Chain Breaking Antioxidant Activity of Heavy (S, Se, Te) Chalcogens Substituted Polyphenols



Abstract
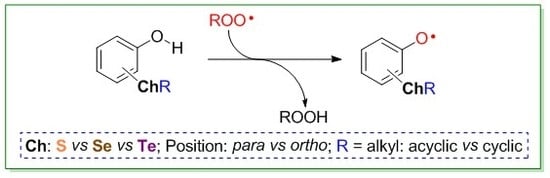
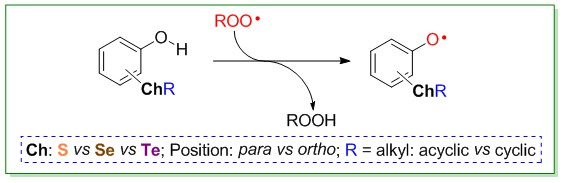
:
{kind=link}
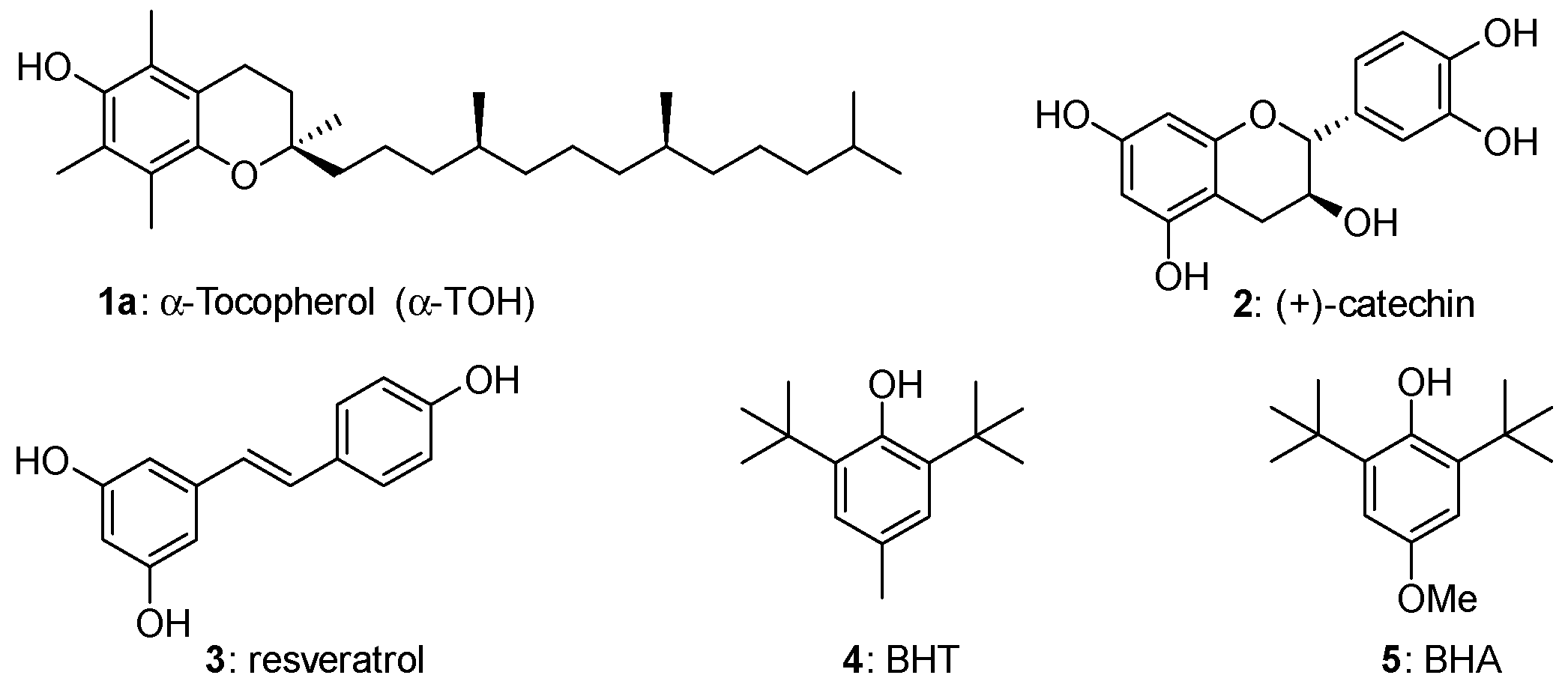{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
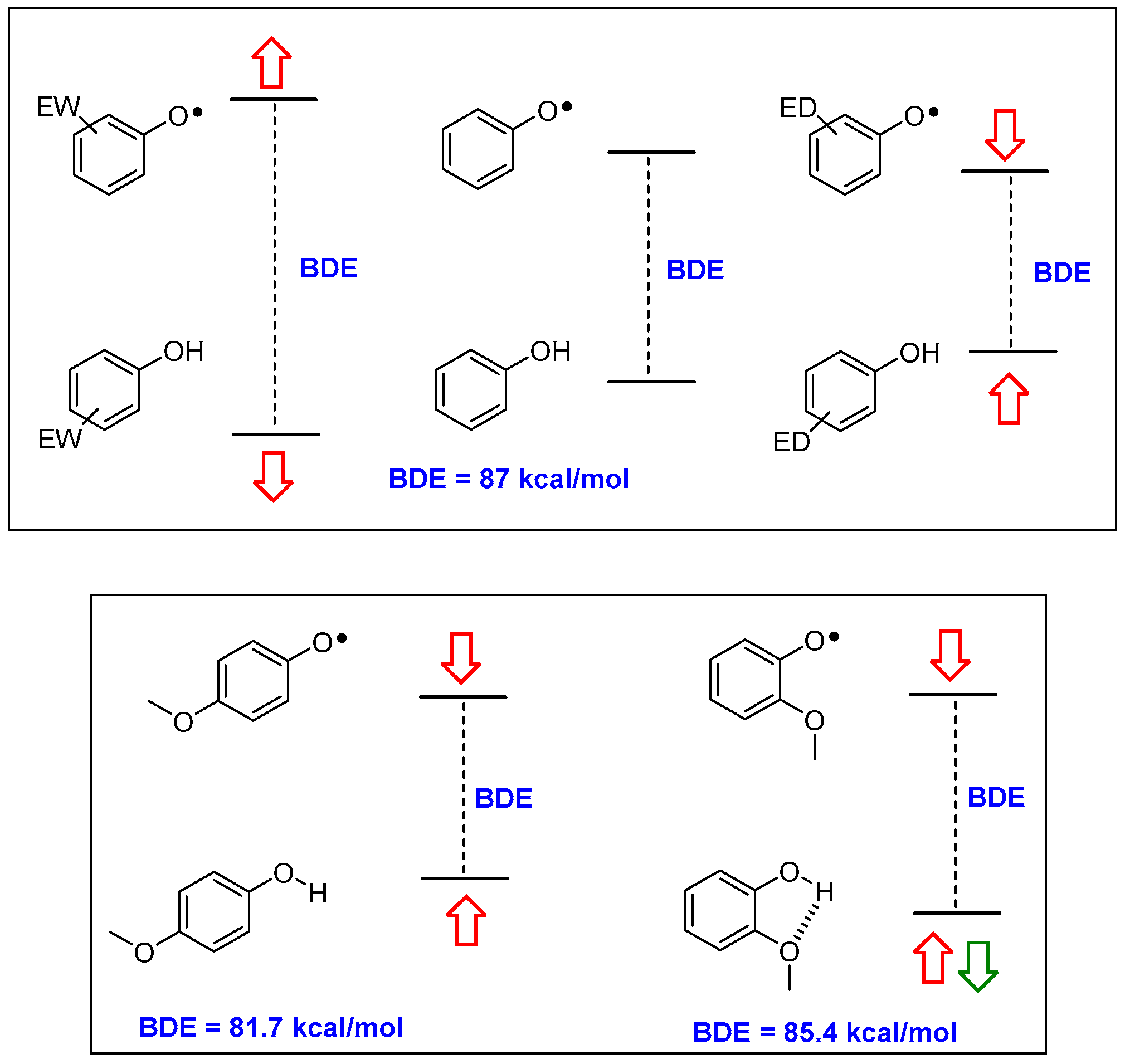
1. Introduction
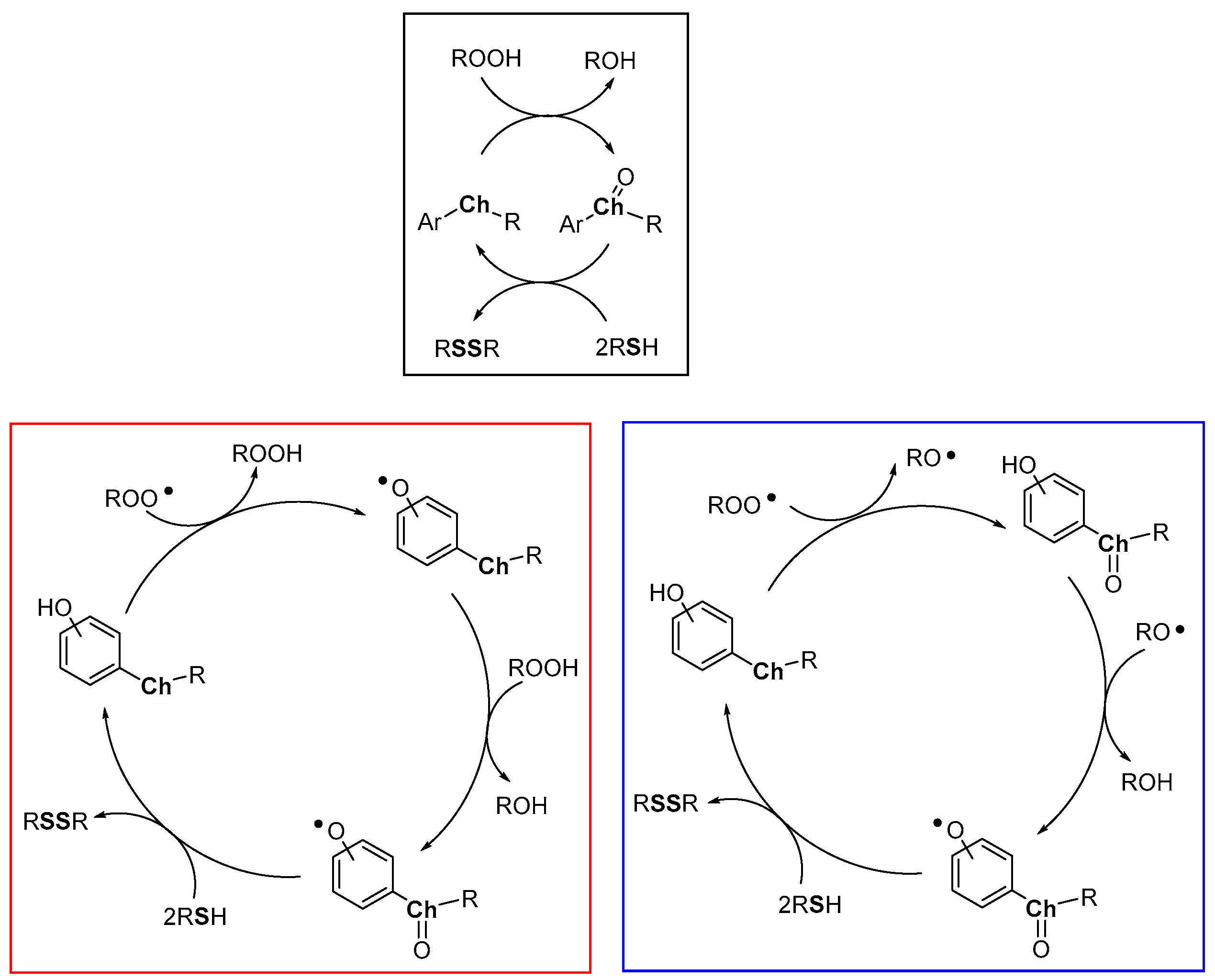
2. Discussion
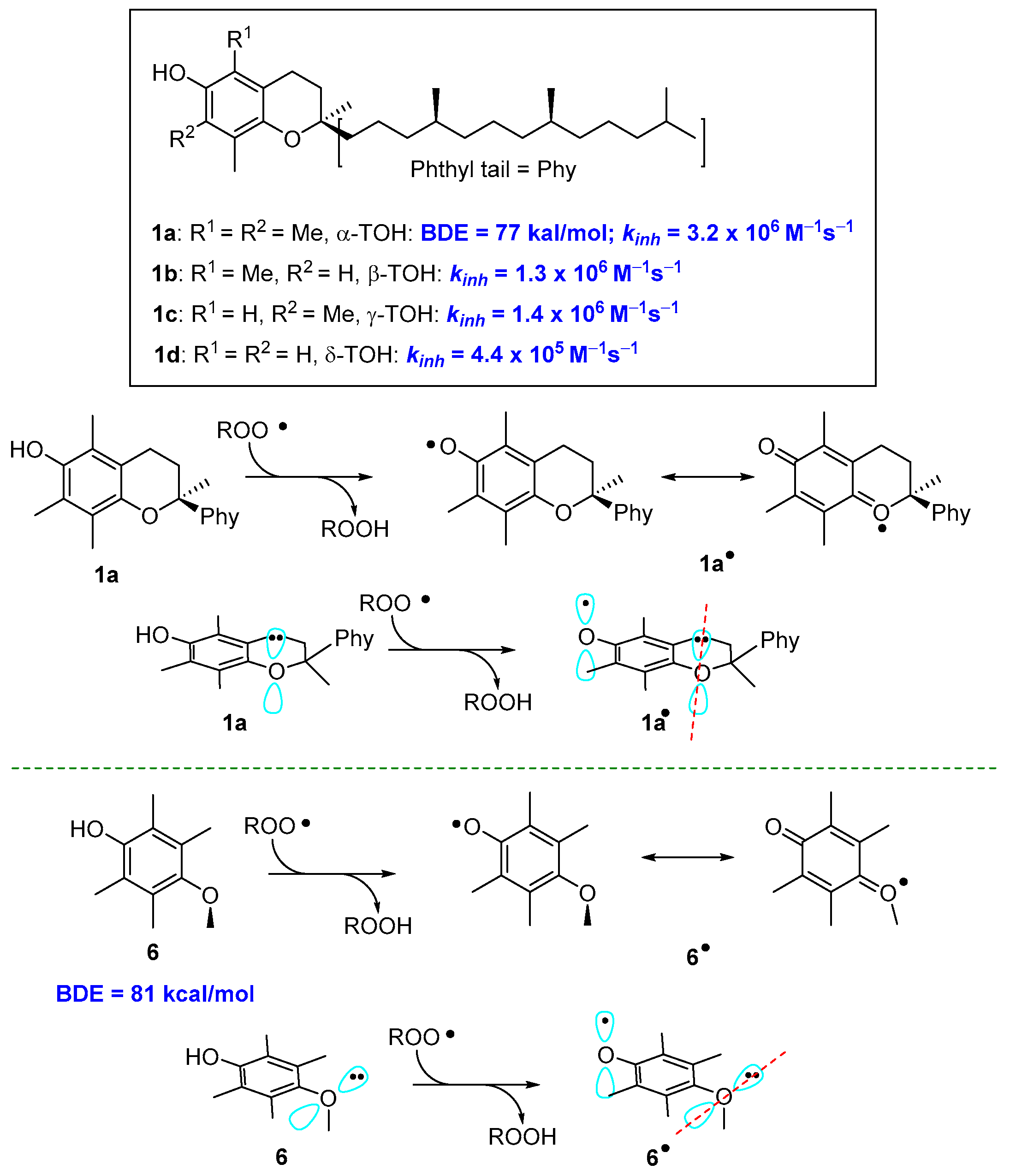
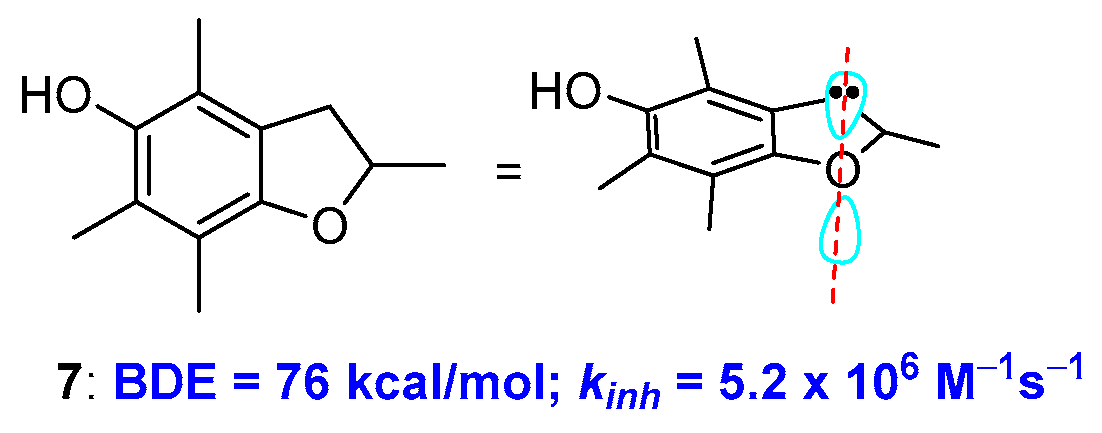
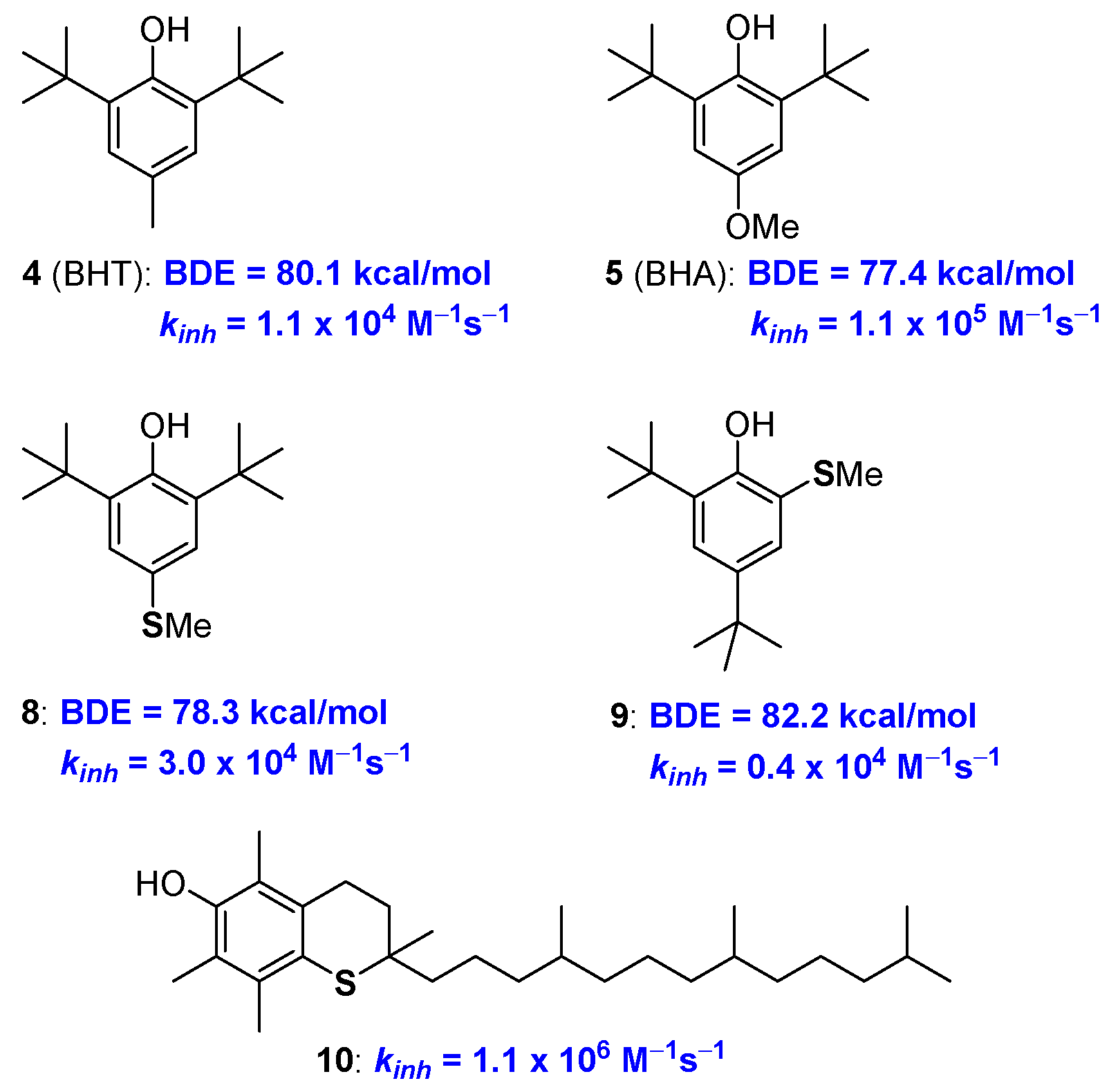
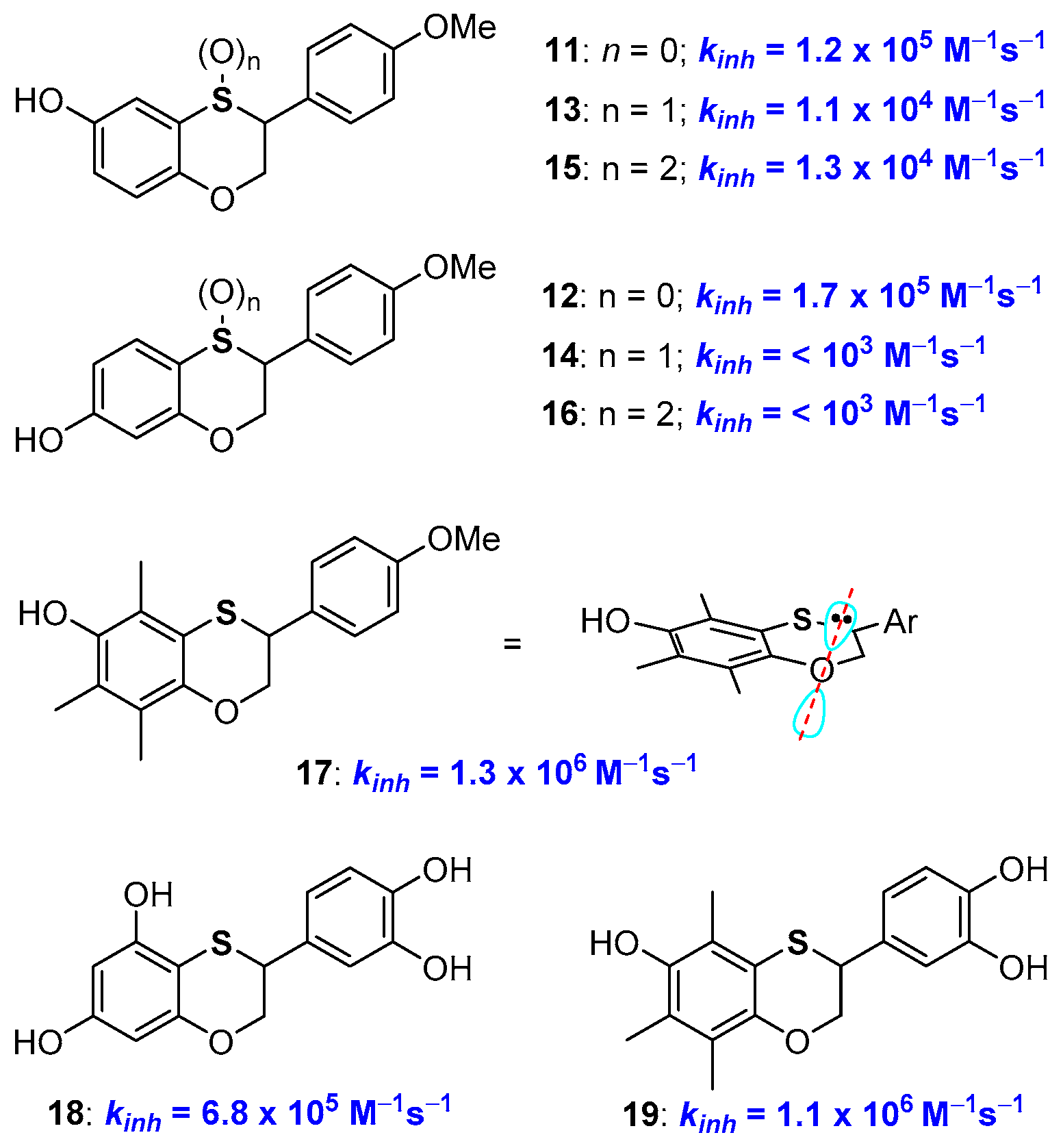
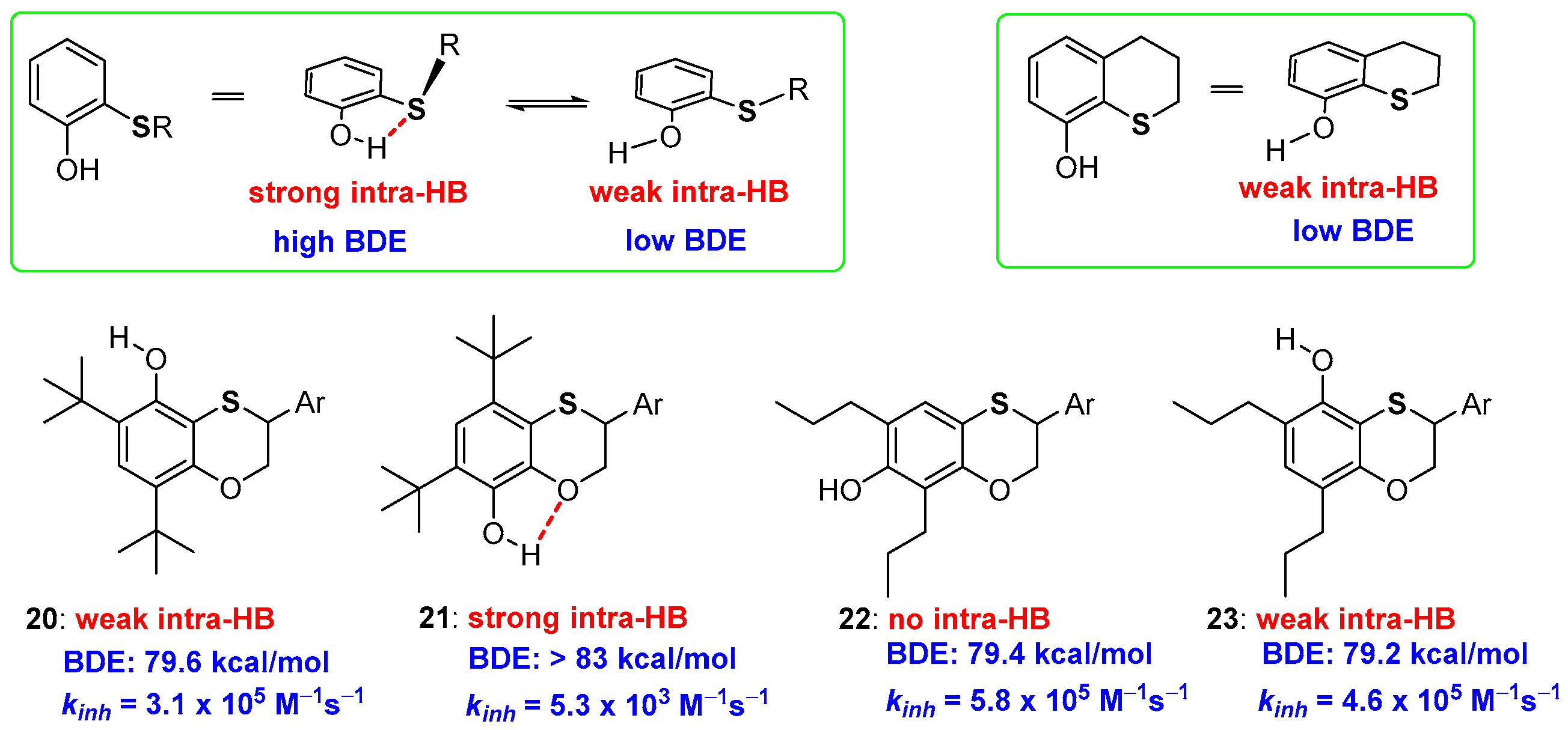
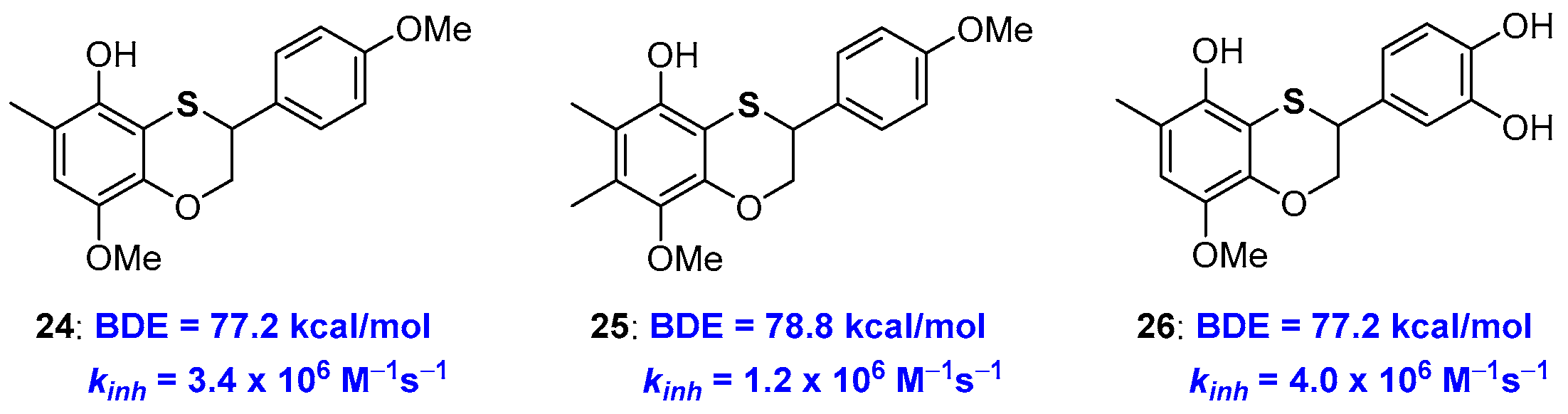
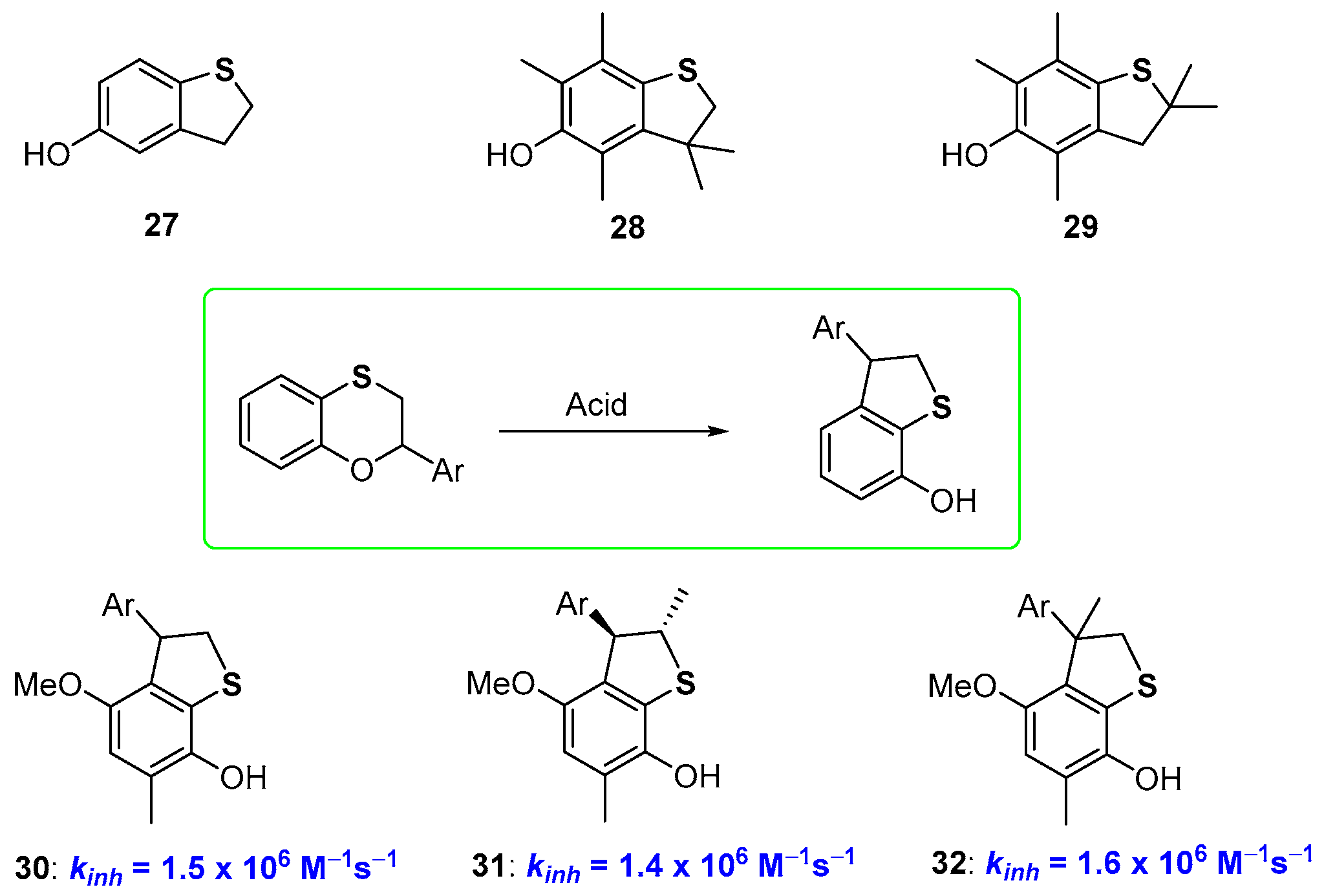
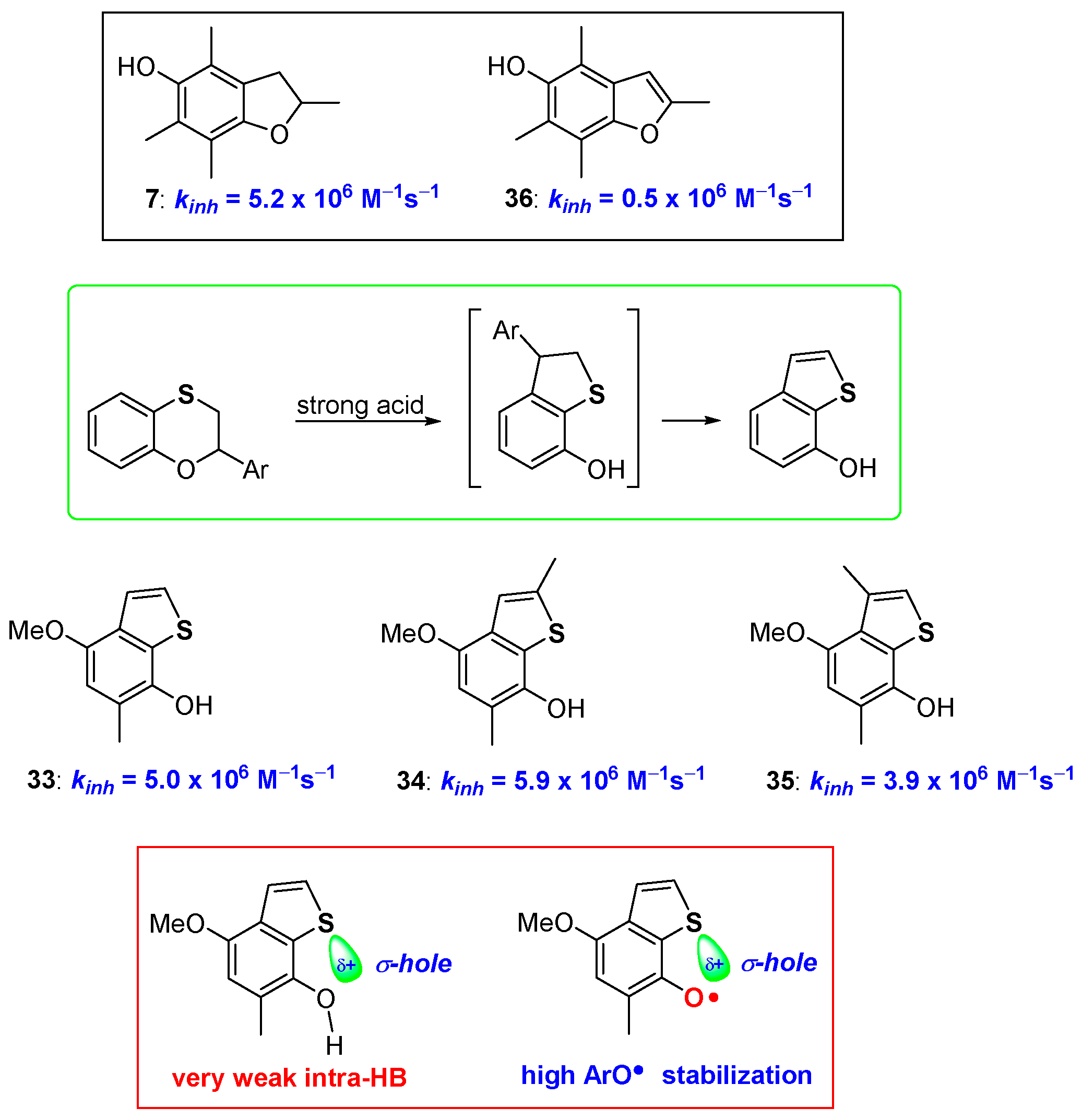
2.1. Sulfur Containing Phenolic Antioxidants
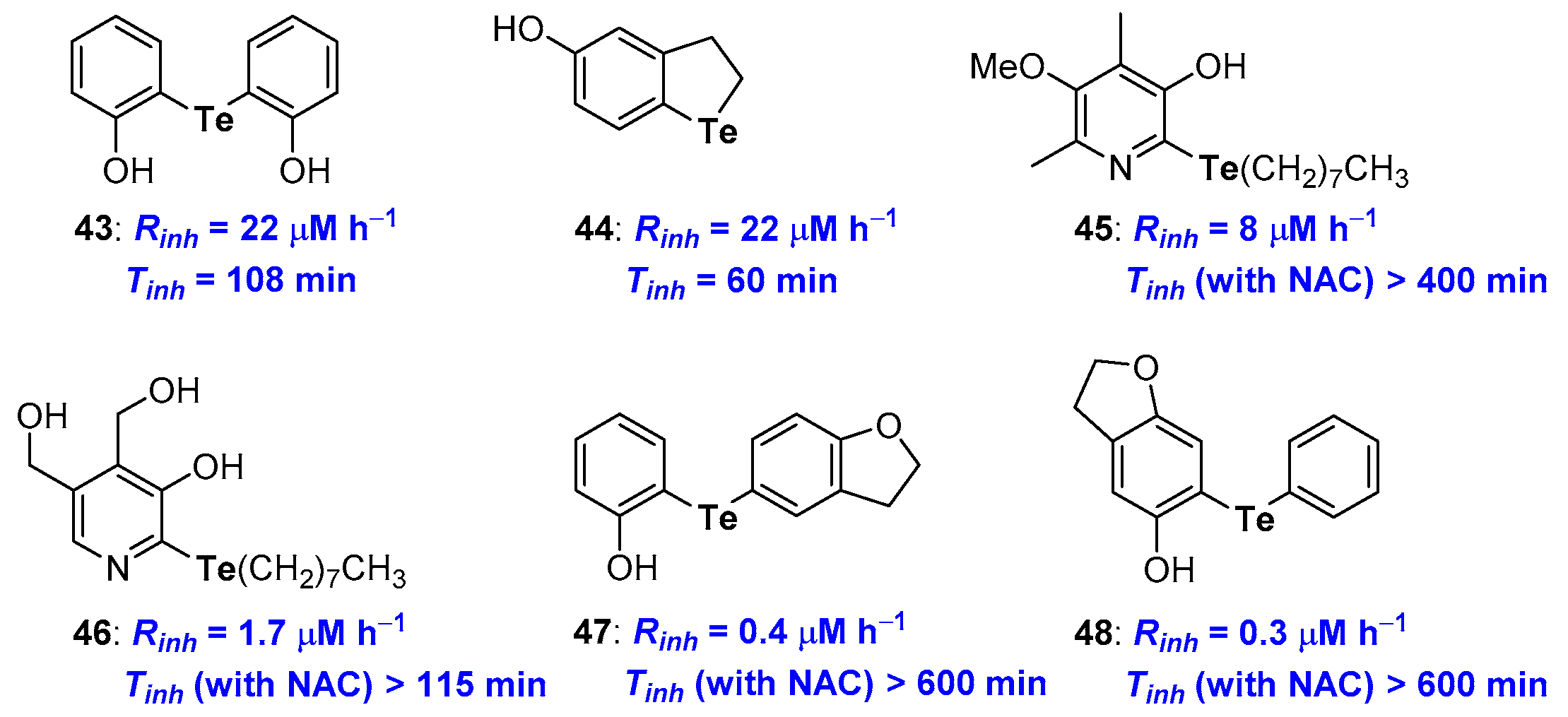
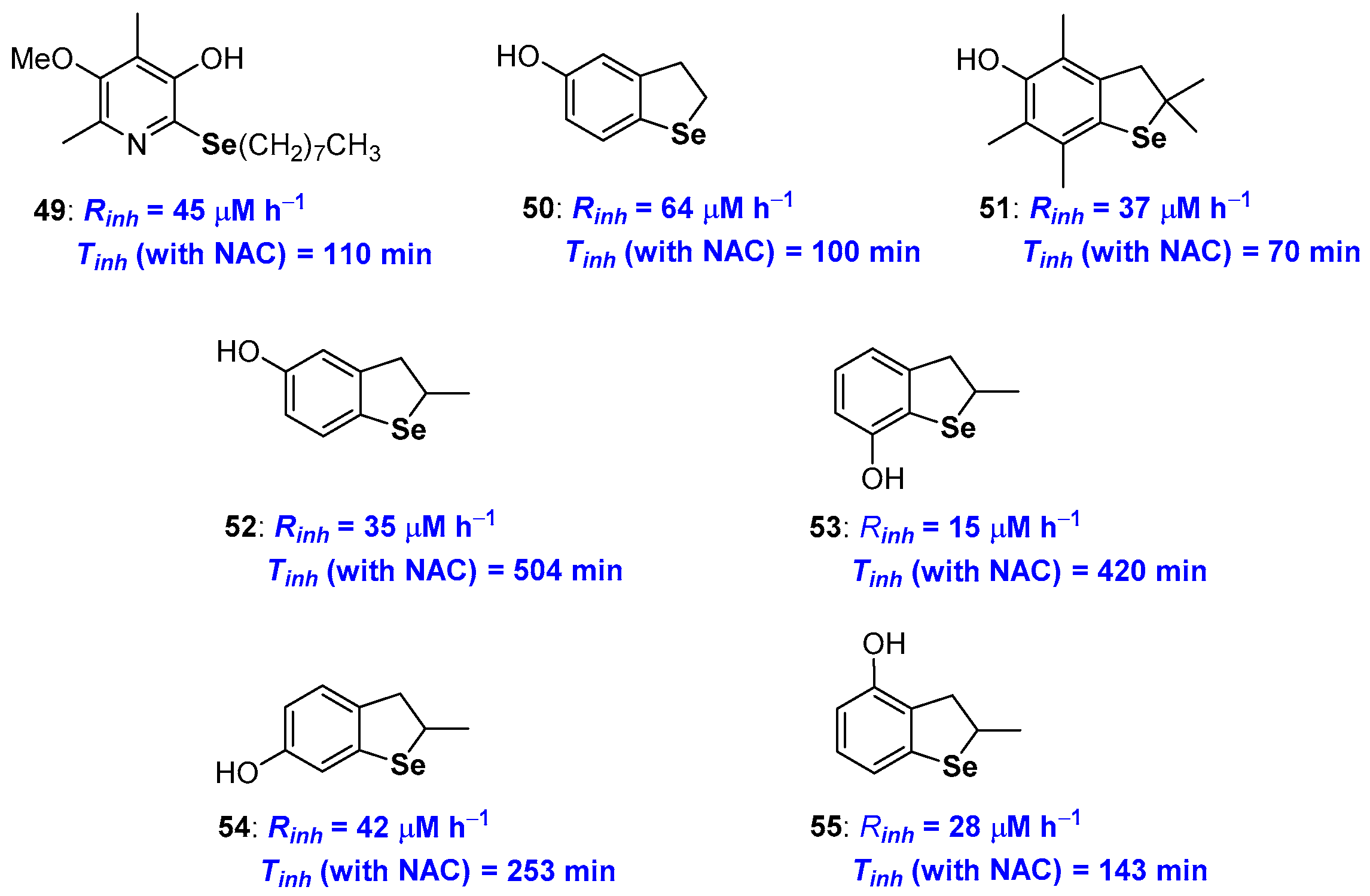
2.2. Selenium and Tellurium Containing Phenolic Antioxidants
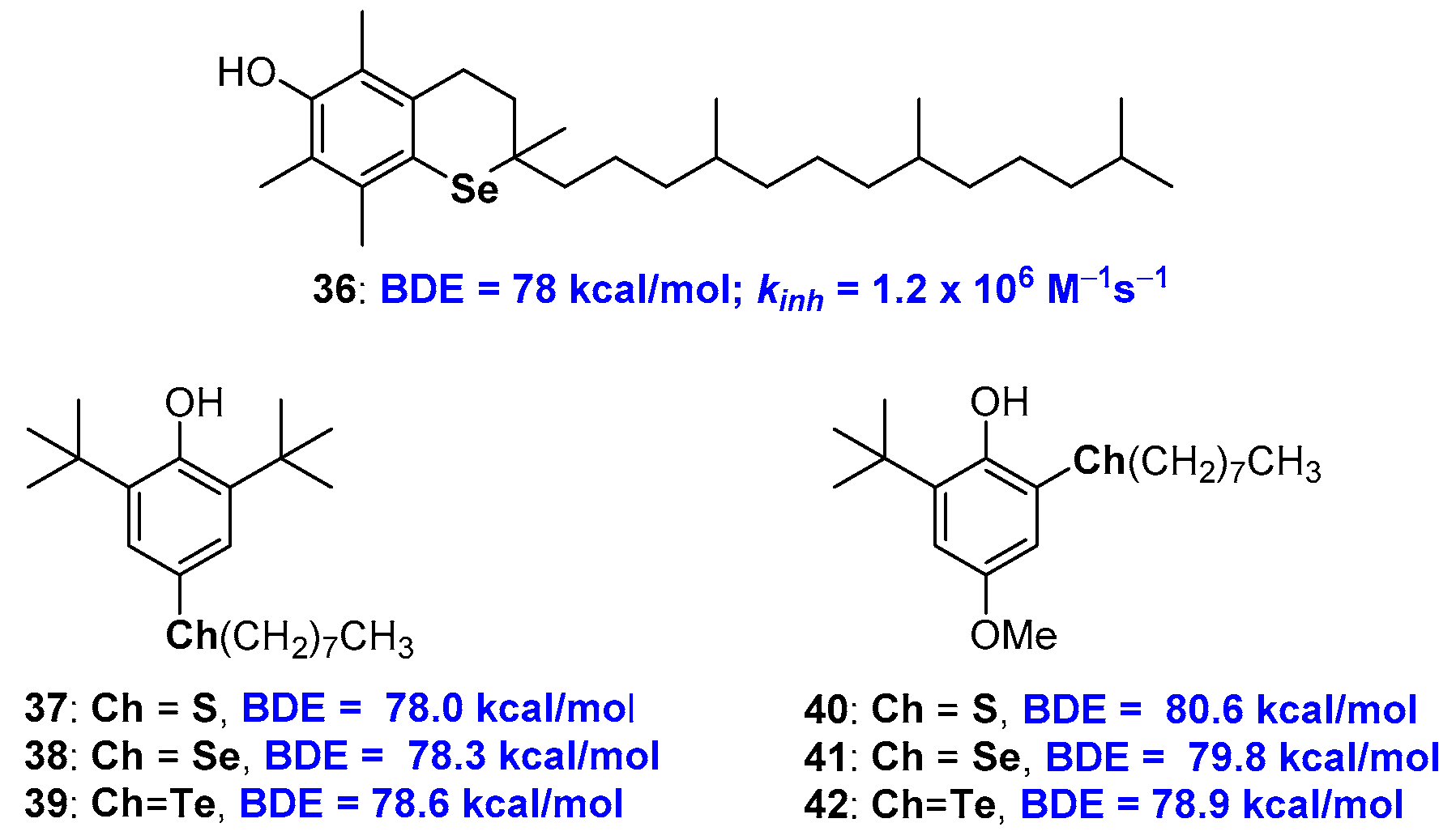
2.3. Chalcogens in Tocopherol Skeletons
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ingold, K.U.; Pratt, D.A. Advances in Radical-Trapping Antioxidant Chemistry in the 21st Century: A Kinetics and Mechanisms Perspective. Chem. Rev. 2014, 114, 9022–9046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggersdorfer, M.; Laudert, D.; Létinois, U.; McClymont, T.; Medlock, J.; Netscher, T.; Bonrath, W. One hundred years of vitamins—A success story of the natural sciences. Angew. Chem. Int. Ed. 2012, 51, 12960–12990. [Google Scholar] [CrossRef] [PubMed]
- Ingold, K.U. Inhibition of the Autoxidation of Organic Substances in the Liquid Phase. Chem. Rev. 1961, 61, 563–589. [Google Scholar] [CrossRef]
- Noguchi, N.; Niki, E. Phenolic antioxidants: A rational for design and evaluation of novel drug for atherosclerosis. Free Radic. Biol. Med. 2000, 28, 1538–1546. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.M.; Bishop, K.S. On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science 1922, 56, 650–651. [Google Scholar] [CrossRef] [PubMed]
- Niki, E.; Traber, M.G. A History of Vitamin E. Ann. Nutr. Metab. 2012, 61, 207–212. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R. Vitamin E: The shrew waiting to be tamed. Free. Radic. Biol. Med. 2009, 46, 543–554. [Google Scholar] [CrossRef]
- Burton, G.W.; Ingold, K.U. Autoxidation of biological molecules. 1. Antioxidant activity of vitamin E and related chain-breaking phenolic antioxidants in vitro. J. Am. Chem. Soc. 1981, 103, 6472–6477. [Google Scholar] [CrossRef]
- Burton, G.W.; Ingold, K.U. Vitamin E: Application of the principles of physical organic chemistry to the exploration of its structure and function. Acc. Chem. Res. 1986, 19, 194–201. [Google Scholar] [CrossRef]
- Böhm, V. Vitamin E. Antioxidants 2018, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Lotito, S.B.; Frei, B. Consumption of flavonoid-rich foods and increased plasma antioxidant capacity in humans: Cause, consequence, or epiphenomenon? Free Radic. Biol. Med. 2006, 41, 1727–1746. [Google Scholar] [CrossRef] [PubMed]
- Dorta, J.D.; Pigoso, A.A.; Mingatto, F.E.; Rodrigues, T.; Prado, I.M.R.; Helena, A.F.C.; Uyemura, S.A.; Santos, A.C.; Curti, C. The interaction of flavonoids with mitochondria: Effects on energetic processes. Chem. Biol. Interact. 2005, 152, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Pietta, P.-G. Flavonoids as Antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Marchesi, P.; Passamonti, S.; Pirillo, A.; Violi, F.; Catapano, A.L. Anti-inflammatory and anti-atherogenic effects of cathechin, caffeic acid and trans-resveratrol in apolipoprotein E deficient mice. Atherosclerosis 2007, 191, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Wang, X.-N.; Lou, H.-X. Natural stilbenes: An overview. Nat. Prod. Rep. 2009, 26, 916–935. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Singh, R.; Verma, S.S.; Rai, V.; Kaschula, C.H.; Maiti, P.; Gupta, S.C. Health benefits of resveratrol: Evidence from clinical studies. Med. Res. Rev. 2019, 39, 1851–1891. [Google Scholar] [CrossRef]
- Keylor, M.H.; Matsuura, B.S.; Stephenson, C.R.J. Chemistry and Biology of Resveratrol-Derived Natural Products. Chem. Rev. 2015, 115, 8976–9027. [Google Scholar] [CrossRef]
- Kuczowsky, J.A.; Gillick, J.G. Polymer-bound antioxidants. Rubber Chem. Technol. 1984, 57, 621–651. [Google Scholar] [CrossRef]
- Menichetti, S.; Viglianisi, C.; Liguori, F.; Cogliati, C.; Boragno, L.; Stagnaro, P.; Losio, S.; Sacchi, M.C. Ethylene-based copolymers with tunable content of polymerizable hindered phenols as nonreleasing macromolecular additives. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6393–6406. [Google Scholar] [CrossRef]
- Richaud, E.; Fayolle, B.; Verdu, J. Polypropylene stabilization by hindered phenols—Kinetic aspects. Polym. Degrad. Stab. 2011, 96, 1–11. [Google Scholar] [CrossRef]
- Lucarini, M.; Pedulli, G.F. Free radical intermediates in the inhibition of the autoxidation reaction. Chem. Soc. Rev. 2010, 39, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the Activity of Phenolic Antioxidants: Theoretical Method, Analysis of Substituent Effects, and Application to Major Families of Antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Doba, T.; Burton, G.W.; Ingold, K.U. EPR spectra of soma a-tocopherol model compounds. Polar and conformational effects on their relation to antioxidant activity. J. Am. Chem. Soc. 1983, 105, 6505–6506. [Google Scholar] [CrossRef]
- Burton, G.W.; Le Page, Y.; Gabe, E.J.; Ingold, K.U. Antioxidant activity of Vitamin E and related phenols. Importance of stereoelectroniuc factors. J. Am. Chem. Soc. 1980, 102, 7791–7792. [Google Scholar] [CrossRef]
- Zhang, H.-Y. Structure-activity relationship and rational design strategies for radical-scavenging antioxidants. Curr. Comput. Aided Drug Des. 2005, 1, 65–72. [Google Scholar] [CrossRef]
- Malmström, J.; Gupta, V.; Engman, L. Novel Antioxidants: Unexpected Rearrangements in the Radical Cyclization Approach to 2,3-Dihydrobenzo[b]thiophene-5-ol Derivatives. J. Org. Chem. 1998, 63, 3318–3323. [Google Scholar] [CrossRef]
- Malmström, J.; Jonsson, M.; Cotgreave, I.A.; Hammarström, L.; Sjödin, M.; Engman, L. The Antioxidant Profile of 2,3-Dihydrobenzo[b]furan-5-ol and Its 1-Thio, 1-Seleno, and 1-Telluro Analogues. J. Am. Chem. Soc. 2001, 123, 3434–3440. [Google Scholar] [CrossRef]
- Hussain, H.H.; Babic, G.; Durst, T.; Wright, J.S.; Flueraru, M.; Chichirau, A.; Chepelev, L.L. Development of Novel Antioxidants: Design, Synthesis, and Reactivity. J. Org. Chem. 2003, 68, 7023–7032. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Pratt, D.A.; Seal, J.R.; Wijtmans, M.; Porter, N.A. Lipid-Soluble 3-Pyridinol Antioxidants Spare α-Tocopherol and Do Not Efficiently Mediate Peroxidation of Cholesterol Esters in Human Low-Density Lipoprotein. J. Med. Chem. 2005, 48, 6787–6789. [Google Scholar] [CrossRef]
- Nam, T.-G.; Rector, C.L.; Kim, H.-Y.; Sonnen, A.F.-P.; Meyer, R.; Nau, W.M.; Atkinson, J.; Rintoul, J.; Pratt, D.A.; Porter, N.A. Tetrahydro-1,8-naphthyridinol Analogues of α-Tocopherol as Antioxidants in Lipid Membranes and Low-Density Lipoproteins. J. Am. Chem. Soc. 2007, 129, 10211–10219. [Google Scholar] [CrossRef] [PubMed]
- Serwa, R.; Nam, T.-G.; Valgimigli, L.; Culbertson, S.; Rector, C.L.; Jeong, B.-S.; Pratt, D.A.; Porter, N.A. Preparation and Investigation of Vitamin B6-Derived Aminopyridinol Antioxidants. Chem. Eur. J. 2010, 16, 14106–14114. [Google Scholar] [CrossRef] [PubMed]
- Ku, J.-M.; Nam, T.-G.; Park, H.-G.; Porter, N.A.; Jeong, B.-S. New synthetic route to N-tocopherol derivatives: Synthesis of pyrrolopyridinol analogue of α-tocopherol from pyridoxine. Org. Biomol. Chem. 2011, 9, 1749–1755. [Google Scholar] [CrossRef]
- Li, B.; Harjani, J.R.; Cormier, N.S.; Madarati, H.; Atkinson, J.; Cosa, G.; Pratt, D.A. Besting Vitamin E: Sidechain Substitution is Key to the Reactivity of Naphthyridinol Antioxidants in Lipid Bilayers. J. Am. Chem. Soc. 2013, 135, 1394–1405. [Google Scholar] [CrossRef]
- Barclay, L.R.C.; Edwards, C.E.; Vinqvist, M.R. Media Effects on Antioxidant Activities of Phenols and Catechols. J. Am. Chem. Soc. 1999, 121, 6226–6231. [Google Scholar] [CrossRef]
- Foti, M.C.; Ross, L.; Barclay, C.; Ingold, K.U. The Role of Hydrogen Bonding on the H-Atom-Donating Abilities of Catechols and Naphthalene Diols and on a Previously Overlooked Aspect of Their Infrared Spectra. J. Am. Chem. Soc. 2002, 124, 12881–12888. [Google Scholar] [CrossRef]
- Lucarini, M.; Mugnaini, V.; Pedulli, G.F.; Guerra, M. Hydrogen-Bonding Effects on the Properties of Phenoxyl Radicals. An EPR, Kinetic, and Computational Study. J. Am. Chem. Soc. 2003, 125, 8318–8329. [Google Scholar] [CrossRef]
- Amorati, R.; Franchi, P.; Pedulli, G.F. Intermolecular Hydrogen Bonding Modulates the Hydrogen-Atom-Donating Ability of Hydroquinones. Angew. Chem. Int. Ed. 2007, 46, 6336–6338. [Google Scholar] [CrossRef]
- Amorati, R.; Valgimigli, L. Modulation of the antioxidant activity of phenols by non-covalent interactions. Org. Biomol. Chem. 2012, 10, 4147–4158. [Google Scholar] [CrossRef]
- Liu, Z.-Q. Chemical methods to evaluate antioxidant activity. Chem. Rev. 2010, 110, 5675–5691. [Google Scholar] [CrossRef]
- Foti, M.C. Use and abuse of DPPH radical. J. Agric. Food Chem. 2015, 63, 8765–8776. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ou, B.; Prior, R.L. The chemistry behind antioxidant capacity assays. J. Agric. Food Chem. 2005, 53, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Amorati, R.; Fumo, M.G.; Menichetti, S.; Mugnaini, V.; Pedulli, G.F. Electronic and Hydrogen Bonding Effects on the Chain-Breaking Activity of Sulfur-Containing Phenolic Antioxidants. J. Org. Chem. 2006, 71, 6325–6332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robillard, B.; Hughes, L.; Slaby, M.; Lindsay, D.A.; Ingold, K.U. Synthesis of 2-Substituted 5,7,8-Trimethyl-6-hydroxythiochromans and Purported Syntheses of Sulfur-Containing Analogues of Vitamin E. J. Org. Chem. 1986, 51, 1700–1704. [Google Scholar] [CrossRef]
- Zahalka, H.A.; Robillard, B.; Hughes, L.; Lusztyk, J.; Burton, G.W.; Janzen, E.G.; Kotake, Y.; Ingold, K.U. Antioxidant Activity of 1-Thio-a-tocopherol and Related Compounds. EPR, ENDOR, and UV-Visible Absorption Spectra of Some of the Derived Phenoxy1 Radicals. J. Org. Chem. 1988, 53, 3739–3745. [Google Scholar] [CrossRef]
- Capozzi, G.; Nativi, C.; Sarri, P.; Nostro, P.L.; Menichetti, S. Easy synthesis of polyphenolic 4-thiaflavans with a ‘double-faced’ antioxidant activity. Chem. Commun. 2001, 551–552. [Google Scholar] [CrossRef]
- Menichetti, S.; Aversa, M.C.; Cimino, F.; Contini, A.; Viglianisi, C.; Tomaino, A. Synthesis and “double-faced” antioxidant activity of polyhydroxylated 4-thiaflavans. Org. Biomol. Chem. 2005, 3, 3066–3072. [Google Scholar] [CrossRef]
- Amorati, A.; Fumo, M.G.; Pedulli, G.F.; Menichetti, S.; Pagliuca, C.; Viglianisi, C. Antioxidant and Antiradical Activity of Hydroxy-Substituted 4-Thiaflavanes. Helv. Chim. Acta 2006, 89, 2462–2472. [Google Scholar] [CrossRef]
- Amorati, R.; Attanasi, O.A.; Favi, G.; Menichetti, S.; Pedulli, G.F.; Viglianisi, C. Amphiphilic antioxidants from “cashew nut shell liquid” (CNSL) waste. Org. Biomol. Chem. 2011, 9, 1352–1355. [Google Scholar] [CrossRef]
- Buzzini, P.; Menichetti, S.; Pagliuca, C.; Viglianisi, C.; Branda, E.; Turchetti, B. Antimycotic activity of 4-thioisosteres of flavonoids towards yeast and yeast-like microorganisms. Bioorg. Med. Chem. Lett. 2008, 18, 3731–3733. [Google Scholar] [CrossRef]
- Amorati, A.; Cavalli, A.; Fumo, M.G.; Masetti, M.; Menichetti, S.; Pagliuca, C.; Pedulli, G.F.; Viglianisi, C. Kinetic and Thermochemical Study of the Antioxidant Activity of Sulfur-Containing Analogues of Vitamin E. Chem. Eur. J. 2007, 13, 8223–8230. [Google Scholar] [CrossRef] [PubMed]
- Böhm, F.; Edge, R.; Land, E.J.; McGarvey, D.J.; Truscott, G. Carotenoids Enhance Vitamin E Antioxidant Efficiency. J. Am. Chem. Soc. 1997, 119, 621–622. [Google Scholar] [CrossRef]
- Collins, C.A.; Fry, F.H.; Holme, A.L.; Yiakouvaki, A.; Al-Qenaei, A.; Pourzand, C.; Jacob, C. Towards multifunctional antioxidants: Synthesis, electrochemistry, in vitro and cell culture evaluation of compounds with ligand/catalytic properties. Org. Biomol. Chem. 2005, 3, 1541–1546. [Google Scholar] [CrossRef] [PubMed]
- Goupy, P.; Vulcain, E.; Caris-Veyrat, C.; Dangles, O. Dietary antioxidants as inhibitors of the heme-induced peroxidation of linoleic acid: Mechanism of action and synergism. Free Radic. Biol. Med. 2007, 43, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Cavalli, A.; Bergamini, C.; Fato, R.; Lenaz, G.; Rosini, M.; Bartolini, M.; Andrisano, V.; Melchiorre, C. Toward a Rational Design of Multitarget-Directed Antioxidants: Merging Memoquin and Lipoic Acid Molecular Frameworks. J. Med. Chem. 2009, 52, 7883–7886. [Google Scholar] [CrossRef] [PubMed]
- Amorati, R.; Valgimigli, L.; Panzella, L.; Napolitano, A.; D’Ischia, M. 5-S-Lipoylhydroxytyrosol, a Multidefense Antioxidant Featuring a Solvent-Tunable Peroxyl Radical-Scavenging 3-Thio-1,2-dihydroxybenzene Motif. J. Org. Chem. 2013, 78, 9857–9864. [Google Scholar] [CrossRef] [PubMed]
- Tassano, E.; Alama, A.; Basso, A.; Dondo, G.; Galatini, A.; Riva, R.; Banfi, L. Conjugation of Hydroxytyrosol with Other Natural Phenolic Fragments: From Waste to Antioxidants and Antitumour Compounds. Eur. J. Org. Chem. 2015, 6710–6726. [Google Scholar] [CrossRef]
- Lodovici, M.; Menichetti, S.; Viglianisi, C.; Caldini, S.; Giuliani, E. Polyhydroxylated 4-thiaflavans as multipotent antioxidants: Protective effect on oxidative DNA damage in vitro. Bioorg. Med. Chem. Lett. 2006, 16, 1957–1960. [Google Scholar] [CrossRef]
- Viglianisi, C.; Menichetti, S.; Morelli, P.; Baschieri, A.; Amorati, R. From catechol-tocopherol to catechol-hydroquinone polyphenolic antioxidant hybrids. Heteroat. Chem. 2018, e21466. [Google Scholar] [CrossRef]
- Amorati, R.; Catarzi, F.; Menichetti, S.; Pedulli, G.F.; Viglianisi, C. Effect of ortho-SR Groups on O-H Bond Strength and H-Atom Donating Ability of Phenols: A Possible Role for the Tyr-Cys Link in Galactose Oxidase Active Site? J. Am. Chem. Soc. 2008, 130, 237–244. [Google Scholar] [CrossRef]
- Schaefer, T.; Wildman, T.A.; Salman, R.S. The perpendicular conformation of 2-hydroxythiophenol. Intramolecular hydrogen bonding to a specific lone pair. J. Am. Chem. Soc. 1980, 102, 107–110. [Google Scholar] [CrossRef]
- Schaefer, T.; Salman, R.S.; Wildman, T.A.; Clark, P.D. Conformational consequences of intramolecular hydrogen bonding by OH to the directional lone-pair of sulfur in derivatives of methyl phenyl sulfide, diphenyl sulfide, and diphenyl disulfide. Can. J. Chem. 1982, 60, 342–348. [Google Scholar] [CrossRef]
- Schaefer, T.; McKinnon, D.M.; Sebastian, R.; Peeling, J.; Penner, G.H.; Veregin, R.P. Concerning lone-pair stereospecificity of intramolecular OH hydrogen bonds to oxygen and sulfur in solution. Can. J. Chem. 1987, 65, 908–914. [Google Scholar] [CrossRef]
- Himo, F.; Eriksson, L.A.; Blomberg, M.R.A.; Siegbahn, P.E.M. Substituent effects on OH bond strength and hyperfine properties of phenol, as model for modified tyrosyl radicals in proteins. Int. J. Quantum Chem. 2000, 76, 714–723. [Google Scholar] [CrossRef]
- Schaefer, T.; Penner, G.H. Mechanisms of long-range 13C, 13C spin–spin coupling in thioanisole and its derivatives. Conformational applications. Can. J. Chem. 1988, 66, 1229–1238. [Google Scholar] [CrossRef]
- Viglianisi, C.; Bartolozzi, M.G.; Pedulli, G.F.; Amorati, R.; Menichetti, S. Optimization of the Antioxidant Activity of Hydroxy-Substituted 4-Thiaflavanes: A Proof-of-Concept Study. Chem. Eur. J. 2011, 17, 12396–12404. [Google Scholar] [CrossRef]
- Viglianisi, C.; Amorati, R.; Di Pietro, L.; Menichetti, S. A Straightforward Route to Potent Phenolic Chain-Breaking Antioxidants by Acid-Promoted Transposition of 1,4-Benzo[b]oxathiines to Dihydrobenzo[b]thiophenes. Chem. Eur. J. 2015, 21, 16639–16645. [Google Scholar] [CrossRef]
- Viglianisi, C.; Di Pietro, L.; Meoni, V.; Amorati, R.; Menichetti, S. From simple phenols to potent chain-breaking antioxidants by transposition of benzo[1,4]oxathiines to benzo[b]thiophenes. Arkivoc 2019, 2019, 65–85. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Politzer, P. Sigma-hole bonding: Molecules containing group VI atoms. J. Mol. Model. 2007, 13, 1033–1038. [Google Scholar] [CrossRef]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef] [PubMed]
- Fick, R.J.; Kroner, G.M.; Nepal, B.; Magnani, R.; Horowitz, S.; Houtz, R.L.; Scheiner, S.; Trievel, R.C. Sulfur–Oxygen Chalcogen Bonding Mediates AdoMet Recognition in the Lysine Methyltransferase SET7/9. ACS Chem. Biol. 2016, 11, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Politzer, P. Simultaneous σ-hole and hydrogen bonding by sulfur- and selenium-containing heterocycles. Int. J. Quantum Chem. 2008, 108, 2770–2781. [Google Scholar] [CrossRef]
- Fanfrlik, J.; Prada, A.; Padelkova, Z.; Pecina, A.; Machacek, J.; Lepsik, M.; Holub, J.; Ruzicka, A.; Hnyk, D.; Hobza, P. The Dominant Role of Chalcogen Bonding in the Crystal Packing of 2D/3D Aromatics. Angew. Chem. Int. Ed. 2014, 53, 10139–10142. [Google Scholar] [CrossRef]
- Nziko, V.P.; Scheiner, S. Intramolecular S···O Chalcogen Bond as Stabilizing Factor in Geometry of Substituted Phenyl-SF3 Molecules. J. Org. Chem. 2015, 80, 2356–2363. [Google Scholar] [CrossRef]
- Rittner, R.; Ducati, L.C.; Tormena, C.F.; Fiorin, B.C.; Braga, C.B. Conformational preferences for some 5-substituted 2-acetylthiophenes through infrared spectroscopy and theoretical calculations. Spectrochim. Acta Part A 2011, 79, 1071–1076. [Google Scholar] [CrossRef]
- Shanks, D.; Amorati, R.; Fumo, M.G.; Pedulli, G.F.; Valgimigli, L.; Engman, L. Synthesis and Antioxidant Profile of all-rac-α-Selenotocopherol. J. Org. Chem. 2006, 71, 1033–1038. [Google Scholar] [CrossRef]
- Amorati, R.; Pedulli, G.F.; Valgimigli, L.; Johansson, H.; Engman, L. Organochalcogen Substituents in Phenolic Antioxidants. Org. Lett. 2010, 12, 2326–2329. [Google Scholar] [CrossRef]
- Johansson, H.; Shanks, D.; Engman, L.; Amorati, R.; Pedulli, G.F.; Valgimigli, L. Long-Lasting Antioxidant Protection: A Regenerable BHA Analogue. J. Org. Chem. 2010, 75, 7535–7541. [Google Scholar] [CrossRef]
- Mugesh, G.; Du Mont, W.-W.; Sies, H. Chemistry of biologically important synthetic organoselenium compounds. Chem. Rev. 2001, 101, 2125–2179. [Google Scholar] [CrossRef]
- Reich, H.J.; Hondal, R.J. Why Nature Chose Selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- Bhuyan, B.J.; Mugesh, G. Biological and biochemical aspects of selenium compounds. In Organoselenium Chemistry: Synthesis and Reactions; Wirth, T., Ed.; Wiley-VCH Verlag & Co.: Weinheim, Germany, 2012. [Google Scholar]
- Nogueira, C.W.; Zeni, G.; Rocha, J.B.T. Organoselenium and Organotellurium Compounds: Toxicology and Pharmacology. Chem. Rev. 2004, 104, 6255–6286. [Google Scholar] [CrossRef] [PubMed]
- Orian, L.; Toppo, S. Organochalcogen peroxidase mimetics as potential drugs: A long story of a promise still unfulfilled. Free Radic. Biol. Med. 2014, 66, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Mugesh, G.; Singh, H.B. Synthetic organoselenium compounds as antioxidants: Glutathione peroxidase activity. Chem. Soc. Rev. 2000, 29, 347–357. [Google Scholar] [CrossRef]
- Vessman, K.; Ekstrom, M.; Berglund, M.; Andersson, C.-M. Catalytic Antioxidant Activity of Diaryl Tellurides in a Two-Phase Lipid Peroxidation Model. J. Org. Chem. 1995, 60, 4461–4467. [Google Scholar] [CrossRef]
- Singh, V.P.; Poon, J.-F.; Engman, L. Turning Pyridoxine into a Catalytic Chain-Breaking and Hydroperoxide-Decomposing Antioxidant. J. Org. Chem. 2013, 78, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Amorati, R.; Valgimigli, L.; Dinér, P.; Bakhtiari, K.; Saeedi, M.; Engman, L. Multi-faceted Reactivity of Alkyltellurophenols Towards Peroxyl Radicals: Catalytic Antioxidant versus Thiol-Depletion Effect. Chem. Eur. J. 2013, 19, 7510–7522. [Google Scholar] [CrossRef]
- Poon, J.-F.; Yan, J.; Jorner, K.; Ottosson, H.; Donau, C.; Singh, V.P.; Gates, P.J.; Engman, L. Substituent Effects in Chain-Breaking Aryltellurophenol Antioxidants. Chem. Eur. J. 2018, 24, 3520–3527. [Google Scholar] [CrossRef]
- Kumar, S.; Johansson, H.; Kanda, T.; Engman, L.; Müller, T.; Jonsson, M.; Pedulli, G.F.; Petrucci, S.; Valgimigli, L. Catalytic Chain-Breaking Pyridinol Antioxidants. Org. Lett. 2008, 10, 4895–4898. [Google Scholar] [CrossRef]
- Kumar, S.; Johansson, H.; Kanda, T.; Engman, L.; Müller, T.; Bergenudd, H.; Jonsson, M.; Pedulli, G.F.; Amorati, R.; Valgimigli, L. Catalytic Chain-Breaking Pyridinol Antioxidants. J. Org. Chem. 2010, 75, 716–725. [Google Scholar] [CrossRef]
- Kumar, S.; Johansson, H.; Engman, L.; Valgimigli, L.; Amorati, R.; Fumo, M.G.; Pedulli, G.F. Regenerable Chain-Breaking 2,3-Dihydrobenzo[b]selenophene-5-ol Antioxidants. J. Org. Chem. 2007, 72, 2583–2595. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Svartström, O.; Phadnis, P.; Engman, L.; Ott, M.K. Exploring a synthetic organoselenium compound for antioxidant pharmacotherapy—Toxicity and effects on ROS-production. Bioorg. Med. Chem. 2010, 18, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Yan, J.; Poon, J.-F.; Gates, P.G.; Butcher, R.J.; Engman, L. Chain-Breaking Phenolic 2,3-Dihydrobenzo[b]selenophene Antioxidants: Proximity Effects and Regeneration Studies. Chem. Eur. J. 2017, 23, 15080–15088. [Google Scholar] [CrossRef] [PubMed]
- Tanini, D.; Panzella, L.; Amorati, R.; Capperucci, A.; Pizzo, E.; Napolitano, A.; Menichetti, S.; D’Ischia, M. Resveratrol-based benzoselenophenes with an enhanced antioxidant and chain breaking capacity. Org. Biomol. Chem. 2015, 13, 5757–5764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paegle, E.; Domracheva, I.; Turovska, B.; Petrova, M.; Kanepe-Lapsa, I.; Gulbe, A.; Liepinsh, E.; Arsenyan, P. Natural-Antioxidant-Inspired Benzo[b]selenophenes: Synthesis, Redox Properties, and Antiproliferative Activity. Chem. Asian J. 2016, 11, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Rad. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzi, A. Molecular mechanism of α-tocopherol action. Free Rad. Biol. Med. 2007, 43, 16–21. [Google Scholar] [CrossRef]
- Bowry, V.W.; Ingold, K.U. The Unexpected Role of Vitamin E (α-Tocopherol) in the Peroxidation of Human Low-Density Lipoprotein. Acc. Chem. Res. 1999, 32, 27–34. [Google Scholar] [CrossRef]
- Shanks, D.; Frisell, H.; Ottosson, H.; Engman, L. Design principles for α-tocopherol analogues. Org. Biomol. Chem. 2006, 4, 846–852. [Google Scholar] [CrossRef]
- Menichetti, S.; Amorati, R.; Bartolozzi, M.G.; Pedulli, G.F.; Salvini, A.; Viglianisi, C. A Straightforward Hetero-Diels–Alder Approach to (2-ambo,4′R,8′R)-α/β/γ/δ-4-Thiatocopherol. Eur. J. Org. Chem. 2010, 2218–2225. [Google Scholar] [CrossRef]
- Singh, V.P.; Poon, J.-F.; Engman, L. Catalytic Antioxidants: Regenerable Tellurium Analogues of Vitamin E. Org. Lett. 2013, 15, 6274–6277. [Google Scholar] [CrossRef] [PubMed]
- Poon, J.-F.; Singh, V.G.; Yan, J.; Engman, L. Regenerable Antioxidants-Introduction of Chalcogen Substituents into Tocopherols. Chem. Eur. J. 2015, 21, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Menichetti, S.; Amorati, R.; Meoni, V.; Tofani, L.; Caminati, G.; Viglianisi, C. Role of Noncovalent Sulfur···Oxygen Interactions in Phenoxyl Radical Stabilization: Synthesis of Super Tocopherol-like Antioxidants. Org. Lett. 2016, 18, 5464–5467. [Google Scholar] [CrossRef] [PubMed]
- Poon, J.-F.; Yan, J.; Singh, V.P.; Gates, P.J.; Engman, L. Regenerable Radical-Trapping Tellurobistocopherol Antioxidants. J. Org. Chem. 2016, 81, 12540–12544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viglianisi, C.; Vasa, K.; Tanini, D.; Capperucci, A.; Amorati, R.; Valgimigli, L.; Baschieri, A.; Menichetti, S. Ditocopheryl Sulfides and Disulfides: Synthesis and Antioxidant Profile. Chem. Eur. J. 2019, 25, 9108–9116. [Google Scholar] [CrossRef]


















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viglianisi, C.; Menichetti, S. Chain Breaking Antioxidant Activity of Heavy (S, Se, Te) Chalcogens Substituted Polyphenols. Antioxidants 2019, 8, 487. https://doi.org/10.3390/antiox8100487
Viglianisi C, Menichetti S. Chain Breaking Antioxidant Activity of Heavy (S, Se, Te) Chalcogens Substituted Polyphenols. Antioxidants. 2019; 8(10):487. https://doi.org/10.3390/antiox8100487
Chicago/Turabian StyleViglianisi, Caterina, and Stefano Menichetti. 2019. "Chain Breaking Antioxidant Activity of Heavy (S, Se, Te) Chalcogens Substituted Polyphenols" Antioxidants 8, no. 10: 487. https://doi.org/10.3390/antiox8100487