Inhibition of Xanthine Oxidoreductase Enhances the Potential of Tyrosine Kinase Inhibitors against Chronic Myeloid Leukemia


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Proliferation Analysis
2.3. Analysis of Drug Interactions
2.4. Cell Viability Analysis
2.5. Colony Forming Unit Assays
2.6. Detection of Intracellular ROS Levels
2.7. Immunoblotting
2.8. Statistical Analysis
3. Results
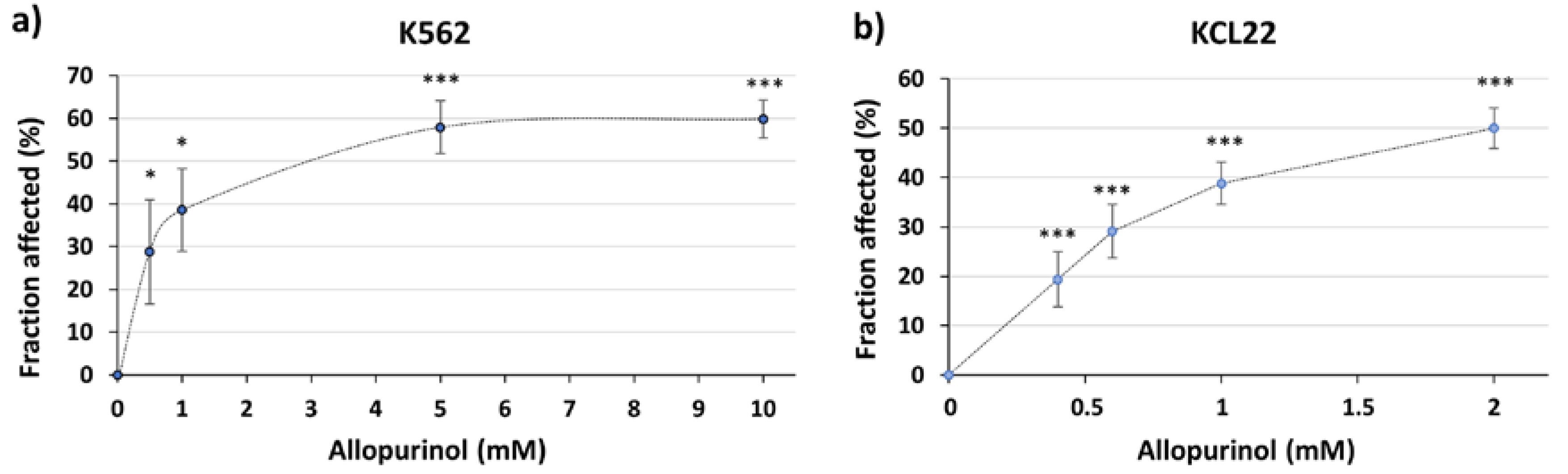
3.1. The XOR Inhibitor Allopurinol Inhibits K562 Cells Proliferation
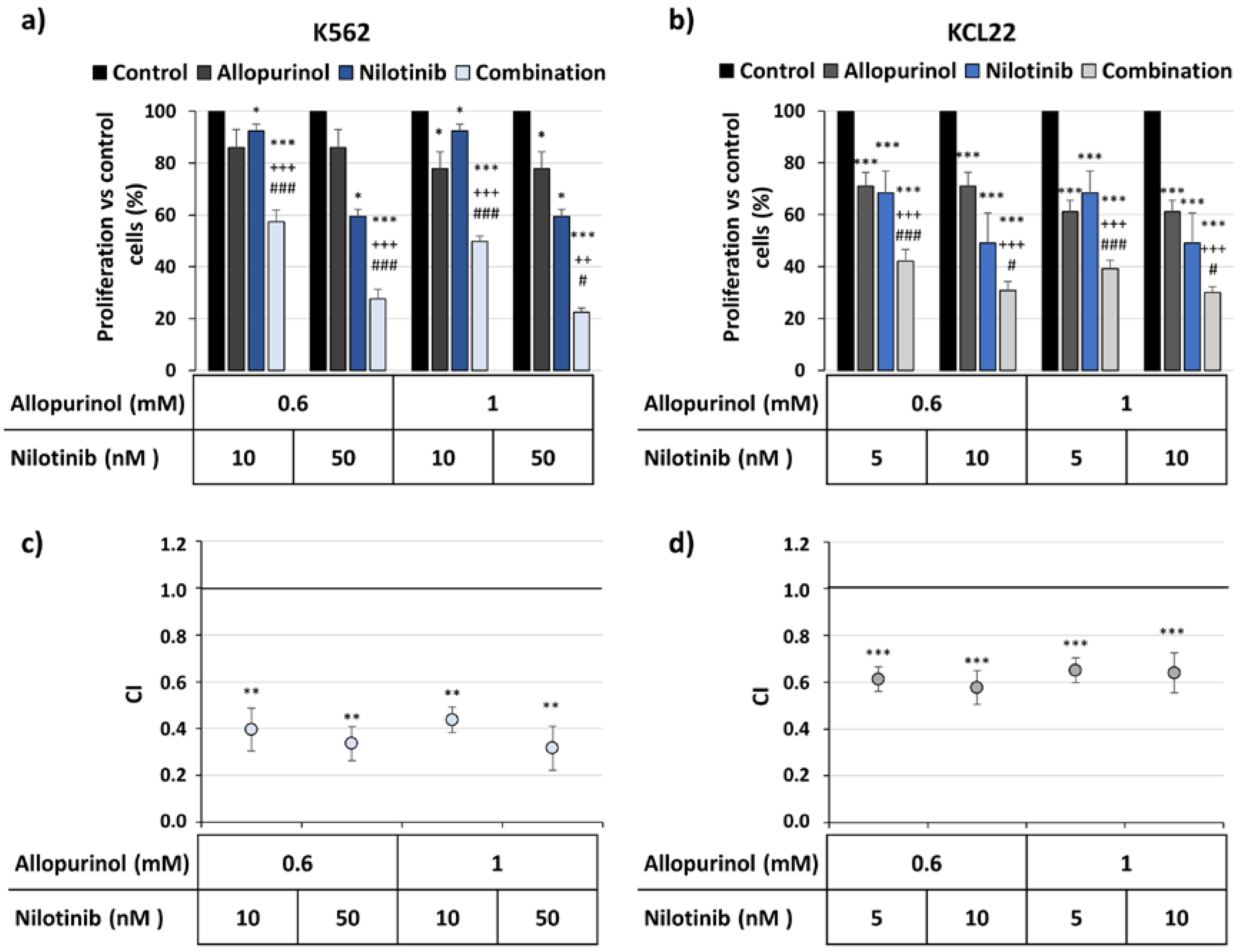
3.2. Allopurinol and TKIs Inhibits K562 and KCL22 Cells Proliferation in a Synergistic Manner
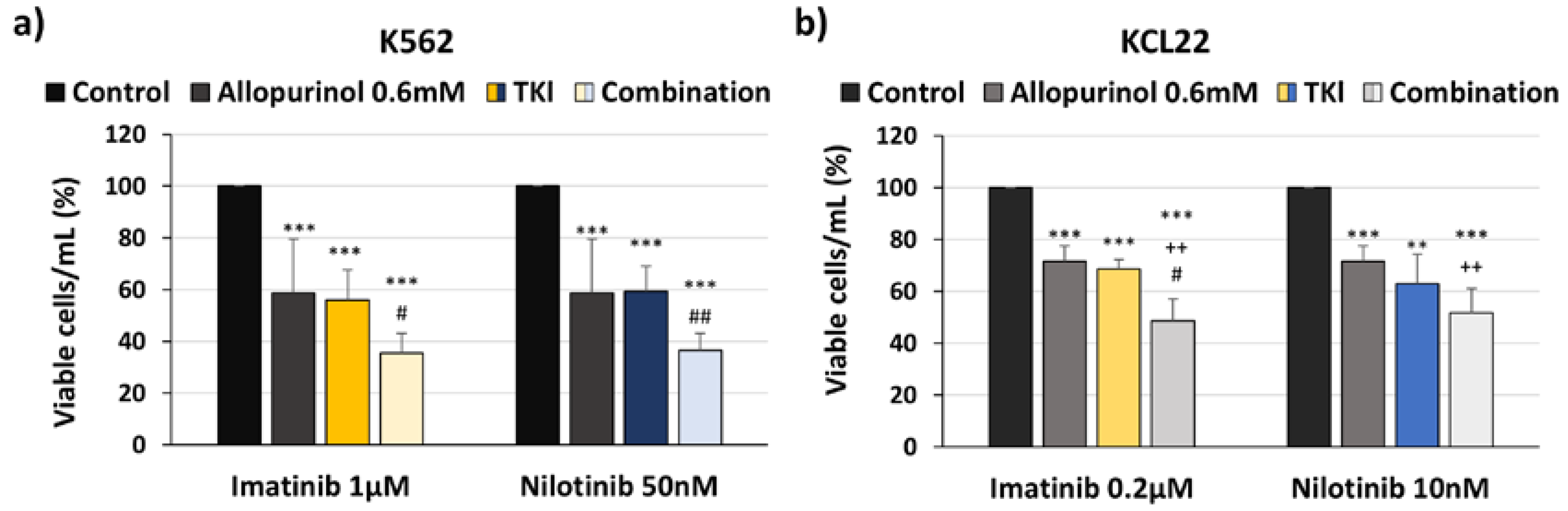
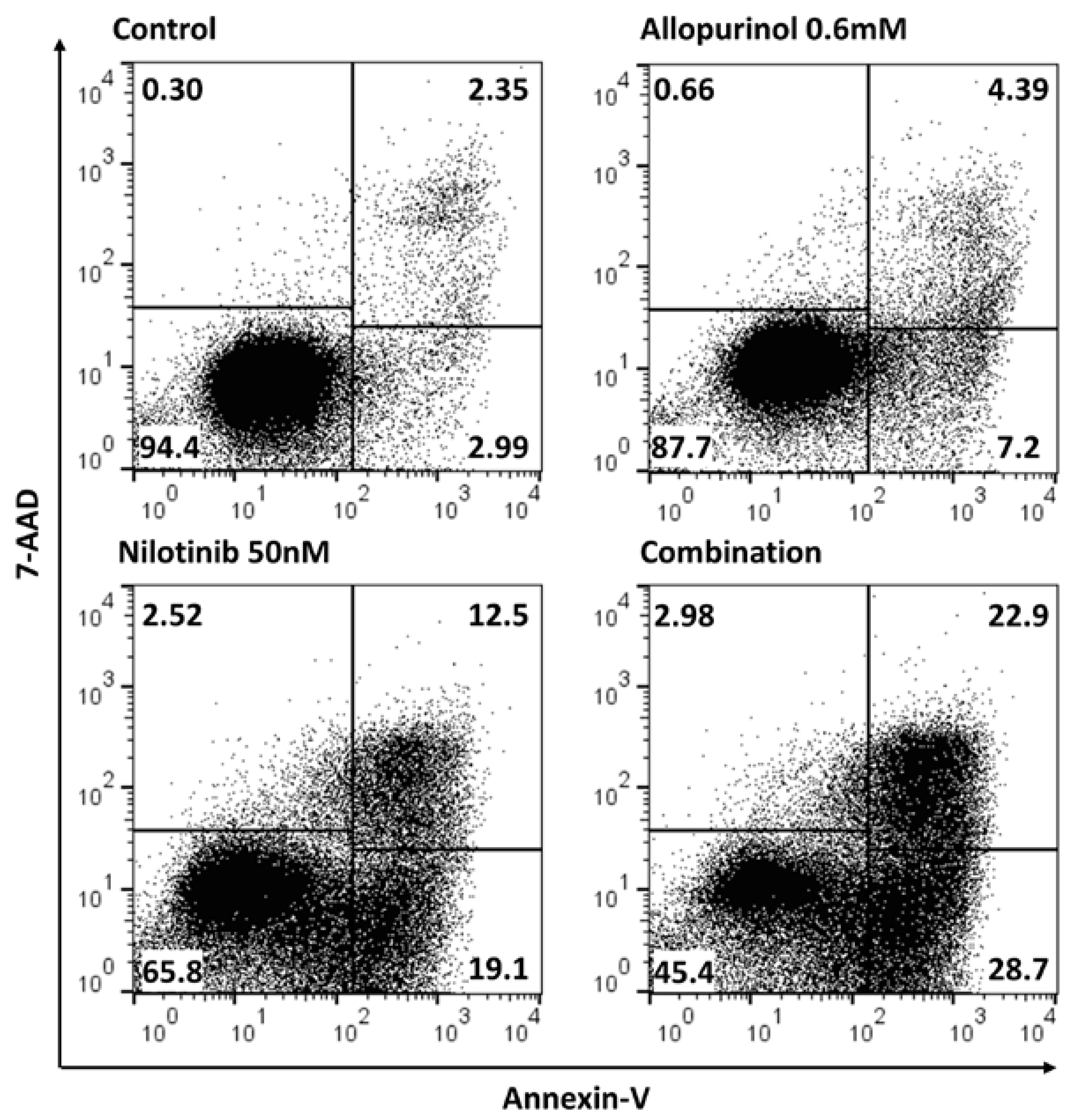
3.3. Allopurinol and TKIs Co-Treatment Induces Cell Death More Efficiently Than Individual Treatments
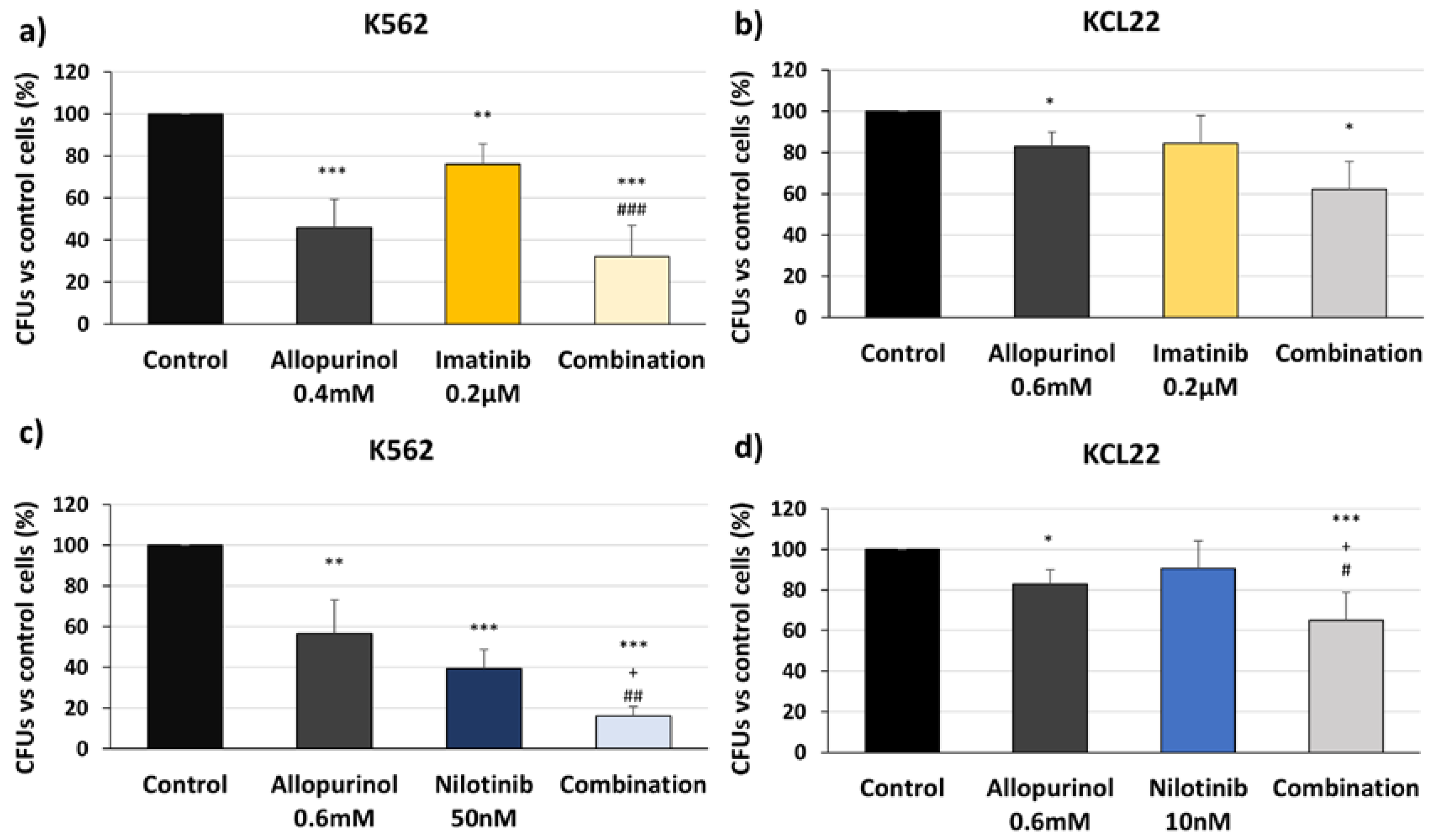
3.4. Allopurinol and TKIs Combination Reduces K562 and KCL22 Cells Clonogenic Capacity
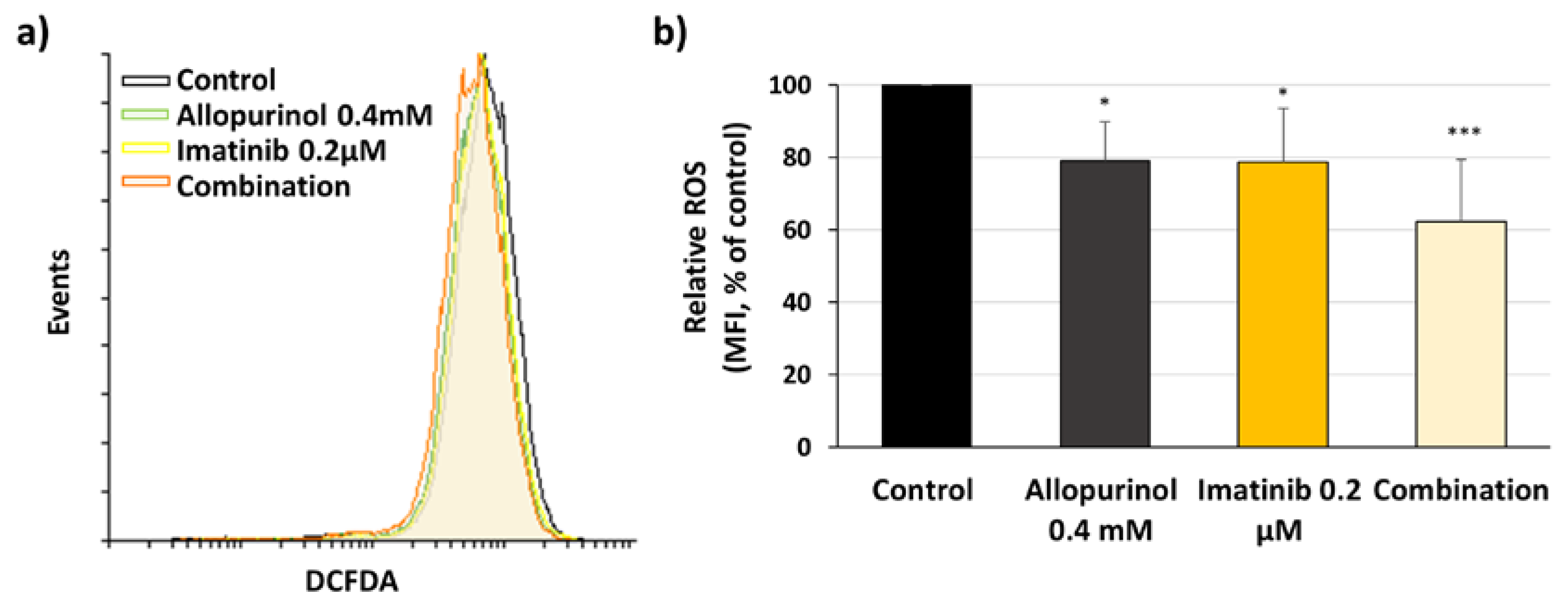
3.5. Imatinib, Allopurinol, and Their Combination Reduce Intracellular ROS Levels
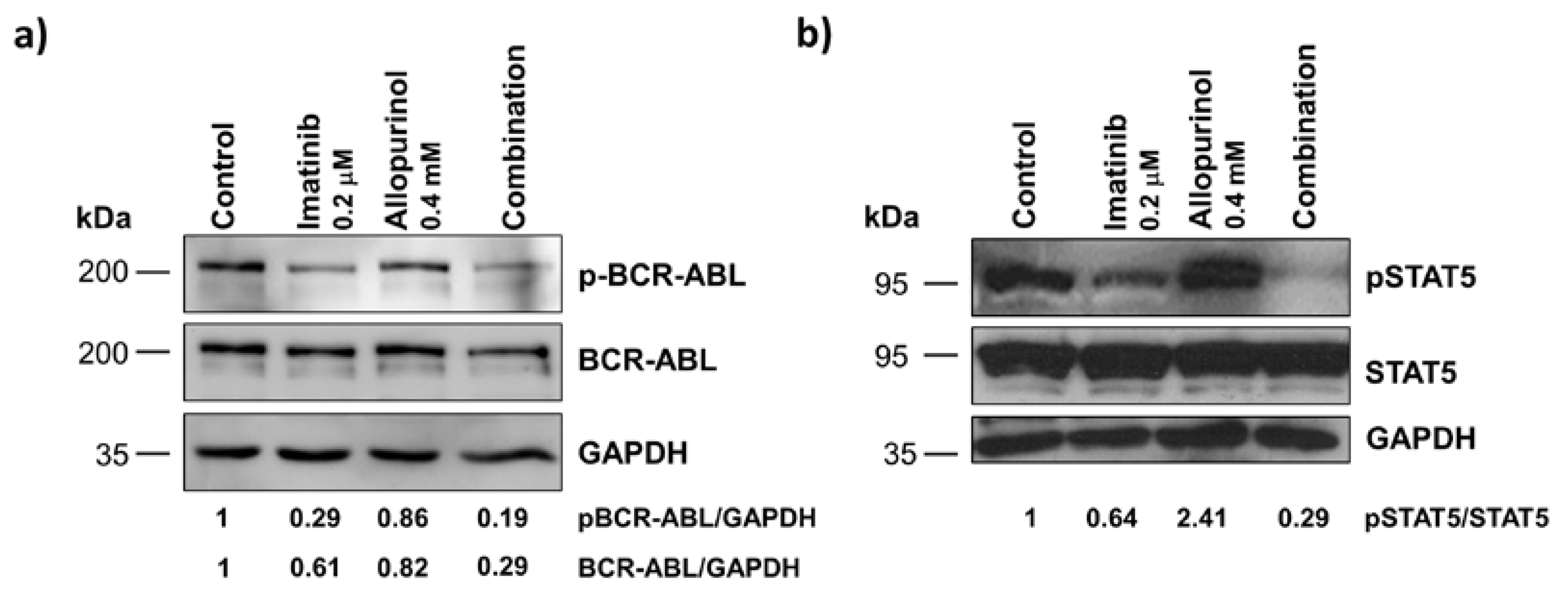
3.6. Imatinib, Allopurinol and Their Combination Attenuate BCR-ABL Signaling
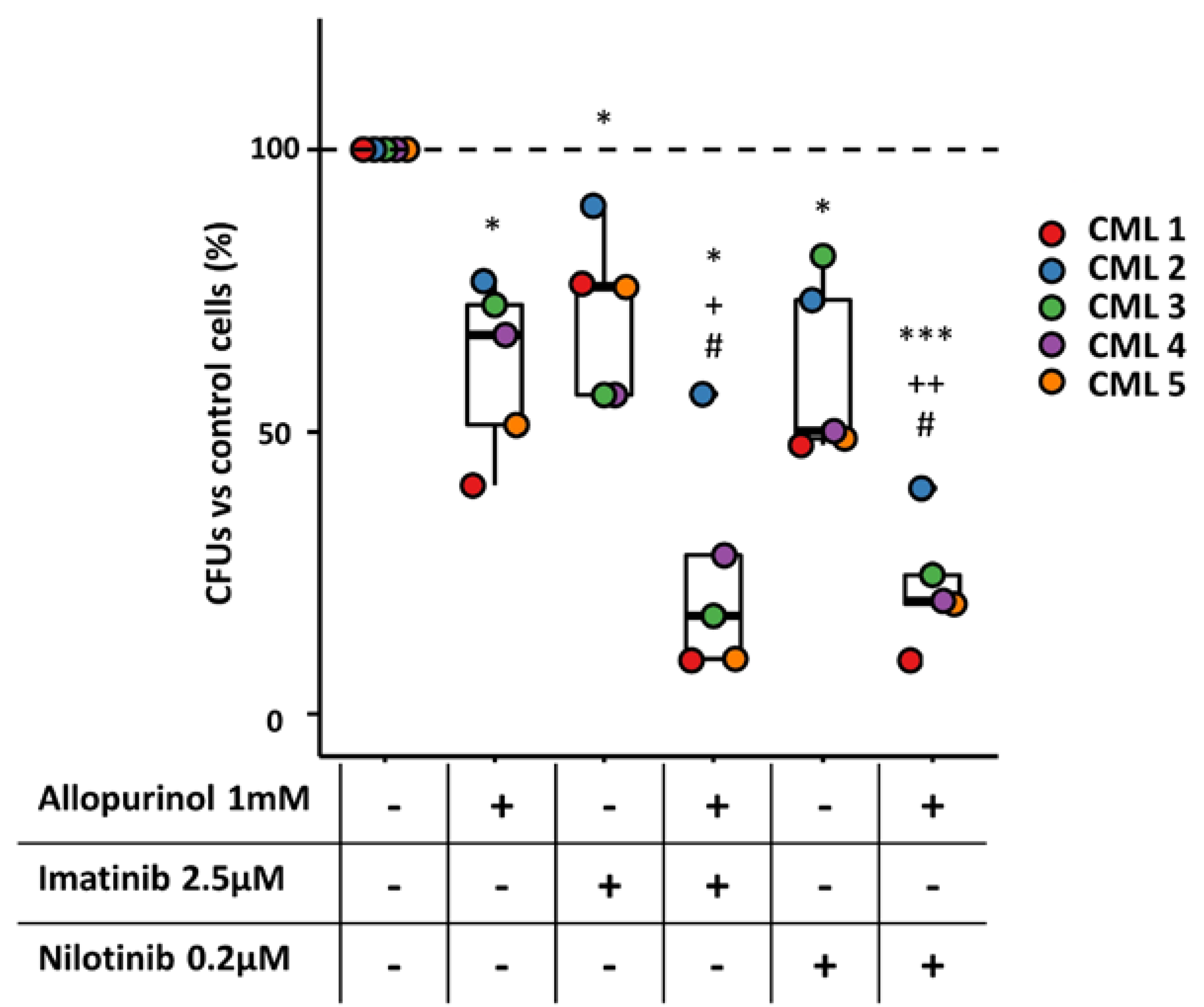
3.7. The Combination of Allopurinol and TKIs Reduces the Clonogenic Ability of CML Primary Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Melo, J.V.; Barnes, D.J. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat. Rev. Cancer 2007, 7, 441–453. [Google Scholar] [CrossRef]
- Chereda, B.; Melo, J.V. Natural course and biology of CML. Ann. Hematol. 2015, 94, 107–121. [Google Scholar] [CrossRef]
- García-Gutiérrez, V.; Hernández-Boluda, J.C. Tyrosine Kinase Inhibitors Available for Chronic Myeloid Leukemia: Efficacy and Safety. Front. Oncol. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.B.; O’Hare, T.; Deininger, M.W. Mechanisms of Resistance to ABL Kinase Inhibition in Chronic Myeloid Leukemia and the Development of Next Generation ABL Kinase Inhibitors. Hematol. Oncol. Clin. N. Am. 2017, 31, 589–612. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive oxygen species in haematopoiesis: Leukaemic cells take a walk on the wild side. J. Exp. Clin. Cancer Res. 2018, 37, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.E.; Rivera-Del Valle, N.; Chandra, J. Redox control of leukemia: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 1349–1383. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Sardina, J.L.; Lopez-Ruano, G.; Sanchez-Sanchez, B.; Llanillo, M.; Hernandez-Hernandez, A.; López-Ruano, G.; Sánchez-Sánchez, B.; Llanillo, M.; Hernández-Hernández, A.; Lopez-Ruano, G.; et al. Reactive oxygen species: Are they important for haematopoiesis? Crit. Rev. Oncol. Hematol. 2012, 81, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Bermejo, R.; Hernandez-Hernandez, A.; Hernández-Hernández, A. The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis. Antioxidants 2017, 6, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem. 2000, 275, 24273–24278. [Google Scholar] [CrossRef] [Green Version]
- Reddy, M.M.; Fernandes, M.S.; Salgia, R.; Levine, R.L.; Griffin, J.D.; Sattler, M. NADPH oxidases regulate cell growth and migration in myeloid cells transformed by oncogenic tyrosine kinases. Leukemia 2011, 25, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, M.O.; Falinski, R.; Koptyra, M.; Slupianek, A.; Stoklosa, T.; Gloc, E.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood 2004, 104, 3746–3753. [Google Scholar] [CrossRef]
- Naughton, R.; Quiney, C.; Turner, S.D.; Cotter, T.G. Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway. Leukemia 2009, 23, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Sanchez, B.; Gutierrez-Herrero, S.; Lopez-Ruano, G.; Prieto-Bermejo, R.; Romo-Gonzalez, M.; Llanillo, M.; Pandiella, A.; Guerrero, C.; Miguel, J.F.S.; Sanchez-Guijo, F.; et al. NADPH oxidases as therapeutic targets in chronic myelogenous leukemia. Clin. Cancer Res. 2014, 20, 4014–4025. [Google Scholar] [CrossRef] [Green Version]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef]
- Nanduri, J.; Vaddi, D.R.; Khan, S.A.; Wang, N.; Makarenko, V.; Semenza, G.L.; Prabhakar, N.R. HIF-1alpha activation by intermittent hypoxia requires NADPH oxidase stimulation by xanthine oxidase. PLoS ONE 2015, 10, e0119762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, G.; Dolzhenko, A.V.; Mancera, R.L. Inhibitors of Xanthine Oxidase: Scaffold Diversity and Structure-Based Drug Design. ChemMedChem 2019, 14, 714–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardina, J.L.L.; Lopez-Ruano, G.; Sanchez-Abarca, L.I.; Perez-Simon, J.A.; Gaztelumendi, A.; Trigueros, C.; Llanillo, M.; Sanchez-Yague, J.; Hernandez-Hernandez, A.; López-Ruano, G.; et al. p22phox-dependent NADPH oxidase activity is required for megakaryocytic differentiation. Cell Death. Differ. 2010, 17, 1842–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Guièze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernández-Sánchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384.e13. [Google Scholar] [CrossRef]
- Abbotts, R.; Topper, M.J.; Biondi, C.; Fontaine, D.; Goswami, R.; Stojanovic, L.; Choi, E.Y.; McLaughlin, L.; Kogan, A.A.; Xia, L.; et al. DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 22609–22618. [Google Scholar] [CrossRef]
- Lorenzo-Herrero, S.; Sordo-Bahamonde, C.; Bretones, G.; Payer, Á.R.; González-Rodríguez, A.P.; González-García, E.; Pérez-Escuredo, J.; Villa-Álvarez, M.; Núñez, L.E.; Morís, F.; et al. The Mithralog EC-7072 Induces Chronic Lymphocytic Leukemia Cell Death by Targeting Tonic B-Cell Receptor Signaling. Front. Immunol. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Daunys, S.; Matulis, D.; Petrikaitė, V. Synergistic activity of Hsp90 inhibitors and anticancer agents in pancreatic cancer cell cultures. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Nie, L.; Wei, Y.; Zhang, F.; Hsu, Y.H.; Chan, L.C.; Xia, W.; Ke, B.; Zhu, C.; Deng, R.; Tang, J.; et al. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Jiménez-Solas, T.; López-Cadenas, F.; Aires-Mejía, I.; Caballero-Berrocal, J.C.; Ortega, R.; Redondo, A.M.; Sánchez-Guijo, F.; Muntión, S.; García-Martín, L.; Albarrán, B.; et al. Deferasirox reduces oxidative DNA damage in bone marrow cells from myelodysplastic patients and improves their differentiation capacity. Br. J. Haematol. 2019, 187, 93–104. [Google Scholar] [CrossRef]
- Yadav, N.K.; Shukla, P.; Omer, A.; Singh, P.; Singh, R.K. Alternative methods in toxicology: CFU assays application, limitation and future prospective. Drug Chem. Toxicol. 2016, 39, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Nivorozhkin, A.; Szabo, C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Landry, W.D.; Woolley, J.F.; Cotter, T.G. Imatinib and Nilotinib inhibit Bcr-Abl-induced ROS through targeted degradation of the NADPH oxidase subunit p22phox. Leuk. Res. 2013, 37, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Warsch, W.; Walz, C.; Sexl, V. JAK of all trades: JAK2-STAT5 as novel therapeutic targets in BCR-ABL1 + chronic myeloid leukemia. Blood 2013, 122, 2167–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Behrend, L.; Henderson, G.; Zwacka, R.M. Reactive oxygen species in oncogenic transformation. Biochem. Soc. Trans. 2003, 31, 1441–1444. [Google Scholar] [CrossRef]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Jackson, A.L.; Loeb, L.A. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res. 2001, 477, 7–21. [Google Scholar] [CrossRef]
- Hitchler, M.J.; Domann, F.E. Metabolic defects provide a spark for the epigenetic switch in cancer. Free Radic. Biol. Med. 2009, 47, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraman, L.; Murthy, K.G.; Zhu, C.; Curran, T.; Xanthoudakis, S.; Prives, C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev. 1997, 11, 558–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, M.Y.; Arbiser, J.L. The antioxidant paradox: What are antioxidants and how should they be used in a therapeutic context for cancer. Future Med. Chem. 2014, 6, 1413–1422. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.S.; Huang, M.; Medeiros, B.C.; Mitchell, B.S. Selective Toxicity of Investigational Ixazomib for Human Leukemia Cells Expressing Mutant Cytoplasmic NPM1: Role of Reactive Oxygen Species. Clin. Cancer Res. 2016, 22, 1978–1988. [Google Scholar] [CrossRef] [Green Version]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH oxidases: A perspective on reactive oxygen species production in tumor biology. Antioxid. Redox Signal. 2014, 20, 2873–2889. [Google Scholar] [CrossRef] [Green Version]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase in cancer: More than a differentiation marker. Cancer Med. 2016, 5, 546–557. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase-derived reactive species: Physiological and pathological effects. Oxid. Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.L.; Zhang, W.G.; Wei, Y.C.; Meng, S.; Bai, G.G.; Wang, B.Y.; Yang, H.Y.; Tian, W.; Meng, X.; Zhang, H.; et al. Involvement of oxidative stress in the relapse of acute myeloid leukemia. J. Biol. Chem. 2010, 285, 15010–15015. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.; Kantarjian, H. How I treat newly diagnosed chronic phase CML. Blood 2012, 120, 1390–1397. [Google Scholar] [CrossRef]
- Berger, A.; Sexl, V.; Valent, P.; Moriggl, R. Inhibition of STAT5: A therapeutic option in BCR-ABL1-driven leukemia. Oncotarget 2014, 5, 9564–9576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Mao, X.; Li, H.; Qiao, S.; Xu, A.; Wang, J.; Lei, S.; Liu, Z.; Ng, K.F.J.; Wong, G.T.; et al. N-Acetylcysteine and allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic. Biol. Med. 2013, 63, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Qian, P.; Jackson, F.R.; Qian, G.; Wu, G.; Padi, S.K.R.; Luevano, L.A.; An, N.; Pandey, R.; Singh, N.; et al. Targeting the PIM protein kinases for the treatment of a T-cell acute lymphoblastic leukemia subset. Oncotarget 2013, 8, 291–303. [Google Scholar]
- Wang, G.; Qian, P.; Jackson, F.R.; Qian, G.; Wu, G. Sequential activation of JAKs, STATs and xanthine dehydrogenase/oxidase by hypoxia in lung microvascular endothelial cells. Int. J. Biochem. Cell Biol. 2008, 40, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battelli, M.G.; Bortolotti, M.; Polito, L.; Bolognesi, A. Metabolic syndrome and cancer risk: The role of xanthine oxidoreductase. Redox Biol. 2019, 21, 101070. [Google Scholar] [CrossRef] [PubMed]
- Mirrakhimov, A.E. Tumor lysis syndrome: A clinical review. World J. Crit. Care Med. 2015, 4, 130. [Google Scholar] [CrossRef]
- Cuenca, J.A.; Balda, J.; Palacio, A.; Young, L.; Pillinger, M.H.; Tamariz, L. Febuxostat and Cardiovascular Events: A Systematic Review and Meta-Analysis. Int. J. Rheumatol. 2019, 2019, 1076189. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | K562 | KCL22 | ||||||
|---|---|---|---|---|---|---|---|---|
| Content | Control | Allopurinol 0.4 mM | Imatinib 0.2 µM | Allopurinol + Imatinib | Control | Allopurinol 0.6 mM | Imatinib 0.2 µM | Allopurinol + Imatinib |
| Viable cells | 84.6 ± 6.7 | 78.7 ± 5.6 | 68.8 ± 5.1 ** | 49.5 ± 9.3 ***/+++/### | 91.8 ± 2.0 | 85.7 ± 2.6 *** | 89.7 ± 1.0 * | 77.4 ± 2.2 ***/+++/### |
| Early apoptosis | 6.3 ± 1.2 | 12.6 ± 5.0 | 15.6 ± 4.0 * | 27.3 ± 8.1 ***/++/### | 4.3 ± 0.4 | 8.8 ± 1.2 *** | 6 ± 0.9 ** | 15.0 ± 2.5 ***/++/### |
| Late apoptosis | 2.3 ± 2.6 | 1.6 ± 0.6 | 4.8 ± 2.0 | 5.2 ± 1.5 */## | 3.0 ± 1.2 | 4.5 ± 1.7 | 3.5 ± 0.6 | 6.7 ± 1.6 ***/+/## |
| Necrosis | 6.8 ± 4.2 | 7.1 ± 2.5 | 10.8 ± 2.9 | 18.1 ± 6.1 ***/+/### | 1.0 ± 0.8 | 0.9 ± 0.8 | 0.9 ± 1.0 | 0.9 ± 0.8 |
| Cell Lines | K562 | KCL22 | ||||||
|---|---|---|---|---|---|---|---|---|
| Content | Control | Allopurinol 0.6 mM | Nilotinib 50 nM | Allopurinol + Nilotinib | Control | Allopurinol 0.6 mM | Nilotinib 10 nM | Allopurinol + Nilotinib |
| Viable cells | 93.3 ± 1.7 | 86.1 ± 2.0 *** | 65.3 ± 2.4 *** | 45.5 ± 3.0 ***/+++/### | 91.8 ± 2.0 | 85.7 ± 2.6 *** | 82.8 ± 2.2 *** | 75.6 ± 2.2 ***/+++/### |
| Early apoptosis | 3.7 ± 1.1 | 8.4 ± 1.5 *** | 22.5 ± 1.5 *** | 32.1 ± 2.2 **/++/## | 4.3 ± 0.4 | 8.8 ± 1.2 *** | 10.9 ± 1.9 *** | 16.1 ± 2.2 ***/+++/## |
| Late apoptosis | 2.2 ± 0.6 | 4.0 ± 0.7 ** | 9.9 ± 2.1 *** | 19.9 ± 1.9 ***/+++/### | 3.0 ± 1.2 | 4.5 ± 1.7 | 5.6 ± 1.6 ** | 7.4 ± 2 ** |
| Necrosis | 0.7 ± 0.4 | 1.5 ± 0.3 ** | 2.3 ± 0.8 ** | 2.5 ± 0.4 ***/+ | 1.0 ± 0.8 | 0.9 ± 0.8 | 0.8 ± 0.7 | 0.9 ± 0.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romo-González, M.; Moreno-Paz, S.; García-Hernández, V.; Sánchez-Guijo, F.; Hernández-Hernández, Á. Inhibition of Xanthine Oxidoreductase Enhances the Potential of Tyrosine Kinase Inhibitors against Chronic Myeloid Leukemia. Antioxidants 2020, 9, 74. https://doi.org/10.3390/antiox9010074
Romo-González M, Moreno-Paz S, García-Hernández V, Sánchez-Guijo F, Hernández-Hernández Á. Inhibition of Xanthine Oxidoreductase Enhances the Potential of Tyrosine Kinase Inhibitors against Chronic Myeloid Leukemia. Antioxidants. 2020; 9(1):74. https://doi.org/10.3390/antiox9010074
Chicago/Turabian StyleRomo-González, Marta, Sara Moreno-Paz, Violeta García-Hernández, Fermín Sánchez-Guijo, and Ángel Hernández-Hernández. 2020. "Inhibition of Xanthine Oxidoreductase Enhances the Potential of Tyrosine Kinase Inhibitors against Chronic Myeloid Leukemia" Antioxidants 9, no. 1: 74. https://doi.org/10.3390/antiox9010074