

Hypoxic Inducible Factor Stabilization in Pericytes beyond Erythropoietin Production: The Good and the Bad
 , and
, and
Abstract
:1. Introduction
2. Pericytes (PCs)—Endothelial Cells (ECs) Crosstalk
3. Ischemia–Reperfusion Injury and HIF
3.1. HIF-1α and Cerebral IRI
3.2. HIF-1α and Renal IRI
3.3. HIF-1α and Hepatic IRI
4. Ischemia–Reperfusion Injury and Pericytes
5. HIF Stabilization in Pericytes
6. Hints of Potential Therapeutic Strategies
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ahmed, T.A.; El-Badri, N. Pericytes: The Role of Multipotent Stem Cells in Vascular Maintenance and Regenerative Medicine. Adv. Exp. Med. Biol. 2018, 1079, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Holm, A.; Heumann, T.; Augustin, H.G. Microvascular Mural Cell Organotypic Heterogeneity and Functional Plasticity. Trends Cell Biol. 2018, 28, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Geevarghese, A.; Herman, I.M. Pericyte-endothelial crosstalk: Implications and opportunities for advanced cellular therapies. Transl. Res. 2014, 163, 296–306. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cowled, P.; Fitridge, R. Pathophysiology of Reperfusion Injury. In Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists [Internet]; Fitridge, R., Thompson, M., Eds.; University of Adelaide Press: Adelaide, AU, USA, 2011; p. 18. Available online: https://www.ncbi.nlm.nih.gov/books/NBK534267/# (accessed on 22 February 2024).
- Wang, Y.; Yu, X. Stabilizing Hypoxia-Inducible Factor to Manage Anemia in Chronic Kidney Disease: From Basic Theory to Clinical Study. Kidney Dis. 2024, 10, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Kisseleva, T.; Brenner, D.A.; Duffield, J.S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008, 173, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Gao, J.; Ding, P.; Gao, Y. The role of endothelial cell-pericyte interactions in vascularization and diseases. J. Adv. Res. 2024. [Google Scholar] [CrossRef]
- Cai, J.; Kehoe, O.; Smith, G.M.; Hykin, P.; Boulton, M.E. The angiopoietin/Tie-2 system regulates pericyte survival and recruitment in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2163–2171. [Google Scholar] [CrossRef]
- Feng, Y.; vom Hagen, F.; Pfister, F.; Djokic, S.; Hoffmann, S.; Back, W.; Wagner, P.; Lin, J.; Deutsch, U.; Hammes, H.P. Impaired pericyte recruitment and abnormal retinal angiogenesis because of angiopoietin-2 overexpression. Thromb. Haemost. 2007, 97, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Antonelli-Orlidge, A.; Saunders, K.B.; Smith, S.R.; D’Amore, P.A. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc. Natl. Acad. Sci. USA 1989, 86, 4544–4548. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N. TGF-beta: From latent to active. Microbes Infect. 1999, 1, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Oshima, H.; Taketo, M.M. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996, 179, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.P.; Seki, T.; Goss, K.A.; Imamura, T.; Yi, Y.; Donahoe, P.K.; Li, L.; Miyazono, K.; ten Dijke, P.; Kim, S.; et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.; Goumans, M.J.; Sjöstrand, L.J.; van Rooijen, M.A.; Ward, D.; Levéen, P.; Xu, X.; ten Dijke, P.; Mummery, C.L.; Karlsson, S. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001, 20, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Kalén, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar] [CrossRef]
- Kemp, S.S.; Lin, P.K.; Sun, Z.; Castaño, M.A.; Yrigoin, K.; Penn, M.R.; Davis, G.E. Molecular basis for pericyte-induced capillary tube network assembly and maturation. Front. Cell Dev. Biol. 2022, 10, 943533. [Google Scholar] [CrossRef]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 2001, 153, 543–553. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Ghalehbandi, S.; Yuzugulen, J.; Pranjol, M.Z.I.; Pourgholami, M.H. The role of VEGF in cancer-induced angiogenesis and research progress of drugs targeting VEGF. Eur. J. Pharmacol. 2023, 949, 175586. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Xue, Y.; Hedlund, E.M.; Zhong, Z.; Tritsaris, K.; Tondelli, B.; Lucchini, F.; Zhu, Z.; Dissing, S.; Cao, Y. VEGFR1-mediated pericyte ablation links VEGF and PlGF to cancer-associated retinopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 856–861. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kunej, T. Integrative Map of HIF1A Regulatory Elements and Variations. Genes 2021, 12, 1526. [Google Scholar] [CrossRef] [PubMed]
- Troise, D.; Infante, B.; Mercuri, S.; Netti, G.S.; Ranieri, E.; Gesualdo, L.; Stallone, G.; Pontrelli, P. Hypoxic State of Cells and Immunosenescence: A Focus on the Role of the HIF Signaling Pathway. Biomedicines 2023, 11, 2163. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhong, Z.F.; Wang, S.P.; Vong, C.T.; Yu, B.; Wang, Y.T. HIF-1: Structure, biology, and natural modulators. Chin. J. Nat. Med. 2021, 19, 521–527. [Google Scholar] [CrossRef]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Haase, V.H. The VHL tumor suppressor and HIF: Insights from genetic studies in mice. Cell Death Differ. 2008, 15, 650–659. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Meng, Y.M.; Jiang, X.; Zhao, X.; Meng, Q.; Wu, S.; Chen, Y.; Kong, X.; Qiu, X.; Su, L.; Huang, C.; et al. Hexokinase 2-driven glycolysis in pericytes activates their contractility leading to tumor blood vessel abnormalities. Nat. Commun. 2021, 12, 6011. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Tian, R.; Abel, E.D. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation 2001, 103, 2961–2966. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Ikeda, Y.; Oka, S.; Fong, G.H.; Tian, R.; Sadoshima, J. Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1α and upregulation of peroxisome proliferator-activated receptor-α. Circ. Res. 2013, 112, 1135–1149. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jang, S.; Lewis, T.S.; Powers, C.; Khuchua, Z.; Baines, C.P.; Wipf, P.; Javadov, S. Elucidating Mitochondrial Electron Transport Chain Supercomplexes in the Heart During Ischemia-Reperfusion. Antioxid. Redox Signal. 2017, 27, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, G.; Alasmari, A.; Mouli, S.; Eldoumani, H.; Quindry, J.; McGinnis, G.; Fu, X.; Berlin, A.; Peters, B.; Zhong, J.; et al. Cardioprotective HIF-1α-frataxin signaling against ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H867–H879. [Google Scholar] [CrossRef] [PubMed]
- Kandilci, H.B.; Richards, M.A.; Fournier, M.; Şimşek, G.; Chung, Y.J.; Lakhal-Littleton, S.; Swietach, P. Cardiomyocyte Na+/H+ Exchanger-1 Activity Is Reduced in Hypoxia. Front. Cardiovasc. Med. 2021, 7, 617038. [Google Scholar] [CrossRef]
- Aldakkak, M.; Stowe, D.F.; Heisner, J.S.; Spence, M.; Camara, A.K. Enhanced Na+/H+ exchange during ischemia and reperfusion impairs mitochondrial bioenergetics and myocardial function. J. Cardiovasc. Pharmacol. 2008, 52, 236–244. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shimoda, L.A.; Fallon, M.; Pisarcik, S.; Wang, J.; Semenza, G.L. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L941–L949. [Google Scholar] [CrossRef]
- Liu, Y.; Zou, J.; Liu, X.; Zhang, Q. MicroRNA-138 attenuates myocardial ischemia reperfusion injury through inhibiting mitochondria-mediated apoptosis by targeting HIF1-α. Exp. Ther. Med. 2019, 18, 3325–3332. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Song, W.; Liang, Q.; Cai, M.; Tian, Z. HIF-1α-induced up-regulation of microRNA-126 contributes to the effectiveness of exercise training on myocardial angiogenesis in myocardial infarction rats. J. Cell. Mol. Med. 2020, 24, 12970–12979. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef]
- Quiles, J.M.; Gustafsson, Å.B. The role of mitochondrial fission in cardiovascular health and disease. Nat. Rev. Cardiol. 2022, 19, 723–736. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pedriali, G.; Ramaccini, D.; Bouhamida, E.; Wieckowski, M.R.; Giorgi, C.; Tremoli, E.; Pinton, P. Perspectives on mitochondrial relevance in cardiac ischemia/reperfusion injury. Front. Cell Dev. Biol. 2022, 10, 1082095. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chen, P.; Zhong, J.; Cheng, Y.; Chen, H.; He, Y.; Chen, C. HIF-1α in myocardial ischemia-reperfusion injury (Review). Mol. Med. Rep. 2021, 23, 352. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, S.F.; Pan, M.X.; Tang, J.C.; Cheng, J.; Zhao, D.; Zhang, Y.; Liao, H.B.; Liu, R.; Zhuang, Y.; Zhang, Z.F.; et al. Arginine is neuroprotective through suppressing HIF-1α/LDHA-mediated inflammatory response after cerebral ischemia/reperfusion injury. Mol. Brain 2020, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liao, X.Y.; Pan, M.X.; Tang, J.C.; Chen, S.F.; Zhang, Y.; Lu, P.X.; Lu, L.J.; Zou, Y.Y.; Qin, X.P.; et al. Glycine Exhibits Neuroprotective Effects in Ischemic Stroke in Rats through the Inhibition of M1 Microglial Polarization via the NF-κB p65/Hif-1α Signaling Pathway. J. Immunol. 2019, 202, 1704–1714. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Wang, R.H.; Wang, H.; Long, C.L.; Wang, H. Brain protection against ischemic stroke using choline as a new molecular bypass treatment. Acta Pharmacol. Sin. 2015, 36, 1416–1425. [Google Scholar] [CrossRef]
- Jin, W.; Zhao, J.; Yang, E.; Wang, Y.; Wang, Q.; Wu, Y.; Tong, F.; Tan, Y.; Zhou, J.; Kang, C. Neuronal STAT3/HIF-1α/PTRF axis-mediated bioenergetic disturbance exacerbates cerebral ischemia-reperfusion injury via PLA2G4A. Theranostics 2022, 12, 3196–3216. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, N.; Yang, X.; Fang, Y.; Huang, Y.; Shi, W.; Li, W.; Ding, M.; An, Q.; Zhao, Y. Nitric oxide promotes cerebral ischemia/reperfusion injury through upregulating hypoxia-inducible factor1-α-associated inflammation and apoptosis in rats. Neurosci. Lett. 2023, 795, 137034. [Google Scholar] [CrossRef]
- Liu, H.; Li, Y.; Xiong, J. The Role of Hypoxia-Inducible Factor-1 Alpha in Renal Disease. Molecules 2022, 27, 7318. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fu, Z.J.; Wang, Z.Y.; Xu, L.; Chen, X.H.; Li, X.X.; Liao, W.T.; Ma, H.K.; Jiang, M.D.; Xu, T.T.; Xu, J.; et al. HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020, 36, 101671. [Google Scholar] [CrossRef] [PubMed]
- Chun, N.; Coca, S.G.; He, J.C. A protective role for microRNA-688 in acute kidney injury. J. Clin. Investig. 2018, 128, 5216–5218. [Google Scholar] [CrossRef]
- Xu, X.; Song, N.; Zhang, X.; Jiao, X.; Hu, J.; Liang, M.; Teng, J.; Ding, X. Renal Protection Mediated by Hypoxia Inducible Factor-1α Depends on Proangiogenesis Function of miR-21 by Targeting Thrombospondin 1. Transplantation 2017, 101, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Ji, J.L.; Wen, Y.; Cao, J.Y.; Kharbuja, N.; Ni, W.J.; Yin, D.; Feng, S.T.; Liu, H.; Lv, L.L.; et al. HIF-1α is transcriptionally regulated by NF-κB in acute kidney injury. Am. J. Physiol. Renal Physiol. 2021, 321, F225–F235. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Wakabayashi, G.; Takayanagi, A.; Shimazu, M.; Matsumoto, K.; Obara, H.; Shimizu, N.; Kitajima, M. Transfer of the interleukin-1 receptor antagonist gene into rat liver abrogates hepatic ischemia-reperfusion injury. Transplantation 2002, 74, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Sun, H.; Peng, Y.; Liang, C.; Zheng, C.; Wang, L.; Yang, J. Mu-opioid receptor alleviated ferroptosis in hepatic ischemia-reperfusion injury via the HIF-1α/KCNQ1OT1 axis. Am. J. Physiol. Cell Physiol. 2023, 324, C927–C940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Du, P.; Luo, K.; Li, Y.; Liu, Z.; Wang, W.; Zeng, C.; Ye, Q.; Xiao, Q. Hypoxia-inducible factor-1alpha protects the liver against ischemia-reperfusion injury by regulating the A2B adenosine receptor. Bioengineered 2021, 12, 3737–3752. [Google Scholar] [CrossRef]
- Khairoun, M.; van der Pol, P.; de Vries, D.K.; Lievers, E.; Schlagwein, N.; de Boer, H.C.; Bajema, I.M.; Rotmans, J.I.; van Zonneveld, A.J.; Rabelink, T.J.; et al. Renal ischemia-reperfusion induces a dysbalance of angiopoietins, accompanied by proliferation of pericytes and fibrosis. Am. J. Physiol. Renal Physiol. 2013, 305, F901–F910. [Google Scholar] [CrossRef]
- de Vries, D.K.; Khairoun, M.; Lindeman, J.H.; Bajema, I.M.; de Heer, E.; Roest, M.; van Zonneveld, A.J.; van Kooten, C.; Rabelink, T.J.; Schaapherder, A.F.; et al. Renal ischemia-reperfusion induces release of angiopoietin-2 from human grafts of living and deceased donors. Transplantation 2013, 96, 282–289. [Google Scholar] [CrossRef]
- Smith, S.W.; Eardley, K.S.; Croft, A.P.; Nwosu, J.; Howie, A.J.; Cockwell, P.; Isacke, C.M.; Buckley, C.D.; Savage, C.O. CD248+ stromal cells are associated with progressive chronic kidney disease. Kidney Int. 2011, 80, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Pai, C.H.; Lin, S.R.; Liu, C.H.; Pan, S.Y.; Hsu, H.; Chen, Y.T.; Yen, C.T.; Yu, I.S.; Wu, H.L.; Lin, S.L.; et al. Targeting fibroblast CD248 attenuates CCL17-expressing macrophages and tissue fibrosis. Sci. Rep. 2020, 10, 16772. [Google Scholar] [CrossRef] [PubMed]
- Shaw, I.; Rider, S.; Mullins, J.; Hughes, J.; Péault, B. Pericytes in the renal vasculature: Roles in health and disease. Nat. Rev. Nephrol. 2018, 14, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Freitas, F.; Attwell, D. Pericyte-mediated constriction of renal capillaries evokes no-reflow and kidney injury following ischaemia. eLife 2022, 11, e74211. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, L.; Li, X.; Deng, Y.; Chen, J.; Huang, M.; Zhu, F.; Gao, Z.; Wu, L.; Hong, Q.; Feng, Z.; et al. The PI3K-Akt-mTOR pathway mediates renal pericyte-myofibroblast transition by enhancing glycolysis through HKII. J. Transl. Med. 2023, 21, 323. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, C.; Hong, Q.; Zhuang, K.; Ren, X.; Cui, S.; Dong, Z.; Wang, Q.; Bai, X.; Chen, X. Regulation of pericyte metabolic reprogramming restricts the AKI to CKD transition. Metabolism 2023, 145, 155592. [Google Scholar] [CrossRef]
- Castellano, G.; Franzin, R.; Stasi, A.; Divella, C.; Sallustio, F.; Pontrelli, P.; Lucarelli, G.; Battaglia, M.; Staffieri, F.; Crovace, A.; et al. Complement Activation During Ischemia/Reperfusion Injury Induces Pericyte-to-Myofibroblast Transdifferentiation Regulating Peritubular Capillary Lumen Reduction Through pERK Signaling. Front. Immunol. 2018, 9, 1002. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chou, Y.H.; Pan, S.Y.; Shao, Y.H.; Shih, H.M.; Wei, S.Y.; Lai, C.F.; Chiang, W.C.; Schrimpf, C.; Yang, K.C.; Lai, L.C.; et al. Methylation in pericytes after acute injury promotes chronic kidney disease. J. Clin. Investig. 2020, 130, 4845–4857. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Shrouder, J.J.; Calandra, G.M.; Filser, S.; Varga, D.P.; Besson-Girard, S.; Mamrak, U.; Dorok, M.; Bulut-Impraim, B.; Seker, F.B.; Gesierich, B.; et al. Continued dysfunction of capillary pericytes promotes no-reflow after experimental stroke in vivo. Brain 2023, 147, 1057–1074. [Google Scholar] [CrossRef]
- Attwell, D.; Mishra, A.; Hall, C.N.; O’Farrell, F.M.; Dalkara, T. What is a pericyte? J. Cereb. Blood Flow. Metab. 2016, 36, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Pericyte-specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol. Neurodegener. 2010, 5, 32. [Google Scholar] [CrossRef]
- Zhou, S.Y.; Guo, Z.N.; Zhang, D.H.; Qu, Y.; Jin, H. The Role of Pericytes in Ischemic Stroke: Fom Cellular Functions to Therapeutic Targets. Front. Mol. Neurosci. 2022, 15, 866700. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hausenloy, D.J.; Botker, H.E.; Engstrom, T.; Erlinge, D.; Heusch, G.; Ibanez, B.; Kloner, R.A.; Ovize, M.; Yellon, D.M.; Garcia-Dorado, D. Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: Trials and tribulations. Eur. Heart J. 2017, 38, 935–941. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, F.M.; Mastitskaya, S.; Hammond-Haley, M.; Freitas, F.; Wah, W.R.; Attwell, D. Capillary pericytes mediate coronary no-reflow after myocardial ischaemia. eLife 2017, 6, e29280. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Beltrami, A.P.; Thomas, A.C.; Spinetti, G.; Alvino, V.V.; Avolio, E.; Veneziano, C.; Rolle, I.G.; Sponga, S.; Sangalli, E.; et al. The longevity-associated BPIFB4 gene supports cardiac function and vascularization in ageing cardiomyopathy. Cardiovasc. Res. 2023, 119, 1583–1595. [Google Scholar] [CrossRef]
- Rolle, I.G.; Crivellari, I.; Zanello, A.; Mazzega, E.; Dalla, E.; Bulfoni, M.; Avolio, E.; Battistella, A.; Lazzarino, M.; Cellot, A. Heart failure impairs the mechanotransduction properties of human cardiac pericytes. J. Mol. Cell. Cardiol. 2021, 151, 15–30. [Google Scholar] [CrossRef]
- Quijada, P.; Park, S.; Zhao, P.; Kolluri, K.S.; Wong, D.; Shih, K.D.; Fang, K.; Pezhouman, A.; Wang, L.; Daraei, A.; et al. Cardiac pericytes mediate the remodeling response to myocardial infarction. J. Clin. Investig. 2023, 133, e162188. [Google Scholar] [CrossRef]
- Avolio, E.; Campagnolo, P.; Katare, R.; Madeddu, P. The role of cardiac pericytes in health and disease: Therapeutic targets for myocardial infarction. Nat. Rev. Cardiol. 2024, 21, 106–118. [Google Scholar] [CrossRef]
- Shih, H.M.; Pan, S.Y.; Wu, C.J.; Chou, Y.H.; Chen, C.Y.; Chang, F.C.; Chen, Y.T.; Chiang, W.C.; Tsai, H.C.; Chen, Y.M.; et al. Transforming growth factor-β1 decreases erythropoietin production through repressing hypoxia-inducible factor 2α in erythropoietin-producing cells. J. Biomed. Sci. 2021, 28, 73. [Google Scholar] [CrossRef] [PubMed]
- Souma, T.; Nezu, M.; Nakano, D.; Yamazaki, S.; Hirano, I.; Sekine, H.; Dan, T.; Takeda, K.; Fong, G.H.; Nishiyama, A.; et al. Erythropoietin Synthesis in Renal Myofibroblasts Is Restored by Activation of Hypoxia Signaling. J. Am. Soc. Nephrol. 2016, 27, 428–438. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pan, S.Y.; Tsai, P.Z.; Chou, Y.H.; Chang, Y.T.; Chang, F.C.; Chiu, Y.L.; Chiang, W.C.; Hsu, T.; Chen, Y.M.; Chu, T.S.; et al. Kidney pericyte hypoxia-inducible factor regulates erythropoiesis but not kidney fibrosis. Kidney Int. 2021, 99, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Storti, F.; Santambrogio, S.; Crowther, L.M.; Otto, T.; Abreu-Rodríguez, I.; Kaufmann, M.; Hu, C.J.; Dame, C.; Fandrey, J.; Wenger, R.H. A novel distal upstream hypoxia response element regulating oxygen-dependent erythropoietin gene expression. Haematologica 2014, 99, e45-8. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hörl, W.H. Anaemia management and mortality risk in chronic kidney disease. Nat. Rev. Nephrol. 2013, 9, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Yang, C.C.; Pan, S.Y.; Chou, Y.H.; Chang, F.C.; Lai, C.F.; Tsai, M.H.; Hsu, H.L.; Lin, C.H.; Chiang, W.C.; et al. DNA methyltransferase inhibition restores erythropoietin production in fibrotic murine kidneys. J. Clin. Investig. 2016, 126, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Schrimpf, C.; Duffield, J.S. Mechanisms of fibrosis: The role of the pericyte. Curr. Opin. Nephrol. Hypertens. 2011, 20, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Souma, T.; Suzuki, N.; Yamamoto, M. Renal erythropoietin-producing cells in health and disease. Front. Physiol. 2015, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Tanaka, T.; Nangaku, M. Prolyl hydroxylase domain inhibitors as a novel therapeutic approach against anemia in chronic kidney disease. Kidney Int. 2017, 92, 306–312. [Google Scholar] [CrossRef]
- Kawakami, T.; Mimura, I.; Shoji, K.; Tanaka, T.; Nangaku, M. Hypoxia and fibrosis in chronic kidney disease: Crossing at pericytes. Kidney Int. Suppl. 2014, 4, 107–112. [Google Scholar] [CrossRef]
- Herbek, S.; Edmonston, D.L.; Souma, T. Hypoxia signaling in renal pericytes-is it safe to activate? Kidney Int. 2021, 99, 1267–1269. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.C.; Baumann, J.; Huang, S.F.; Kindler, D.; Schroeter, A.; Kachappilly, N.; Gassmann, M.; Rudin, M.; Ogunshola, O.O. Pericyte hypoxia-inducible factor-1 (HIF-1) drives blood-brain barrier disruption and impacts acute ischemic stroke outcome. Angiogenesis 2021, 24, 823–842. [Google Scholar] [CrossRef] [PubMed]
- Baumann, J.; Tsao, C.C.; Patkar, S.; Huang, S.F.; Francia, S.; Magnussen, S.N.; Gassmann, M.; Vogel, J.; Köster-Hegmann, C.; Ogunshola, O.O. Pericyte, but not astrocyte, hypoxia inducible factor-1 (HIF-1) drives hypoxia-induced vascular permeability in vivo. Fluids Barriers CNS 2022, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Mayo, J.N.; Bearden, S.E. Driving the Hypoxia-Inducible Pathway in Human Pericytes Promotes Vascular Density in an Exosome-Dependent Manner. Microcirculation 2015, 22, 711–723. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ogunshola, O.O.; Tsao, C.C. Harnessing the power of pericytes and hypoxia-inducible factor-1 to modulate stroke outcome. Neural Regen. Res. 2024, 19, 473–474. [Google Scholar] [CrossRef]
- Lee, L.L.; Khakoo, A.Y.; Chintalgattu, V. Cardiac pericytes function as key vasoactive cells to regulate homeostasis and disease. FEBS Open Bio 2021, 11, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Cantrell, A.C.; Zeng, H.; Zhu, S.H.; Chen, J.X. Emerging Role of Pericytes and Their Secretome in the Heart. Cells 2021, 10, 548. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Garrison, A.T.; Bignold, R.E.; Wu, X.; Johnson, J.R. Pericytes: The lung-forgotten cell type. Front. Physiol. 2023, 14, 1150028. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.D.; Kim, M.; D’Agati, V.D.; Lee, H.T. Ischemic preconditioning provides both acute and delayed protection against renal ischemia and reperfusion injury in mice. J. Am. Soc. Nephrol. 2006, 17, 3115–3123. [Google Scholar] [CrossRef]
- Lu, N.; Li, X.; Tan, R.; An, J.; Cai, Z.; Hu, X.; Wang, F.; Wang, H.; Lu, C.; Lu, H. HIF-1α/Beclin1-Mediated Autophagy Is Involved in Neuroprotection Induced by Hypoxic Preconditioning. J. Mol. Neurosci. 2018, 66, 238–250. [Google Scholar] [CrossRef]
- Jia, P.; Wu, X.; Dai, Y.; Teng, J.; Fang, Y.; Hu, J.; Zou, J.; Liang, M.; Ding, X. MicroRNA-21 Is Required for Local and Remote Ischemic Preconditioning in Multiple Organ Protection Against Sepsis. Crit. Care Med. 2017, 45, e703–e710. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhong, H.; Bosch-Marce, M.; Fox-Talbot, K.; Wang, L.; Wei, C.; Trush, M.A.; Semenza, G.L. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1 alpha. Cardiovasc. Res. 2008, 77, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guo, Z.; Wu, C.; Tu, Y.; Wu, Y.; Xie, E.; Yu, C.; Sun, W.; Li, X.; Zheng, J.; et al. Ischemia preconditioning alleviates ischemia/reperfusion injury-induced coronary no-reflow and contraction of microvascular pericytes in rats. Microvasc. Res. 2022, 142, 104349. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Liu, Y.; Li, S.P.; Li, J.J.; Zhang, Z.; Xiao, X.C.; Ou, Y.; Wang, H.; Cai, J.Z.; Yang, S. Hypoxia inducible factor 1α promotes interleukin-1 receptor antagonist expression during hepatic ischemia-reperfusion injury. World J. Gastroenterol. 2022, 28, 5573–5588. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Iwano, M.; Higgins, D.F.; Yamaguchi, Y.; Nakatani, K.; Harada, K.; Kubo, A.; Akai, Y.; Rankin, E.B.; Neilson, E.G.; et al. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am. J. Physiol. Renal Physiol. 2008, 295, F1023–F1029. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Tanaka, T.; Ishii, T.; Wakashima, T.; Fukui, K.; Nangaku, M. Prolyl hydroxylase inhibition protects the kidneys from ischemia via upregulation of glycogen storage. Kidney Int. 2020, 97, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Hill, P.; Shukla, D.; Tran, M.G.; Aragones, J.; Cook, H.T.; Carmeliet, P.; Maxwell, P.H. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2008, 19, 39–46. [Google Scholar] [CrossRef]
- Fleming, R.E.; Sly, W.S. Hepcidin: A putative iron-regulatory hormone relevant to hereditary hemochromatosis and the anemia of chronic disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8160–8162. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Liu, B.C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N. Engl. J. Med. 2019, 381, 1011–1022. [Google Scholar] [CrossRef]
- Riopel, M.; Moon, J.S.; Bandyopadhyay, G.K.; You, S.; Lam, K.; Liu, X.; Kisseleva, T.; Brenner, D.; Lee, Y.S. Inhibition of prolyl hydroxylases increases hepatic insulin and decreases glucagon sensitivity by an HIF-2α-dependent mechanism. Mol. Metab. 2020, 41, 101039. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kadowaki, T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 2013, 17, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Coyne, D.W.; Roger, S.D.; Shin, S.K.; Kim, S.G.; Cadena, A.A.; Moustafa, M.A.; Chan, T.M.; Besarab, A.; Chou, W.; Bradley, C.; et al. Roxadustat for CKD-related Anemia in Non-dialysis Patients. Kidney Int. Rep. 2020, 6, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Sadiku, P.; Willson, J.A.; Dickinson, R.S.; Murphy, F.; Harris, A.J.; Lewis, A.; Sammut, D.; Mirchandani, A.S.; Ryan, E.; Watts, E.R.; et al. Prolyl hydroxylase 2 inactivation enhances glycogen storage and promotes excessive neutrophilic responses. J. Clin. Investig. 2017, 127, 3407–3420. [Google Scholar] [CrossRef]
- Walmsley, S.R.; Chilvers, E.R.; Thompson, A.A.; Vaughan, K.; Marriott, H.M.; Parker, L.C.; Shaw, G.; Parmar, S.; Schneider, M.; Sabroe, I.; et al. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J. Clin. Investig. 2011, 121, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.H.; Pan, S.Y.; Lin, S.L. Pleotropic effects of hypoxia-inducible factor-prolyl hydroxylase domain inhibitors: Are they clinically relevant? Kidney Res. Clin. Pract. 2023, 42, 27–38. [Google Scholar] [CrossRef]
- Catrina, S.B.; Zheng, X. Hypoxia and hypoxia-inducible factors in diabetes and its complications. Diabetologia 2021, 64, 709–716. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Warmke, N.; Griffin, K.J.; Cubbon, R.M. Pericytes in diabetes-associated vascular disease. J. Diabetes Complicat. 2016, 30, 1643–1650. [Google Scholar] [CrossRef]
- Thangarajah, H.; Yao, D.; Chang, E.I.; Shi, Y.; Jazayeri, L.; Vial, I.N.; Galiano, R.D.; Du, X.L.; Grogan, R.; Galvez, M.G.; et al. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 13505–13510. [Google Scholar] [CrossRef]
- Khan, M.; Dhammu, T.S.; Sakakima, H.; Shunmugavel, A.; Gilg, A.G.; Singh, A.K.; Singh, I. The inhibitory effect of S-nitrosoglutathione on blood-brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J. Neurochem. 2012, 123 (Suppl. S2), 86–97. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Khan, M.; Khan, H.; Singh, I.; Singh, A.K. Hypoxia inducible factor-1 alpha stabilization for regenerative therapy in traumatic brain injury. Neural Regen. Res. 2017, 12, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Chau, N.M.; Rogers, P.; Aherne, W.; Carroll, V.; Collins, I.; McDonald, E.; Workman, P.; Ashcroft, M. Identification of novel small molecule inhibitors of hypoxia-inducible factor-1 that differentially block hypoxia-inducible factor-1 activity and hypoxia-inducible factor-1alpha induction in response to hypoxic stress and growth factors. Cancer Res. 2005, 65, 4918–4928. [Google Scholar] [CrossRef] [PubMed]
- Karmazyn, M. The role of the myocardial sodium-hydrogen exchanger in mediating ischemic and reperfusion injury. From amiloride to cariporide. Ann. N. Y. Acad. Sci. 1999, 874, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Tani, M. Mechanisms of Ca2+ overload in reperfused ischemic myocardium. Annu. Rev. Physiol. 1990, 52, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Chang, F.C.; Schrimpf, C.; Chen, Y.T.; Wu, C.F.; Wu, V.C.; Chiang, W.C.; Kuhnert, F.; Kuo, C.J.; Chen, Y.M.; et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am. J. Pathol. 2011, 178, 911–923. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, Y.T.; Chang, F.C.; Wu, C.F.; Chou, Y.H.; Hsu, H.L.; Chiang, W.C.; Shen, J.; Chen, Y.M.; Wu, K.D.; Tsai, T.J.; et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011, 80, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Omote, Y.; Deguchi, K.; Kono, S.; Liu, N.; Liu, W.; Kurata, T.; Yamashita, T.; Ikeda, Y.; Abe, K. Neurovascular protection of cilostazol in stroke-prone spontaneous hypertensive rats associated with angiogenesis and pericyte proliferation. J. Neurosci. Res. 2014, 92, 369–374. [Google Scholar] [CrossRef]
- Deguchi, K.; Liu, N.; Liu, W.; Omote, Y.; Kono, S.; Yunoki, T.; Deguchi, S.; Yamashita, T.; Ikeda, Y.; Abe, K. Pericyte protection by edaravone after tissue plasminogen activator treatment in rat cerebral ischemia. J. Neurosci. Res. 2014, 92, 1509–1519. [Google Scholar] [CrossRef]
- Baganha, F.; de Jong, R.C.M.; Peters, E.A.; Voorham, W.; Jukema, J.W.; Delibegovic, M.; de Vries, M.R.; Quax, P.H.A. Atorvastatin pleiotropically decreases intraplaque angiogenesis and intraplaque haemorrhage by inhibiting ANGPT2 release and VE-Cadherin internalization. Angiogenesis 2021, 24, 567–581. [Google Scholar] [CrossRef]





{kind=link}
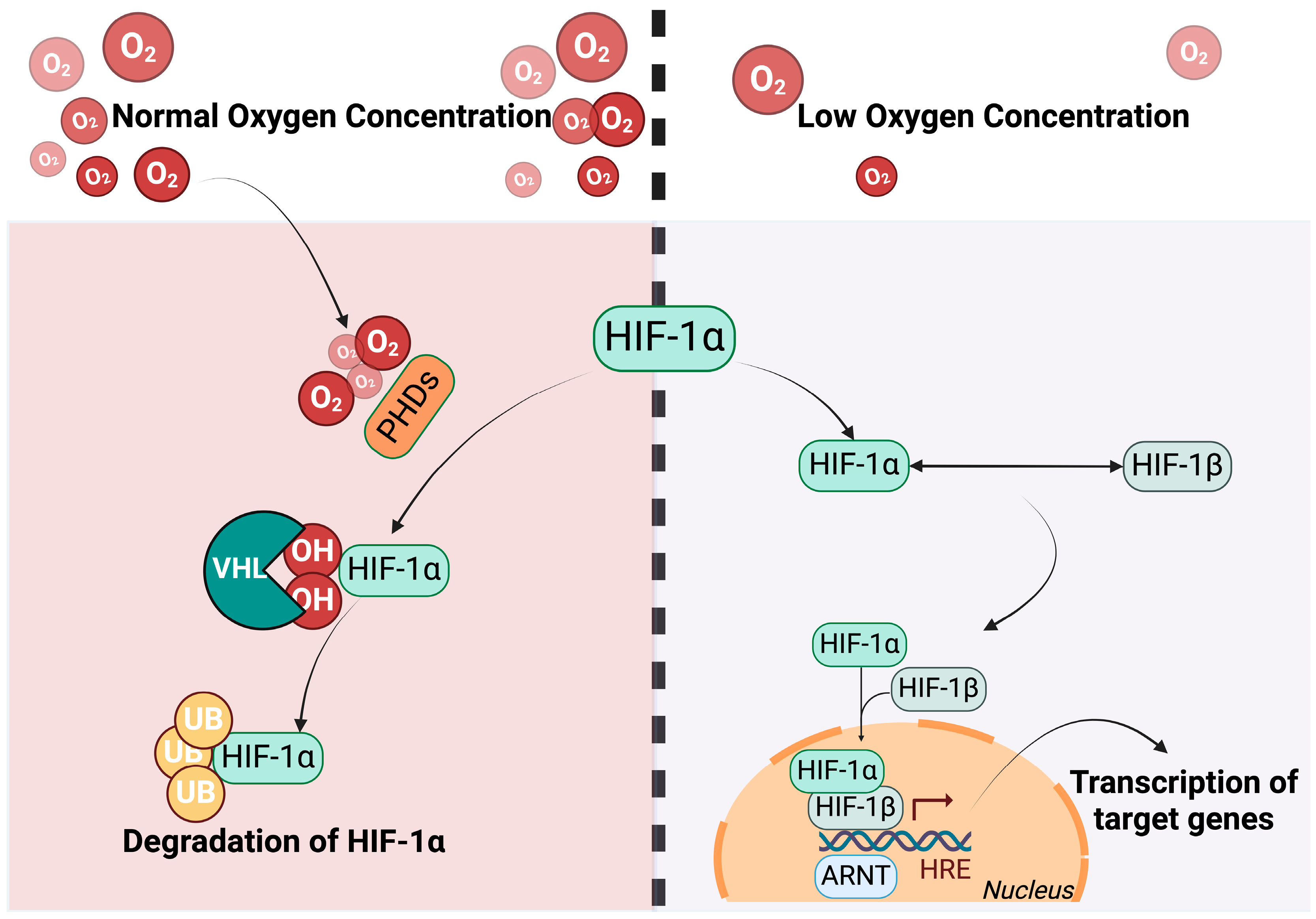{kind=link}
{kind=link}
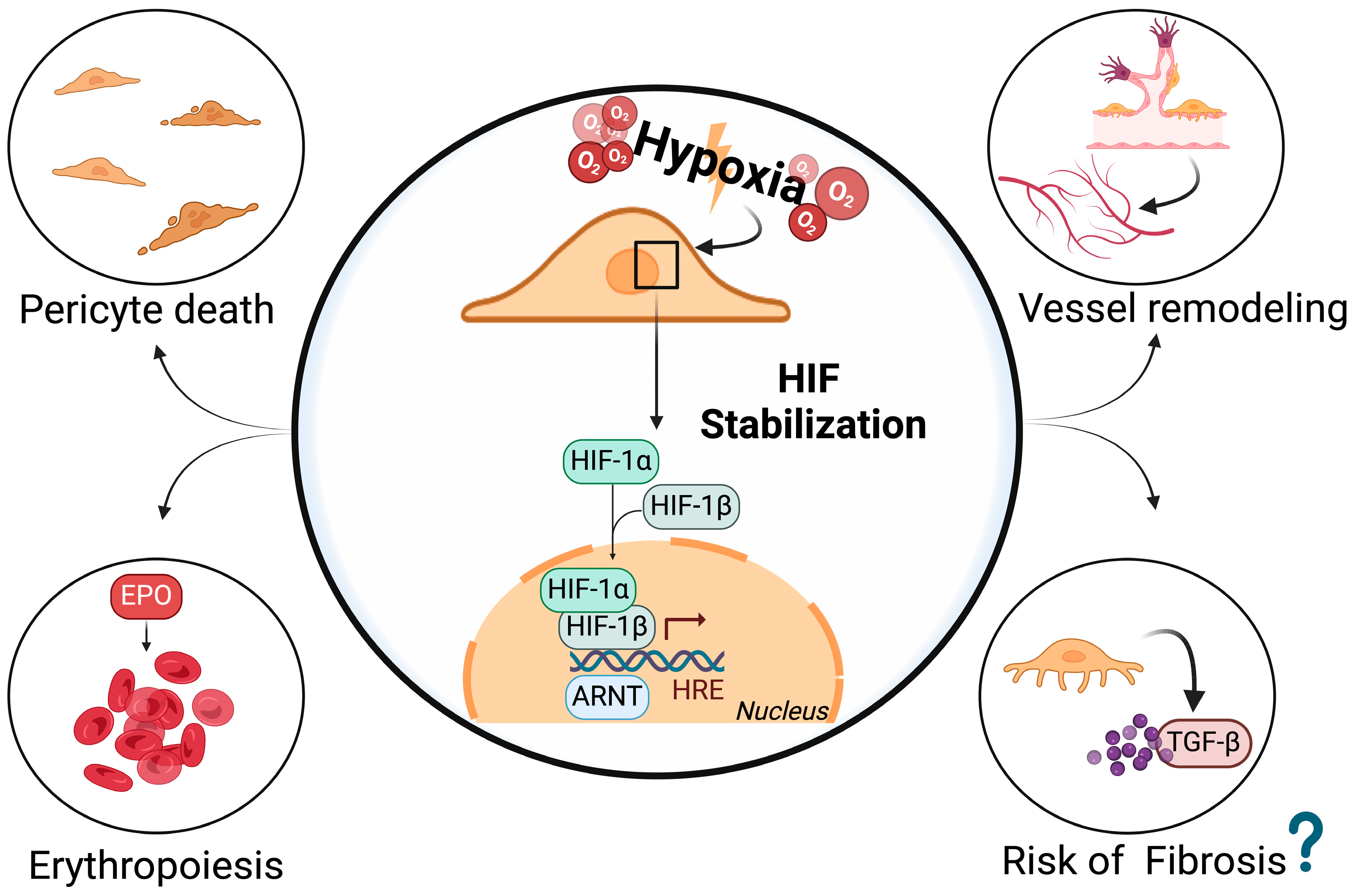{kind=link}
| Organ/Cell Type | Model | Major Findings | References |
|---|---|---|---|
| Kidney | Murine | IRI causes a dysbalance in angiopoietins, loss of ECs, PCs proliferation and fibrosis | Khairoun M et al. [54] |
| Kidney | Human | IRI causes Increased levels of Ang2 and EC activation | de Vries DK et al. [55] |
| Kidney | Murine | IRI causes upregulation of the fibrotic marker CD248 | Pai CH et al. [57] |
| Kidney | Murine | Blocking Rho kinase during IRI reduces AKI by inhibiting PCs contraction and increasing medullary blood flow | Freitas F et al. [59] |
| Kidney | Murine | IRI induces increased expression of PKM2 in PCs, indicating that glycolysis plays a crucial role as a metabolic pathway for PTM | Chen Y et al. [62] |
| Kidney | Murine | Activation of FAO or inhibition of glycolysis during IRI can prevent AKI to CKD progression | Xu C et al. [63] |
| Kidney | Swine | PCs are the target of complement activation leading to a profibrotic maladaptive cellular response. C1-INH may be a therapeutic strategy to counteract the development of PMT | Castellano G et al. [64] |
| Kidney | Murine | PC epigenetic modifications during IRI play a role in their transition towards a profibrotic and proliferative phenotype. | Chou YH et al. [65] |
| Brain | Murine | PDGFRβ is expressed in PCs in the adult brain indicating that genetic disruption of PDGFRβ signaling leads to a PCs specific injury | Winkler EA et al. [69] |
| Heart | Murine | After myocardial infarct PCs regulate the induction of genes associated with vascular permeability, extracellular matrix production, basement membrane degradation, and TGF-β signaling | Quijada P et al. [75] |
| Fibroblasts cell lines | Murine | TGF-β1 can suppress HIF-2α expression by activating ALK5 leading to decreased EPO production | Shih HM et al. [82] |
| Kidney | Murine | The decreased EPO production in myofibroblasts could be partially reversed by the inactivation of PHD | Souma T et al. [83] |
| Kidney | Murine | HIF stabilization resulted in increased EPO production and polycythemia but no noticeable change in renal fibrosis. | Pan SY et al. [84] |
| Therapeutic Intervention | Effects | References |
|---|---|---|
| Ischemic preconditioning | Increase autophagy pathways, decrease proinflammatory cytokine production and prevent microvascular PC constriction and apoptosis | Joo JD et al. [100] Lu N. et al. [101] Jia P. et al. [102] Cai Z. et al. [103] |
| HIF-prolyl hydroxylase inhibitors | Reduce ROS levels and attenuate oxidative stress and lower the levels of hepcidin and ferritin. Regulate immunity systems and metabolic processes. | Ito M et al. [107] Fleming RE et al. [109] Chen N. et al. [111] Riopel M. et al. [112] Sadiku P. et al. [115] Walmsley SR et al. [116] |
| Deferoxamine | Correct the impairment of the HIF-1α signaling pathway induced by hyperglycemia | Thangarajah H et al. [120] |
| S-nitrosoglutathione | Stabilize HIF-1α expression | Khan M. et al. [121] Khan M. et al. [122] |
| Na+/H+ exchanger-1 inhibitors | Cardioprotective role | Karmazyn M. [124] Tani M. [125] |
| PDGFR-β and/or VEGFR2 blocking | Reduce microvascular rarefaction, inflammation, and interstitial fibrosis. Reduce PCs-myofibroblast transition | Lin SL et al. [126] Chen YT et al. [127] |
| Imatinib | Reduce PC–myofibroblast transition | Chen YT et al. [127] |
| Cilostazol | Decrease the expression and activity of MMP-9, elevate the expression of VEGFR3 and increase angiogenesis | Omote Y. et al. [128] |
| Edaravone | Suppress MMP-9 production | Deguchi K. et al. [129] |
| Atorvastatin | Inhibit Ang2 release and increase the expression of VE-cadherin | Baganha F. et al. [130] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Troise, D.; Infante, B.; Mercuri, S.; Piccoli, C.; Lindholm, B.; Stallone, G. Hypoxic Inducible Factor Stabilization in Pericytes beyond Erythropoietin Production: The Good and the Bad. Antioxidants 2024, 13, 537. https://doi.org/10.3390/antiox13050537
Troise D, Infante B, Mercuri S, Piccoli C, Lindholm B, Stallone G. Hypoxic Inducible Factor Stabilization in Pericytes beyond Erythropoietin Production: The Good and the Bad. Antioxidants. 2024; 13(5):537. https://doi.org/10.3390/antiox13050537
Chicago/Turabian StyleTroise, Dario, Barbara Infante, Silvia Mercuri, Claudia Piccoli, Bengt Lindholm, and Giovanni Stallone. 2024. "Hypoxic Inducible Factor Stabilization in Pericytes beyond Erythropoietin Production: The Good and the Bad" Antioxidants 13, no. 5: 537. https://doi.org/10.3390/antiox13050537