001108509.2 |
| β-actin | AGGCACCAGGGCGTGAT | GCCCACATAGGAATCCTTCTGAC | NM_031144.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, N.-C.; Huang, S.-Y.; Yang, C.-Y.; Shen, H.-H.; Lee, Y.-M. Involvement of HO-1 and Autophagy in the Protective Effect of Magnolol in Hepatic Steatosis-Induced NLRP3 Inflammasome Activation In Vivo and In Vitro. Antioxidants 2020, 9, 924. https://doi.org/10.3390/antiox9100924
Kuo N-C, Huang S-Y, Yang C-Y, Shen H-H, Lee Y-M. Involvement of HO-1 and Autophagy in the Protective Effect of Magnolol in Hepatic Steatosis-Induced NLRP3 Inflammasome Activation In Vivo and In Vitro. Antioxidants. 2020; 9(10):924. https://doi.org/10.3390/antiox9100924
Chicago/Turabian StyleKuo, Ni-Chun, Shieh-Yang Huang, Chien-Yi Yang, Hsin-Hsueh Shen, and Yen-Mei Lee. 2020. "Involvement of HO-1 and Autophagy in the Protective Effect of Magnolol in Hepatic Steatosis-Induced NLRP3 Inflammasome Activation In Vivo and In Vitro" Antioxidants 9, no. 10: 924. https://doi.org/10.3390/antiox9100924