

Inflammation in Myocardial Ischemia/Reperfusion Injury: Underlying Mechanisms and Therapeutic Potential

Abstract
:1. Introduction: Myocardial Ischemia/Reperfusion Injury
2. Initiation of Inflammation in Cardiac I/R Injury
3. Neutrophils
3.1. Neutrophil Priming
3.2. Neutrophil Activation
3.3. Neutrophil Polarization and Function in I/R
3.4. Neutrophil Metabolism
4. Macrophages
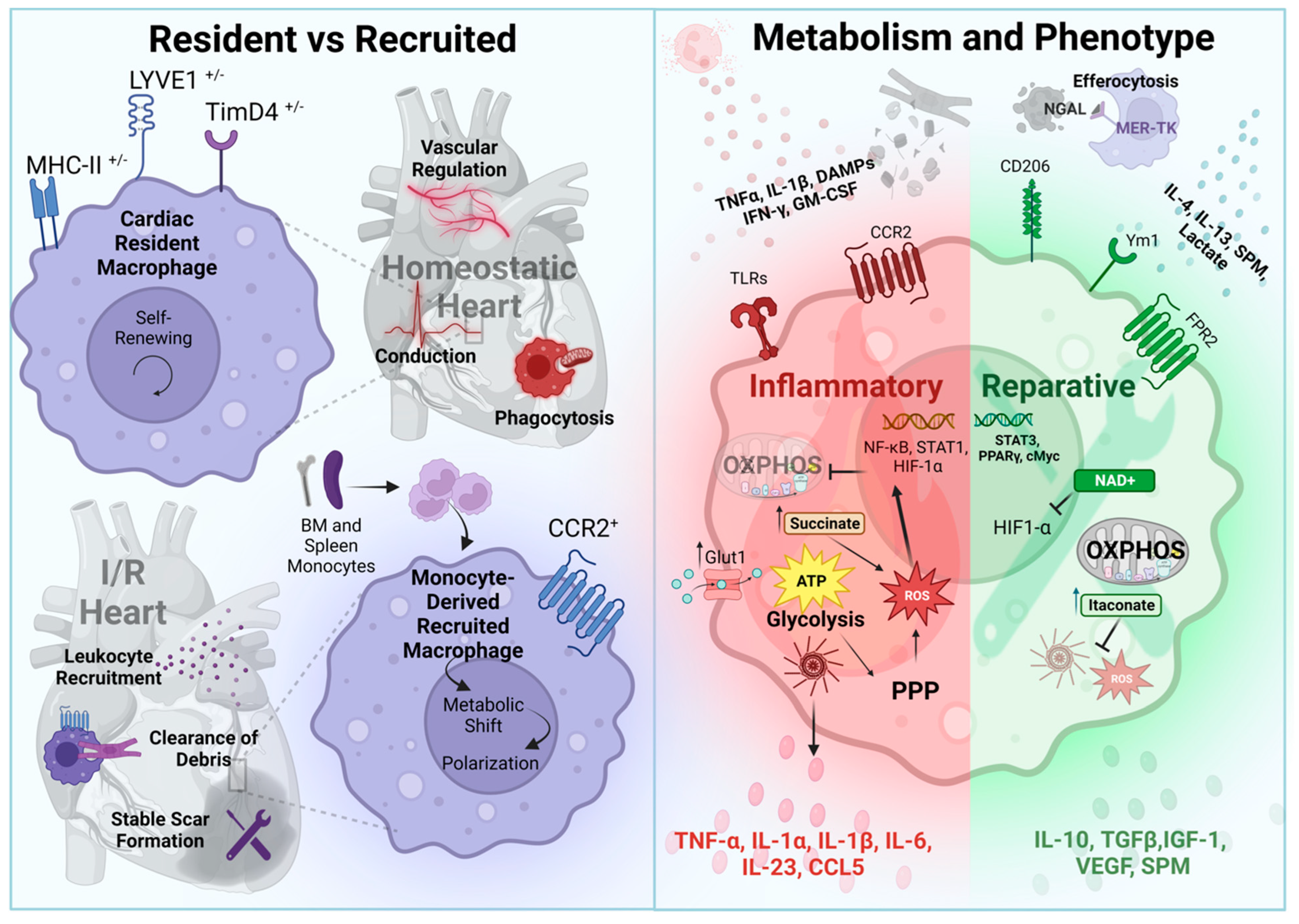
4.1. Cardiac-Resident Macrophages
4.2. Cardiac Recruited Macrophages
4.3. Macrophages in Resolution
4.4. Macrophage Metabolism and Functional Regulation
5. Dendritic Cells
6. T Cells
7. B Cells
8. Translational Potential of Targeting Inflammation during I/R Injury
8.1. Broad Approaches to Inflammatory Inhibition
8.2. Focused Targeting of Inflammatory Cells
8.2.1. The Complement Pathway
8.2.2. Targeting Immune Cell Recruitment and Adhesion
8.2.3. Targeting Immune Cell Function and Inflammatory Mediators
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elgendy, I.Y.; Mahtta, D.; Pepine, C.J. Medical Therapy for Heart Failure Caused by Ischemic Heart Disease. Circ. Res. 2019, 124, 1520–1535. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.; Wang, Z.; Zhang, J.; Zhou, Z. Myocardial ischemia-reperfusion injury; Molecular mechanisms and prevention. Microvasc. Res. 2023, 149, 104565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.Y.K.; Cochran, B.J.; Thomas, S.R.; Rye, K.A. Impact of Reperfusion on Temporal Immune Cell Dynamics after Myocardial Infarction. J. Am. Heart Assoc. 2023, 12, e027600. [Google Scholar] [CrossRef] [PubMed]
- Silvis, M.J.M.; Dengler, S.E.K.G.; Odille, C.A.; Mishra, M.; van der Kaaij, N.P.; Doevendans, P.A.; Sluijter, J.P.G.; de Kleijn, D.P.V.; de Jager, S.C.A.; Bosch, L.; et al. Damage-Associated Molecular Patterns in Myocardial Infarction and Heart Transplantation: The Road to Translational Success. Front. Immunol. 2020, 11, 599511. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.J. Oxygen; oxidative stress; hypoxia; heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Botker, H.E.; Engstrom, T.; Erlinge, D.; Heusch, G.; Ibanez, B.; Kloner, R.A.; Ovize, M.; Yellon, D.M.; Garcia-Dorado, D. Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: Trials and tribulations. Eur. Heart J. 2017, 38, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, B.; Heusch, G.; Ovize, M.; Van de Werf, F. Evolving Therapies for Myocardial Ischemia/Reperfusion Injury. J. Am. Coll. Cardiol. 2015, 65, 1454–1471. [Google Scholar] [CrossRef]
- Schirone, L.; Forte, M.; D’Ambrosio, L.; Valenti, V.; Vecchio, D.; Schiavon, S.; Spinosa, G.; Sarto, G.; Petrozza, V.; Frati, G.; et al. An Overview of the Molecular Mechanisms Associated with Myocardial Ischemic Injury: State of the Art and Translational Perspectives. Cells 2022, 11, 1165. [Google Scholar] [CrossRef]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Olson, E.N.; Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair. Nat. Rev. Cardiol. 2018, 15, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.Z.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020, 577, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Zuurbier, C.J.; Abbate, A.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Collino, M.; De Kleijn, D.P.V.; Downey, J.M.; Pagliaro, P.; Preissner, K.T.; Takahashi, M.; et al. Innate immunity as a target for acute cardioprotection. Cardiovasc. Res. 2019, 115, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Cabrera-Fuentes, H.A.; Devaux, Y.; Frangogiannis, N.G.; Frantz, S.; Guzik, T.; Liehn, E.A.; Gomes, C.P.C.; Schulz, R.; Hausenloy, D.J. Immune cells as targets for cardioprotection: New players and novel therapeutic opportunities. Cardiovasc. Res. 2019, 115, 1117–1130. [Google Scholar] [CrossRef]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple Facets of NF-kB in the Heart. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef]
- Oyama, J.; Blais, C., Jr.; Liu, X.; Pu, M.; Kobzik, L.; Kelly, R.A.; Bourcier, T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation 2004, 109, 784–789. [Google Scholar] [CrossRef]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Abbate, A. Inflammasome; pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H1553–H1568. [Google Scholar] [CrossRef]
- Turner, N.A. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J. Mol. Cell. Cardiol. 2016, 94, 189–200. [Google Scholar] [CrossRef]
- Fujiwara, M.; Matoba, T.; Koga, J.-I.; Okahara, A.; Funamoto, D.; Nakano, K.; Tsutsui, H.; Egashira, K. Nanoparticle incorporating Toll-like receptor 4 inhibitor attenuates myocardial ischaemia–reperfusion injury by inhibiting monocyte-mediated inflammation in mice. Cardiovasc. Res. 2019, 115, 1244–1255. [Google Scholar] [CrossRef]
- Timmers, L.; Sluijter, J.P.; van Keulen, J.K.; Hoefer, I.E.; Nederhoff, M.G.; Goumans, M.J.; Doevendans, P.A.; van Echteld, C.J.; Joles, J.A.; Quax, P.H.; et al. Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ. Res. 2008, 102, 257–264. [Google Scholar] [CrossRef]
- Arslan, F.; Smeets, M.B.; O’Neill, L.A.J.; Keogh, B.; McGuirk, P.; Timmers, L.; Tersteeg, C.; Hoefer, I.E.; Doevendans, P.A.; Pasterkamp, G.; et al. Myocardial Ischemia/Reperfusion Injury Is Mediated by Leukocytic Toll-Like Receptor-2 and Reduced by Systemic Administration of a Novel Anti–Toll-Like Receptor-2 Antibody. Circulation 2010, 121, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C.; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular Endothelial Cells and Innate Immunity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e138–e152. [Google Scholar] [CrossRef] [PubMed]
- Bucciarelli, L.G.; Kaneko, M.; Ananthakrishnan, R.; Harja, E.; Lee, L.K.; Hwang, Y.C.; Lerner, S.; Bakr, S.; Li, Q.; Lu, Y.; et al. Receptor for advanced-glycation end products: Key modulator of myocardial ischemic injury. Circulation 2006, 113, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.H.; Ward, P.A. The phlogistic role of C3 leukotactic fragments in myocardial infarcts of rats. J. Exp. Med. 1971, 133, 885–900. [Google Scholar] [CrossRef]
- Cao, D.J.; Schiattarella, G.G.; Villalobos, E.; Jiang, N.; May, H.I.; Li, T.; Chen, Z.J.; Gillette, T.G.; Hill, J.A. Cytosolic DNA Sensing Promotes Macrophage Transformation and Governs Myocardial Ischemic Injury. Circulation 2018, 137, 2613–2634. [Google Scholar] [CrossRef]
- Lu, C.; Ren, D.; Wang, X.; Ha, T.; Liu, L.; Lee, E.J.; Hu, J.; Kalbfleisch, J.; Gao, X.; Kao, R.; et al. Toll-like receptor 3 plays a role in myocardial infarction and ischemia/reperfusion injury. Biochim. Biophys. Acta 2014, 1842, 22–31. [Google Scholar] [CrossRef]
- Del Re, D.P. Hippo-Yap signaling in cardiac and fibrotic remodeling. Curr. Opin. Physiol. 2022, 26, 100492. [Google Scholar] [CrossRef]
- Schulz, R. TNFα in myocardial ischemia/reperfusion: Damage vs. protection. J. Mol. Cell. Cardiol. 2008, 45, 712–714. [Google Scholar] [CrossRef]
- Margraf, A.; Ley, K.; Zarbock, A. Neutrophil Recruitment: From Model Systems to Tissue-Specific Patterns. Trends Immunol. 2019, 40, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Tallquist, M.D. Fibroblast and Immune Cell Cross-Talk in Cardiac Fibrosis. Curr. Cardiol. Rep. 2023, 25, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.P.; Grisanti, L.A. The Dynamic Interplay Between Cardiac Inflammation and Fibrosis. Front. Physiol. 2020, 11, 529075. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.; Zhang, Y.; Nakada, Y.; Jeong, J.I.; Huang, C.Y.; Ivessa, A.; Oka, S.; Babu, G.J.; Del Re, D.P. AAV-mediated YAP expression in cardiac fibroblasts promotes inflammation and increases fibrosis. Sci. Rep. 2021, 11, 10553. [Google Scholar] [CrossRef]
- Sreejit, G.; Nooti, S.K.; Jaggers, R.M.; Athmanathan, B.; Park, K.H.; Al-Sharea, A.; Johnson, J.; Dahdah, A.; Lee, M.K.S.; Ma, J.; et al. Retention of the NLRP3 Inflammasome-Primed Neutrophils in the Bone Marrow Is Essential for Myocardial Infarction-Induced Granulopoiesis. Circulation 2022, 145, 31–44. [Google Scholar] [CrossRef]
- Vogt, K.L.; Summers, C.; Chilvers, E.R.; Condliffe, A.M. Priming and de-priming of neutrophil responses in vitro and in vivo. Eur. J. Clin. Investig. 2018, 48 (Suppl. S2), e12967. [Google Scholar] [CrossRef]
- Miralda, I.; Uriarte, S.M.; McLeish, K.R. Multiple Phenotypic Changes Define Neutrophil Priming. Front. Cell Infect. Microbiol. 2017, 7, 217. [Google Scholar] [CrossRef]
- Schloss, M.J.; Horckmans, M.; Nitz, K.; Duchene, J.; Drechsler, M.; Bidzhekov, K.; Scheiermann, C.; Weber, C.; Soehnlein, O.; Steffens, S. The time-of-day of myocardial infarction onset affects healing through oscillations in cardiac neutrophil recruitment. EMBO Mol. Med. 2016, 8, 937–948. [Google Scholar] [CrossRef]
- Puhl, S.L.; Steffens, S. Neutrophils in Post-myocardial Infarction Inflammation: Damage vs. Resolution? Front. Cardiovasc. Med. 2019, 6, 25. [Google Scholar] [CrossRef]
- Hoyer, F.F.; Nahrendorf, M. Neutrophil contributions to ischaemic heart disease. Eur. Heart J. 2017, 38, 465–472. [Google Scholar] [CrossRef]
- Ma, Y.; Yabluchanskiy, A.; Iyer, R.P.; Cannon, P.L.; Flynn, E.R.; Jung, M.; Henry, J.; Cates, C.A.; Deleon-Pennell, K.Y.; Lindsey, M.L. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016, 110, 51–61. [Google Scholar] [CrossRef]
- Ma, Y.; Yabluchanskiy, A.; Lindsey, M.L. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis Tissue Repair 2013, 6, 11. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O.A.-O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, S.; Ding, S.; Abudupataer, M.; Zhang, Z.; Zhu, X.; Zhang, W.; Zou, Y.; Yang, X.A.-O.; Ge, J.A.-O.; et al. Excessive Neutrophil Extracellular Trap Formation Aggravates Acute Myocardial Infarction Injury in Apolipoprotein E Deficiency Mice via the ROS-Dependent Pathway. Oxid. Med. Cell. Longev. 2019, 21, 1209307. [Google Scholar] [CrossRef] [PubMed]
- Dang, P.M.; Stensballe, A.; Boussetta, T.; Raad, H.; Dewas, C.; Kroviarski, Y.; Hayem, G.; Jensen, O.N.; Gougerot-Pocidalo, M.A.; El-Benna, J. A specific p47phox-serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. Investig. 2006, 116, 2033–2043. [Google Scholar] [CrossRef]
- Calcagno, D.M.; Zhang, C.; Toomu, A.; Huang, K.; Ninh, V.K.; Miyamoto, S.; Aguirre, A.D.; Fu, Z.; Brown, J.H.; King, K.R. SiglecF(HI) Marks Late-Stage Neutrophils of the Infarcted Heart: A Single-Cell Transcriptomic Analysis of Neutrophil Diversification. J. Am. Heart Assoc. 2021, 10, e019019. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, E.; Rossi, A.G.; Condliffe, A.M.; Haslett, C.; Chilvers, E.R. Demonstration of reversible priming of human neutrophils using platelet-activating factor. Blood 1996, 88, 4330–4337. [Google Scholar] [CrossRef] [PubMed]
- Vafadarnejad, E.; Rizzo, G.; Krampert, L.; Arampatzi, P.; Arias-Loza, A.-P.; Nazzal, Y.; Rizakou, A.; Knochenhauer, T.; Bandi, S.R.; Nugroho, V.A.; et al. Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ. Res. 2020, 127, e232–e249. [Google Scholar] [CrossRef]
- Sano, S.; Wang, Y.; Yura, Y.; Sano, M.; Oshima, K.; Yang, Y.; Katanasaka, Y.; Min, K.D.; Matsuura, S.; Ravid, K.; et al. JAK2V617F—Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC Basic. Transl. Sci. 2019, 4, 684–697. [Google Scholar] [CrossRef]
- Kain, V.; Halade, G.V. Role of neutrophils in ischemic heart failure. Pharmacol. Ther. 2020, 205, 107424. [Google Scholar] [CrossRef]
- Hurtado-Nedelec, M.; Csillag-Grange, M.J.; Boussetta, T.; Belambri, S.A.; Fay, M.; Cassinat, B.; Gougerot-Pocidalo, M.A.; Dang, P.M.; El-Benna, J. Increased reactive oxygen species production and p47phox phosphorylation in neutrophils from myeloproliferative disorders patients with JAK2 (V617F) mutation. Haematologica 2013, 98, 1517–1524. [Google Scholar] [CrossRef]
- Vinten-Johansen, J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 2004, 61, 481–497. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive Oxygen Species and Neutrophil Function. Annu. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef]
- Gierlikowska, B.; Stachura, A.; Gierlikowski, W.; Demkow, U. Phagocytosis, Degranulation and Extracellular Traps Release by Neutrophils-The Current Knowledge, Pharmacological Modulation and Future Prospects. Front. Pharmacol. 2021, 12, 666732. [Google Scholar] [CrossRef]
- Naegelen, I.; Beaume, N.; Plançon, S.; Schenten, V.; Tschirhart, E.J.; Bréchard, S. Regulation of Neutrophil Degranulation and Cytokine Secretion: A Novel Model Approach Based on Linear Fitting. J. Immunol. Res. 2015, 2015, 817038. [Google Scholar] [CrossRef]
- García-Prieto, J.; Villena-Gutiérrez, R.; Gómez, M.; Bernardo, E.; Pun-García, A.; García-Lunar, I.; Crainiciuc, G.; Fernández-Jiménez, R.; Sreeramkumar, V.; Bourio-Martínez, R.; et al. Neutrophil stunning by metoprolol reduces infarct size. Nat. Commun. 2017, 8, 14780. [Google Scholar] [CrossRef]
- Entman, M.L.; Youker, K.; Shoji, T.; Kukielka, G.; Shappell, S.B.; Taylor, A.A.; Smith, C.W. Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18-ICAM-1 adherence. J. Clin. Investig. 1992, 90, 1335–1345. [Google Scholar] [CrossRef]
- Kloner, R.A.; Ganote, C.E.; Jennings, R.B. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J. Clin. Investig. 1974, 54, 1496–1508. [Google Scholar] [CrossRef]
- Halade, G.V.; Lee, D.H. Inflammation and resolution signaling in cardiac repair and heart failure. EBioMedicine 2022, 79, 103992. [Google Scholar] [CrossRef]
- Dehghani, T.; Thai, P.N.; Sodhi, H.; Ren, L.; Sirish, P.; Nader, C.E.; Timofeyev, V.; Overton, J.L.; Li, X.; Lam, K.S.; et al. Selectin-Targeting Glycosaminoglycan-Peptide Conjugate Limits Neutrophil Mediated Cardiac Reperfusion Injury. Cardiovasc. Res. 2020, 118, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Yang, W.; Schmull, S.; Gu, J.; Xue, S. Inhibition of peptidyl arginine deiminase-4 protects against myocardial infarction induced cardiac dysfunction. Int. Immunopharmacol. 2020, 78, 106055. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Tajiri, K.A.-O.; Murakoshi, N.; Xu, D.; Yonebayashi, S.; Li, S.; Okabe, Y.; Feng, D.; Shimoda, Y.; Song, Z.; et al. Neutrophil Elastase Deficiency Ameliorates Myocardial Injury Post Myocardial Infarction in Mice. Int. J. Mol. Sci. 2021, 22, 722. [Google Scholar] [CrossRef] [PubMed]
- Trevelin, S.C.; Shah, A.M. Lombardi, GBeyond bacterial killing: NADPH oxidase 2 is an immunomodulator. Immunol. Lett. 2020, 221, 39–48. [Google Scholar] [CrossRef]
- Ao, L.; Zou, N.; Cleveland, J.C.; Fullerton, D.A.; Meng, X. Myocardial TLR4 is a determinant of neutrophil infiltration after global myocardial ischemia: Mediating KC and MCP-1 expression induced by extracellular HSC70. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H21–H28. [Google Scholar] [CrossRef]
- Mauler, M.; Herr, N.; Schoenichen, C.; Witsch, T.; Marchini, T.; Härdtner, C.; Koentges, C.; Kienle, K.; Ollivier, V.; Schell, M.; et al. Platelet Serotonin Aggravates Myocardial Ischemia/Reperfusion Injury via Neutrophil Degranulation. Circulation 2019, 139, 918–931. [Google Scholar] [CrossRef]
- Daseke, M.J.; Chalise, U.; Becirovic-Agic, M.; Salomon, J.D.; Cook, L.M.; Case, A.J.; Lindsey, M.L. Neutrophil signaling during myocardial infarction wound repair. Cell. Signal. 2021, 77, 109816. [Google Scholar] [CrossRef]
- Ma, Y. Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells 2021, 10, 1676. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Kain, V.; Ingle, K.A.; Kabarowski, J.; Barnes, S.; Limdi, N.A.; Prabhu, S.D.; Halade, G.V. Genetic deletion of 12/15 lipoxygenase promotes effective resolution of inflammation following myocardial infarction. J. Mol. Cell Cardiol. 2018, 118, 70–80. [Google Scholar] [CrossRef]
- Halade, G.V.; Kain, V.; Ingle, K.A.; Prabhu, S.D. Interaction of 12/15-lipoxygenase with fatty acids alters the leukocyte kinetics leading to improved postmyocardial infarction healing. Am. J. Physiol.-Heart Circ. Physiol. 2017, 313, H89–H102. [Google Scholar] [CrossRef]
- Wei, X.; Zou, S.; Xie, Z.; Wang, Z.; Huang, N.; Cen, Z.; Hao, Y.; Zhang, C.; Chen, Z.; Zhao, F.; et al. EDIL3 deficiency ameliorates adverse cardiac remodelling by neutrophil extracellular traps (NET)-mediated macrophage polarization. Cardiovasc. Res. 2022, 118, 2179–2195. [Google Scholar] [CrossRef]
- Zhang, S.; Weinberg, S.; DeBerge, M.; Gainullina, A.; Schipma, M.; Kinchen, J.M.; Ben-Sahra, I.; Gius, D.R.; Yvan-Charvet, L.; Chandel, N.S.; et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019, 29, 443–456.e5. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.E.; O’Neill, C.L.; O’Doherty, T.M.; Medina, R.J.; Stitt, A.W. The role of immune-related myeloid cells in angiogenesis. Immunobiology 2013, 218, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, E.B.; Thorp, E.B.; Sumagin, R. Functional implications of neutrophil metabolism during ischemic tissue repair. Curr. Opin. Pharmacol. 2022, 63, 102191. [Google Scholar] [CrossRef]
- Rodriguez-Espinosa, O.; Rojas-Espinosa, O.; Moreno-Altamirano, M.M.; Lopez-Villegas, E.O.; Sanchez-Garcia, F.J. Metabolic requirements for neutrophil extracellular traps formation. Immunology 2015, 145, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Britt, E.C.; Lika, J.; Giese, M.A.; Schoen, T.J.; Seim, G.L.; Huang, Z.; Lee, P.Y.; Huttenlocher, A.; Fan, J. Switching to the cyclic pentose phosphate pathway powers the oxidative burst in activated neutrophils. Nat. Metab. 2022, 4, 389–403. [Google Scholar] [CrossRef]
- Bao, Y.; Ledderose, C.; Graf, A.F.; Brix, B.; Birsak, T.; Lee, A.; Zhang, J.; Junger, W.G. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J. Cell Biol. 2015, 210, 1153–1164. [Google Scholar] [CrossRef]
- Bao, Y.; Ledderose, C.; Seier, T.; Graf, A.F.; Brix, B.; Chong, E.; Junger, W.G. Mitochondria regulate neutrophil activation by generating ATP for autocrine purinergic signaling. J. Biol. Chem. 2014, 289, 26794–26803. [Google Scholar] [CrossRef]
- Chen, Y.; Corriden, R.; Inoue, Y.; Yip, L.; Hashiguchi, N.; Zinkernagel, A.; Nizet, V.; Insel, P.A.; Junger, W.G. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 2006, 314, 1792–1795. [Google Scholar] [CrossRef]
- Zaman, R.; Hamidzada, H.; Epelman, S. Exploring cardiac macrophage heterogeneity in the healthy and diseased myocardium. Curr. Opin. Immunol. 2021, 68, 54–63. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef] [PubMed]
- Zaman, R.; Epelman, S. Resident cardiac macrophages: Heterogeneity and function in health and disease. Immunity 2022, 55, 1549–1563. [Google Scholar] [CrossRef] [PubMed]
- Kubota, A.; Frangogiannis, N.G. Macrophages in myocardial infarction. Am. J. Physiol. Cell Physiol. 2022, 323, C1304–C1324. [Google Scholar] [CrossRef]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Francisco, J.; Guan, J.; Zhang, Y.; Nakada, Y.; Mareedu, S.; Sung, E.A.; Hu, C.M.; Oka, S.; Zhai, P.; Sadoshima, J.; et al. Suppression of myeloid YAP antagonizes adverse cardiac remodeling during pressure overload stress. J. Mol. Cell Cardiol. 2023, 181, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 2019, 363, eaau0964. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef] [PubMed]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef]
- Leuschner, F.; Rauch, P.J.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y.; et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 2012, 209, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.-M.; Weinheimer, C.; et al. Tissue Resident CCR2− and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Majmudar, M.D.; Keliher, E.J.; Heidt, T.; Leuschner, F.; Truelove, J.; Sena, B.F.; Gorbatov, R.; Iwamoto, Y.; Dutta, P.; Wojtkiewicz, G.; et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 2013, 127, 2038–2046. [Google Scholar] [CrossRef]
- Lantz, C.; Radmanesh, B.; Liu, E.; Thorp, E.B.; Lin, J. Single-cell RNA sequencing uncovers heterogenous transcriptional signatures in macrophages during efferocytosis. Sci. Rep. 2020, 10, 14333. [Google Scholar] [CrossRef] [PubMed]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed]
- DeBerge, M.; Chaudhary, R.; Schroth, S.; Thorp, E.B. Immunometabolism at the Heart of Cardiovascular Disease. JACC Basic. Transl. Sci. 2023, 8, 884–904. [Google Scholar] [CrossRef]
- Zhang, S.; Bories, G.; Lantz, C.; Emmons, R.; Becker, A.; Liu, E.; Abecassis, M.M.; Yvan-Charvet, L.; Thorp, E.B. Immunometabolism of Phagocytes and Relationships to Cardiac Repair. Front. Cardiovasc. Med. 2019, 6, 42. [Google Scholar] [CrossRef]
- Mouton, A.J.; DeLeon-Pennell, K.Y.; Gonzalez, O.J.R.; Flynn, E.R.; Freeman, T.C.; Saucerman, J.J.; Garrett, M.R.; Ma, Y.; Harmancey, R.; Lindsey, M.L. Mapping macrophage polarization over the myocardial infarction time continuum. Basic. Res. Cardiol. 2018, 113, 26. [Google Scholar] [CrossRef] [PubMed]
- DeBerge, M.; Yeap, X.Y.; Dehn, S.; Zhang, S.; Grigoryeva, L.; Misener, S.; Procissi, D.; Zhou, X.; Lee, D.C.; Muller, W.A.; et al. MerTK Cleavage on Resident Cardiac Macrophages Compromises Repair After Myocardial Ischemia Reperfusion Injury. Circ. Res. 2017, 121, 930–940. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Wan, E.; Yeap, X.Y.; Dehn, S.; Terry, R.; Novak, M.; Zhang, S.; Iwata, S.; Han, X.; Homma, S.; Drosatos, K.; et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 2013, 113, 1004–1012. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, S.; Vuckovic, I.; Jeon, R.; Lerman, A.; Folmes, C.D.; Dzeja, P.P.; Herrmann, J. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 2018, 28, 463–475.e4. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Hooftman, A.; Angiari, S.; Hester, S.; Corcoran, S.E.; Runtsch, M.C.; Ling, C.; Ruzek, M.C.; Slivka, P.F.; McGettrick, A.F.; Banahan, K.; et al. The Immunomodulatory Metabolite Itaconate Modifies NLRP3 and Inhibits Inflammasome Activation. Cell Metab. 2020, 32, 468–478.e7. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Oka, S.; Shao, D.; Hariharan, N.; Sadoshima, J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ. Res. 2009, 105, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Byun, J.; Zhai, P.; Ikeda, Y.; Oka, S.; Sadoshima, J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE 2014, 9, e98972. [Google Scholar] [CrossRef]
- Zhai, X.; Han, W.; Wang, M.; Guan, S.; Qu, X. Exogenous supplemental NAD+ protect myocardium against myocardial ischemic/reperfusion injury in swine model. Am. J. Transl. Res. 2019, 11, 6066–6074. [Google Scholar] [PubMed]
- Zhou, B.; Wang, D.D.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’Brien, K.D.; Tian, R. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef]
- Lee, C.F.; Caudal, A.; Abell, L.; Gowda, G.A.N.; Tian, R. Targeting NAD(+) Metabolism as Interventions for Mitochondrial Disease. Sci. Rep. 2019, 9, 3073. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef]
- Cai, S.; Zhao, M.; Zhou, B.; Yoshii, A.; Bugg, D.; Villet, O.; Sahu, A.; Olson, G.S.; Davis, J.; Tian, R. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J. Clin. Investig. 2023, 133, e159498. [Google Scholar] [CrossRef]
- Morioka, S.; Perry, J.S.A.; Raymond, M.H.; Medina, C.B.; Zhu, Y.; Zhao, L.; Serbulea, V.; Onengut-Gumuscu, S.; Leitinger, N.; Kucenas, S.; et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature 2018, 563, 714–718. [Google Scholar] [CrossRef]
- Lai, L.; Zhang, A.; Yang, B.; Charles, E.J.; Kron, I.L.; Yang, Z. Plasmacytoid Dendritic Cells Mediate Myocardial Ischemia/Reperfusion Injury by Secreting Type I Interferons. J. Am. Heart Assoc. 2021, 10, e020754. [Google Scholar] [CrossRef]
- Lee, J.S.; Jeong, S.J.; Kim, S.; Chalifour, L.; Yun, T.J.; Miah, M.A.; Li, B.; Majdoubi, A.; Sabourin, A.; Keler, T.; et al. Conventional Dendritic Cells Impair Recovery after Myocardial Infarction. J. Immunol. 2018, 201, 1784–1798. [Google Scholar] [CrossRef] [PubMed]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012, 125, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Ge, H.; Lin, Z.; Wang, H.; Lin, W.; Liu, Y.; Wu, G.; Xia, J.; Zhao, Q. The role of dendritic cells regulated by HMGB1/TLR4 signalling pathway in myocardial ischaemia reperfusion injury. J. Cell Mol. Med. 2019, 23, 2849–2862. [Google Scholar] [CrossRef]
- Choo, E.H.; Lee, J.-H.; Park, E.-H.; Park, H.E.; Jung, N.-C.; Kim, T.-H.; Koh, Y.-S.; Kim, E.; Seung, K.-B.; Park, C.; et al. Infarcted Myocardium-Primed Dendritic Cells Improve Remodeling and Cardiac Function After Myocardial Infarction by Modulating the Regulatory T Cell and Macrophage Polarization. Circulation 2017, 135, 1444–1457. [Google Scholar] [CrossRef]
- Van der Borght, K.; Scott, C.L.; Nindl, V.; Bouché, A.; Martens, L.; Sichien, D.; Van Moorleghem, J.; Vanheerswynghels, M.; De Prijck, S.; Saeys, Y.; et al. Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells. Cell Rep. 2017, 18, 3005–3017. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Perkins, B.; Sintou, A.; Kalkat, H.S.; Papanikolaou, A.; Jenkins, C.; Alsubaie, M.; Chowdhury, R.A.; Duffy, T.M.; Skelly, D.A.; et al. Cross-Priming Dendritic Cells Exacerbate Immunopathology After Ischemic Tissue Damage in the Heart. Circulation 2021, 143, 821–836. [Google Scholar] [CrossRef]
- Cohen, C.D.; Rousseau, S.T.; Bermea, K.C.; Bhalodia, A.; Lovell, J.P.; Zita, M.D.; Čiháková, D.; Adamo, L. Myocardial Immune Cells: The Basis of Cardiac Immunology. J. Immunol. 2023, 210, 1198–1207. [Google Scholar] [CrossRef]
- Lv, H.; Lipes, M.A. Role of impaired central tolerance to α-myosin in inflammatory heart disease. Trends Cardiovasc. Med. 2012, 22, 113–117. [Google Scholar] [CrossRef]
- Thwe, P.M.; Pelgrom, L.R.; Cooper, R.; Beauchamp, S.; Reisz, J.A.; D’Alessandro, A.; Everts, B.; Amiel, E. Cell-Intrinsic Glycogen Metabolism Supports Early Glycolytic Reprogramming Required for Dendritic Cell Immune Responses. Cell Metab. 2017, 26, 558–567.e5. [Google Scholar] [CrossRef]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef]
- Basit, F.; Mathan, T.; Sancho, D.; de Vries, I.J.M. Human Dendritic Cell Subsets Undergo Distinct Metabolic Reprogramming for Immune Response. Front. Immunol. 2018, 9, 2489. [Google Scholar] [CrossRef] [PubMed]
- Adamo, L.; Rocha-Resende, C.; Lin, C.Y.; Evans, S.; Williams, J.; Dun, H.; Li, W.; Mpoy, C.; Andhey, P.S.; Rogers, B.E.; et al. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight 2020, 5, e134700. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Mendoza, L.H.; Lindsey, M.L.; Ballantyne, C.M.; Michael, L.H.; Smith, C.W.; Entman, M.L. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J. Immunol. 2000, 165, 2798–2808. [Google Scholar] [CrossRef] [PubMed]
- Boag, S.E.; Das, R.; Shmeleva, E.V.; Bagnall, A.; Egred, M.; Howard, N.; Bennaceur, K.; Zaman, A.; Keavney, B.; Spyridopoulos, I. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J. Clin. Investig. 2015, 125, 3063–3076. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Day, Y.J.; Toufektsian, M.C.; Xu, Y.; Ramos, S.I.; Marshall, M.A.; French, B.A.; Linden, J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 2006, 114, 2056–2064. [Google Scholar] [CrossRef]
- Santos-Zas, I.; Lemarié, J.; Zlatanova, I.; Cachanado, M.; Seghezzi, J.C.; Benamer, H.; Goube, P.; Vandestienne, M.; Cohen, R.; Ezzo, M.; et al. Cytotoxic CD8(+) T cells promote granzyme B-dependent adverse post-ischemic cardiac remodeling. Nat. Commun. 2021, 12, 1483. [Google Scholar] [CrossRef]
- Kino, T.; Khan, M.; Mohsin, S. The Regulatory Role of T Cell Responses in Cardiac Remodeling Following Myocardial Infarction. Int. J. Mol. Sci. 2020, 21, 5013. [Google Scholar] [CrossRef]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Zhou, G.; Rokosh, G.; Hamid, T.; Prabhu, S.D. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation 2019, 139, 206–221. [Google Scholar] [CrossRef]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef]
- Xia, N.; Lu, Y.; Gu, M.; Li, N.; Liu, M.; Jiao, J.; Zhu, Z.; Li, J.; Li, D.; Tang, T.; et al. A Unique Population of Regulatory T Cells in Heart Potentiates Cardiac Protection From Myocardial Infarction. Circulation 2020, 142, 1956–1973. [Google Scholar] [CrossRef]
- Hofmann, U.; Frantz, S. Role of Lymphocytes in Myocardial Injury, Healing, and Remodeling After Myocardial Infarction. Circ. Res. 2015, 116, 354–367. [Google Scholar] [CrossRef]
- Saxena, A.; Dobaczewski, M.; Rai, V.; Haque, Z.; Chen, W.; Li, N.; Frangogiannis, N.G. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am. J. Physiol.-Heart Circ. Physiol. 2014, 307, H1233–H1242. [Google Scholar] [CrossRef]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef]
- Rieckmann, M.; Delgobo, M.; Gaal, C.; Buchner, L.; Steinau, P.; Reshef, D.; Gil-Cruz, C.; Horst, E.N.T.; Kircher, M.; Reiter, T.; et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J. Clin. Investig. 2019, 129, 4922–4936. [Google Scholar] [CrossRef]
- Zacchigna, S.; Martinelli, V.; Moimas, S.; Colliva, A.; Anzini, M.; Nordio, A.; Costa, A.; Rehman, M.; Vodret, S.; Pierro, C.; et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat. Commun. 2018, 9, 2432. [Google Scholar] [CrossRef]
- Matsumoto, K.; Ogawa, M.; Suzuki, J.; Hirata, Y.; Nagai, R.; Isobe, M. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J. 2011, 52, 382–387. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Cretenet, G.; Clerc, I.; Matias, M.; Loisel, S.; Craveiro, M.; Oburoglu, L.; Kinet, S.; Mongellaz, C.; Dardalhon, V.; Taylor, N. Cell surface Glut1 levels distinguish human CD4 and CD8 T lymphocyte subsets with distinct effector functions. Sci. Rep. 2016, 6, 24129. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Mann, D.L. The Emerging Role of B Lymphocytes in Cardiovascular Disease. Annu. Rev. Immunol. 2020, 38, 99–121. [Google Scholar] [CrossRef]
- Rocha-Resende, C.; Pani, F.; Adamo, L. B cells modulate the expression of MHC-II on cardiac CCR2− macrophages. J. Mol. Cell Cardiol. 2021, 157, 98–103. [Google Scholar] [CrossRef]
- Adamo, L.; Staloch, L.J.; Rocha-Resende, C.; Matkovich, S.J.; Jiang, W.; Bajpai, G.; Weinheimer, C.J.; Kovacs, A.; Schilling, J.D.; Barger, P.M.; et al. Modulation of subsets of cardiac B lymphocytes improves cardiac function after acute injury. JCI Insight 2018, 3, e120137. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guerin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, T.; Loveland, P.; Kanellakis, P.; Cao, A.; Kallies, A.; Huang, A.L.; Peter, K.; Toh, B.H.; Bobik, A. Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur. Heart J. 2021, 42, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, T.T.; Robinson, K.A.; Pang, W.; Tondato, F.; Cui, J.; Arrington, J.; Godwin, L.; Ungs, M.; Carlesso, N.; Weich, N.; et al. Bone Marrow-Derived B Cells Preserve Ventricular Function After Acute Myocardial Infarction. JACC Cardiovasc. Interv. 2009, 2, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Dalal, R.; Cao, C.D.; Postoak, J.L.; Yang, G.; Zhang, Q.; Wang, Z.; Lal, H.; Van Kaer, L. IL-10-producing B cells are enriched in murine pericardial adipose tissues and ameliorate the outcome of acute myocardial infarction. Proc. Natl. Acad. Sci. USA 2019, 116, 21673–21684. [Google Scholar] [CrossRef]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J. Immunol. 2014, 192, 3626–3636. [Google Scholar] [CrossRef]
- Kim, D.S.; Woo, J.S.; Min, H.K.; Choi, J.W.; Moon, J.H.; Park, M.J.; Kwok, S.K.; Park, S.H.; Cho, M.L. Short-chain fatty acid butyrate induces IL-10-producing B cells by regulating circadian-clock-related genes to ameliorate Sjögren’s syndrome. J. Autoimmun. 2021, 119, 102611. [Google Scholar] [CrossRef]
- Weisel, F.J.; Mullett, S.J.; Elsner, R.A.; Menk, A.V.; Trivedi, N.; Luo, W.; Wikenheiser, D.; Hawse, W.F.; Chikina, M.; Smita, S.; et al. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat. Immunol. 2020, 21, 331–342. [Google Scholar] [CrossRef]
- Grisanti, L.A.; Traynham, C.J.; Repas, A.A.; Gao, E.; Koch, W.J.; Tilley, D.G. beta2-Adrenergic receptor-dependent chemokine receptor 2 expression regulates leukocyte recruitment to the heart following acute injury. Proc. Natl. Acad. Sci. USA 2016, 113, 15126–15131. [Google Scholar] [CrossRef]
- van Amerongen, M.J.; Harmsen, M.C.; van Rooijen, N.; Petersen, A.H.; van Luyn, M.J. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am. J. Pathol. 2007, 170, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Li, B.; Han, X.; Zhang, X.; Dang, M.; Wang, H.; Du, F.; Zeng, X.; Guo, C. Soluble receptor for advanced glycation end-products promotes angiogenesis through activation of STAT3 in myocardial ischemia/reperfusion injury. Apoptosis 2020, 25, 341–353. [Google Scholar] [CrossRef] [PubMed]
- De Hoog, V.C.; Timmers, L.; Van Duijvenvoorde, A.; De Jager, S.C.; Van Middelaar, B.J.; Smeets, M.B.; Woodruff, T.M.; Doevendans, P.A.; Pasterkamp, G.; Hack, C.E.; et al. Leucocyte expression of complement C5a receptors exacerbates infarct size after myocardial reperfusion injury. Cardiovasc. Res. 2014, 103, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Vakeva, A.P.; Agah, A.; Rollins, S.A.; Matis, L.A.; Li, L.; Stahl, G.L. Myocardial infarction and apoptosis after myocardial ischemia and reperfusion: Role of the terminal complement components and inhibition by anti-C5 therapy. Circulation 1998, 97, 2259–2267. [Google Scholar] [CrossRef]
- Frantz, S.; Tillmanns, J.; Kuhlencordt, P.J.; Schmidt, I.; Adamek, A.; Dienesch, C.; Thum, T.; Gerondakis, S.; Ertl, G.; Bauersachs, J. Tissue-specific effects of the nuclear factor kappaB subunit p50 on myocardial ischemia-reperfusion injury. Am. J. Pathol. 2007, 171, 507–512. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Tang, R.; Li, L.; Szucsik, A.; Javan, H.; Saegusa, N.; Spitzer, K.W.; Selzman, C.H. Cardiomyocyte-specific p65 NF-κB deletion protects the injured heart by preservation of calcium handling. Am. J. Physiol.-Heart Circ. Physiol. 2013, 305, H1089–H1097. [Google Scholar] [CrossRef] [PubMed]
- Moss, N.C.; Stansfield, W.E.; Willis, M.S.; Tang, R.-H.; Selzman, C.H. IKKβ inhibition attenuates myocardial injury and dysfunction following acute ischemia-reperfusion injury. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H2248–H2253. [Google Scholar] [CrossRef]
- Maekawa, N.; Wada, H.; Kanda, T.; Niwa, T.; Yamada, Y.; Saito, K.; Fujiwara, H.; Sekikawa, K.; Seishima, M. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-alpha. J. Am. Coll. Cardiol. 2002, 39, 1229–1235. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic. Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Chojnacki, J.; Toldo, S.; Mezzaroma, E.; Tranchida, N.; Rose, S.W.; Federici, M.; Van Tassell, B.W.; Zhang, S.; Abbate, A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 2014, 63, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Gao, Y.; Dong, Z.; Yang, J.; Gao, R.; Li, X.; Zhang, S.; Ma, L.; Sun, X.; Wang, Z.; et al. GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury. Circ. Res. 2021, 129, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, B.; Yang, X.; Zhang, C.; Jiao, Y.; Li, P.; Liu, Y.; Li, Z.; Qiao, B.; Lau, W.B.; et al. S100a8/a9 Signaling Causes Mitochondrial Dysfunction and Cardiomyocyte Death in Response to Ischemic/Reperfusion Injury. Circulation 2019, 140, 751–764. [Google Scholar] [CrossRef]
- Ge, L.; Zhou, X.; Ji, W.-J.; Lu, R.-Y.; Zhang, Y.; Zhang, Y.-D.; Ma, Y.-Q.; Zhao, J.-H.; Li, Y.-M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: Therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol.-Heart Circ. Physiol. 2015, 308, H500–H509. [Google Scholar] [CrossRef]
- Fan, Q.; Tao, R.; Zhang, H.; Xie, H.; Lu, L.; Wang, T.; Su, M.; Hu, J.; Zhang, Q.; Chen, Q.; et al. Dectin-1 Contributes to Myocardial Ischemia/Reperfusion Injury by Regulating Macrophage Polarization and Neutrophil Infiltration. Circulation 2019, 139, 663–678. [Google Scholar] [CrossRef]
- Xiao, J.; Yu, K.; Li, M.; Xiong, C.; Wei, Y.; Zeng, Q. The IL-2/Anti-IL-2 Complex Attenuates Cardiac Ischaemia-Reperfusion Injury Through Expansion of Regulatory T Cells. Cell Physiol. Biochem. 2017, 44, 1810–1827. [Google Scholar] [CrossRef]
- Zhang, M.; Michael, L.H.; Grosjean, S.A.; Kelly, R.A.; Carroll, M.C.; Entman, M.L. The role of natural IgM in myocardial ischemia–reperfusion injury. J. Mol. Cell. Cardiol. 2006, 41, 62–67. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef]
- Libby, P.; Maroko, P.R.; Bloor, C.M.; Sobel, B.E.; Braunwald, E. Reduction of experimental myocardial infarct size by corticosteroid administration. J. Clin. Investig. 1973, 52, 599–607. [Google Scholar] [CrossRef]
- Roberts, R.; DeMello, V.; Sobel, B.E. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation 1976, 53 (Suppl. S3), I204–I206. [Google Scholar] [PubMed]
- Kloner, R.A.; Fishbein, M.C.; Lew, H.; Maroko, P.R.; Braunwald, E. Mummification of the infarcted myocardium by high dose corticosteroids. Circulation 1978, 57, 56–63. [Google Scholar] [CrossRef]
- Arriza, J.L.; Weinberger, C.; Cerelli, G.; Glaser, T.M.; Handelin, B.L.; Housman, D.E.; Evans, R.M. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 1987, 237, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Usher, M.G.; Duan, S.Z.; Ivaschenko, C.Y.; Frieler, R.A.; Berger, S.; Schutz, G.; Lumeng, C.N.; Mortensen, R.M. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J. Clin. Investig. 2010, 120, 3350–3364. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, P.; Vettorazzi, S.; Hovelmann, J.; Scholz, C.J.; Tuckermann, J.P.; Bauersachs, J.; Fraccarollo, D. The glucocorticoid receptor in monocyte-derived macrophages is critical for cardiac infarct repair and remodeling. FASEB J. 2017, 31, 5122–5132. [Google Scholar] [CrossRef]
- Metz, C.A.; Stubbs, D.F.; Hearron, M.S. Significance of infarct site and methylprednisolone on survival following acute myocardial infarction. J. Int. Med. Res. 1986, 1 (Suppl. S14), 11–14. [Google Scholar] [CrossRef]
- Lefer, A.M.; Polansky, E.W. Beneficial effects of ibuprofen in acute myocardial ischemia. Cardiology 1979, 64, 265–279. [Google Scholar] [CrossRef]
- Abbate, A.; Limana, F.; Capogrossi, M.C.; Santini, D.; Biondi-Zoccai, G.G.; Scarpa, S.; Germani, A.; Straino, S.; Severino, A.; Vasaturo, F.; et al. Cyclo-oxygenase-2 (COX-2) inhibition reduces apoptosis in acute myocardial infarction. Apoptosis Int. J. Program. Cell Death 2006, 11, 1061–1063. [Google Scholar] [CrossRef]
- Brown, E.J., Jr.; Kloner, R.A.; Schoen, F.J.; Hammerman, H.; Hale, S.; Braunwald, E. Scar thinning due to ibuprofen administration after experimental myocardial infarction. Am. J. Cardiol. 1983, 51, 877–883. [Google Scholar] [CrossRef]
- Hammerman, H.; Alker, K.J.; Schoen, F.J.; Kloner, R.A. Morphologic and functional effects of piroxicam on myocardial scar formation after coronary occlusion in dogs. Am. J. Cardiol. 1984, 53, 604–607. [Google Scholar] [CrossRef]
- Timmers, L.; Sluijter, J.P.; Verlaan, C.W.; Steendijk, P.; Cramer, M.J.; Emons, M.; Strijder, C.; Grundeman, P.F.; Sze, S.K.; Hua, L.; et al. Cyclooxygenase-2 inhibition increases mortality, enhances left ventricular remodeling, and impairs systolic function after myocardial infarction in the pig. Circulation 2007, 115, 326–332. [Google Scholar] [CrossRef]
- Gislason, G.H.; Jacobsen, S.; Rasmussen, J.N.; Rasmussen, S.; Buch, P.; Friberg, J.; Schramm, T.K.; Abildstrom, S.Z.; Kober, L.; Madsen, M.; et al. Risk of death or reinfarction associated with the use of selective cyclooxygenase-2 inhibitors and nonselective nonsteroidal antiinflammatory drugs after acute myocardial infarction. Circulation 2006, 113, 2906–2913. [Google Scholar] [CrossRef]
- Brophy, J.M.; Levesque, L.E.; Zhang, B. The coronary risk of cyclo-oxygenase-2 inhibitors in patients with a previous myocardial infarction. Heart 2007, 93, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Lamberts, M.; Olsen, A.M.; Fosboll, E.; Niessner, A.; Tamargo, J.; Rosano, G.; Agewall, S.; Kaski, J.C.; Kjeldsen, K.; et al. Cardiovascular safety of non-aspirin non-steroidal anti-inflammatory drugs: Review and position paper by the working group for Cardiovascular Pharmacotherapy of the European Society of Cardiology. Eur. Heart J. 2016, 37, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Costi, A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur. J. Biochem. 1988, 178, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.Y.; Messow, C.M.; Berry, C. Cyclosporin variably and inconsistently reduces infarct size in experimental models of reperfused myocardial infarction: A systematic review and meta-analysis. Br. J. Pharmacol. 2012, 165, 2034–2043. [Google Scholar] [CrossRef]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guerin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Heusch, G.; Botker, H.E.; Przyklenk, K.; Redington, A.; Yellon, D. Remote ischemic conditioning. J. Am. Coll. Cardiol. 2015, 65, 177–195. [Google Scholar] [CrossRef] [PubMed]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic. Res. Cardiol. 2018, 113, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, G.P.; Xue, F.S.; Wang, S.Y.; Cui, X.L.; Li, R.P.; Yang, G.Z.; Sun, C.; Liao, X. Combined Vagal Stimulation and Limb Remote Ischemic Perconditioning Enhances Cardioprotection via an Anti-inflammatory Pathway. Inflammation 2015, 38, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- El Desoky, E.S.; Hassan, A.K.M.; Salem, S.Y.; Fadil, S.A.; Taha, A.F. Cardioprotective effect of atorvastatin alone or in combination with remote ischemic preconditioning on the biochemical changes induced by ischemic/reperfusion injury in a mutual prospective study with a clinical and experimental animal arm. Int. J. Cardiol. 2016, 222, 866–873. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, J.; Yu, P.; Chen, M.; Peng, Q.; Wang, Z.; Dong, N. Remote Ischaemic Preconditioning and Sevoflurane Postconditioning Synergistically Protect Rats from Myocardial Injury Induced by Ischemia and Reperfusion Partly via Inhibition TLR4/MyD88/NF-kappaB Signaling Pathway. Cell Physiol. Biochem. 2017, 41, 22–32. [Google Scholar] [CrossRef]
- Chen, H.; Jing, X.Y.; Shen, Y.J.; Wang, T.L.; Ou, C.; Lu, S.F.; Cai, Y.; Li, Q.; Chen, X.; Ding, Y.J.; et al. Stat5-dependent cardioprotection in late remote ischaemia preconditioning. Cardiovasc. Res. 2018, 114, 679–689. [Google Scholar] [CrossRef]
- Pilz, P.M.; Hamza, O.; Gidlof, O.; Goncalves, I.F.; Tretter, E.V.; Trojanek, S.; Abraham, D.; Heber, S.; Haller, P.M.; Podesser, B.K.; et al. Remote ischemic perconditioning attenuates adverse cardiac remodeling and preserves left ventricular function in a rat model of reperfused myocardial infarction. Int. J. Cardiol. 2019, 285, 72–79. [Google Scholar] [CrossRef]
- Gaspar, A.; Lourenco, A.P.; Pereira, M.A.; Azevedo, P.; Roncon-Albuquerque, R., Jr.; Marques, J.; Leite-Moreira, A.F. Randomized controlled trial of remote ischaemic conditioning in ST-elevation myocardial infarction as adjuvant to primary angioplasty (RIC-STEMI). Basic Res. Cardiol. 2018, 113, 14. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Kharbanda, R.K.; Moller, U.K.; Ramlall, M.; Aaroe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): A single-blind randomised controlled trial. Lancet 2019, 394, 1415–1424. [Google Scholar] [CrossRef]
- Gong, R.; Wu, Y.Q. Remote ischemic conditioning during primary percutaneous coronary intervention in patients with ST-segment elevation myocardial infarction: A systematic review and meta-analysis. J. Cardiothorac. Surg. 2019, 14, 14. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Brunt, K.R.; Kirk, J.A.; Kleinbongard, P.; Calvert, J.W.; de Castro Bras, L.E.; DeLeon-Pennell, K.Y.; Del Re, D.P.; Frangogiannis, N.G.; Frantz, S.; et al. Guidelines for in vivo mouse models of myocardial infarction. Am. J. Physiol.-Heart Circ. Physiol. 2021, 321, H1056–H1073. [Google Scholar] [CrossRef]
- Gedik, N.; Kottenberg, E.; Thielmann, M.; Frey, U.H.; Jakob, H.; Peters, J.; Heusch, G.; Kleinbongard, P. Potential humoral mediators of remote ischemic preconditioning in patients undergoing surgical coronary revascularization. Sci. Rep. 2017, 7, 12660. [Google Scholar] [CrossRef]
- Nederlof, R.; Weber, N.C.; Juffermans, N.P.; de Mol, B.A.; Hollmann, M.W.; Preckel, B.; Zuurbier, C.J. A randomized trial of remote ischemic preconditioning and control treatment for cardioprotection in sevoflurane-anesthetized CABG patients. BMC Anesthesiol. 2017, 17, 51. [Google Scholar] [CrossRef] [PubMed]
- Ney, J.; Hoffmann, K.; Meybohm, P.; Goetzenich, A.; Kraemer, S.; Benstom, C.; Weber, N.C.; Bickenbach, J.; Rossaint, R.; Marx, G.; et al. Remote Ischemic Preconditioning Does Not Affect the Release of Humoral Factors in Propofol-Anesthetized Cardiac Surgery Patients: A Secondary Analysis of the RIPHeart Study. Int. J. Mol. Sci. 2018, 19, 1094. [Google Scholar] [CrossRef]
- Wang, H.; Lyu, Y.; Liao, Q.; Jin, L.; Xu, L.; Hu, Y.; Yu, Y.; Guo, K. Effects of Remote Ischemic Preconditioning in Patients Undergoing Off-Pump Coronary Artery Bypass Graft Surgery. Front. Physiol. 2019, 10, 495. [Google Scholar] [CrossRef] [PubMed]
- Yasojima, K.; Schwab, C.; McGeer, E.G.; McGeer, P.L. Human heart generates complement proteins that are upregulated and activated after myocardial infarction. Circ. Res. 1998, 83, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Pischke, S.E.; Gustavsen, A.; Orrem, H.L.; Egge, K.H.; Courivaud, F.; Fontenelle, H.; Despont, A.; Bongoni, A.K.; Rieben, R.; Tonnessen, T.I.; et al. Complement factor 5 blockade reduces porcine myocardial infarction size and improves immediate cardiac function. Basic. Res. Cardiol. 2017, 112, 20. [Google Scholar] [CrossRef]
- Mahaffey, K.W.; Granger, C.B.; Nicolau, J.C.; Ruzyllo, W.; Weaver, W.D.; Theroux, P.; Hochman, J.S.; Filloon, T.G.; Mojcik, C.F.; Todaro, T.G.; et al. Effect of pexelizumab, an anti-C5 complement antibody, as adjunctive therapy to fibrinolysis in acute myocardial infarction: The COMPlement inhibition in myocardial infarction treated with thromboLYtics (COMPLY) trial. Circulation 2003, 108, 1176–1183. [Google Scholar] [CrossRef]
- Granger, C.B.; Mahaffey, K.W.; Weaver, W.D.; Theroux, P.; Hochman, J.S.; Filloon, T.G.; Rollins, S.; Todaro, T.G.; Nicolau, J.C.; Ruzyllo, W.; et al. Pexelizumab, an anti-C5 complement antibody, as adjunctive therapy to primary percutaneous coronary intervention in acute myocardial infarction: The COMplement inhibition in Myocardial infarction treated with Angioplasty (COMMA) trial. Circulation 2003, 108, 1184–1190. [Google Scholar] [CrossRef]
- Investigators, A.A.; Armstrong, P.W.; Granger, C.B.; Adams, P.X.; Hamm, C.; Holmes, D., Jr.; O’Neill, W.W.; Todaro, T.G.; Vahanian, A.; Van de Werf, F. Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: A randomized controlled trial. JAMA 2007, 297, 43–51. [Google Scholar]
- Hayashidani, S.; Tsutsui, H.; Shiomi, T.; Ikeuchi, M.; Matsusaka, H.; Suematsu, N.; Wen, J.; Egashira, K.; Takeshita, A. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 2003, 108, 2134–2140. [Google Scholar] [CrossRef]
- Montecucco, F.; Braunersreuther, V.; Lenglet, S.; Delattre, B.M.; Pelli, G.; Buatois, V.; Guilhot, F.; Galan, K.; Vuilleumier, N.; Ferlin, W.; et al. CC chemokine CCL5 plays a central role impacting infarct size and post-infarction heart failure in mice. Eur. Heart J. 2012, 33, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Xia, Y.; Bujak, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am. J. Pathol. 2010, 176, 2177–2187. [Google Scholar] [CrossRef]
- Simpson, P.J.; Todd, R.F.; Fantone, J.C.; Mickelson, J.K.; Griffin, J.D.; Lucchesi, B.R. Reduction of experimental canine myocardial reperfusion injury by a monoclonal antibody (anti-Mo1, anti-CD11b) that inhibits leukocyte adhesion. J. Clin. Investig. 1988, 81, 624–629. [Google Scholar] [CrossRef]
- Ma, X.L.; Tsao, P.S.; Lefer, A.M. Antibody to CD-18 exerts endothelial and cardiac protective effects in myocardial ischemia and reperfusion. J. Clin. Investig. 1991, 88, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Aversano, T.; Zhou, W.; Nedelman, M.; Nakada, M.; Weisman, H. A chimeric IgG4 monoclonal antibody directed against CD18 reduces infarct size in a primate model of myocardial ischemia and reperfusion. J. Am. Coll. Cardiol. 1995, 25, 781–788. [Google Scholar] [CrossRef]
- Sager, H.B.; Dutta, P.; Dahlman, J.E.; Hulsmans, M.; Courties, G.; Sun, Y.; Heidt, T.; Vinegoni, C.; Borodovsky, A.; Fitzgerald, K.; et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci. Transl. Med. 2016, 8, 342ra80. [Google Scholar] [CrossRef]
- Baran, K.W.; Nguyen, M.; McKendall, G.R.; Lambrew, C.T.; Dykstra, G.; Palmeri, S.T.; Gibbons, R.J.; Borzak, S.; Sobel, B.E.; Gourlay, S.G.; et al. Limitation of Myocardial Infarction Following Thrombolysis in Acute Myocardial Infarction Study, Double-blind, randomized trial of an anti-CD18 antibody in conjunction with recombinant tissue plasminogen activator for acute myocardial infarction: Limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study. Circulation 2001, 104, 2778–2783. [Google Scholar]
- Faxon, D.P.; Gibbons, R.J.; Chronos, N.A.; Gurbel, P.A.; Sheehan, F.; Investigators, H.-M. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: The results of the HALT-MI study. J. Am. Coll. Cardiol. 2002, 40, 1199–1204. [Google Scholar] [CrossRef]
- Rusnak, J.M.; Kopecky, S.L.; Clements, I.P.; Gibbons, R.J.; Holland, A.E.; Peterman, H.S.; Martin, J.S.; Saoud, J.B.; Feldman, R.L.; Breisblatt, W.M.; et al. An anti-CD11/CD18 monoclonal antibody in patients with acute myocardial infarction having percutaneous transluminal coronary angioplasty (the FESTIVAL study). Am. J. Cardiol. 2001, 88, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Tanguay, J.F.; Wright, S.R.; Duchatelle, V.; Petroni, T.; Gregoire, J.C.; Ibrahim, R.; Heinonen, T.M.; Robb, S.; Bertrand, O.F.; et al. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-ST-segment elevation myocardial infarction: Results of the SELECT-ACS trial. J. Am. Coll. Cardiol. 2013, 61, 2048–2055. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Salloum, F.N.; Vecile, E.; Das, A.; Hoke, N.N.; Straino, S.; Biondi-Zoccai, G.G.; Houser, J.E.; Qureshi, I.Z.; Ownby, E.D.; et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 2008, 117, 2670–2683. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; Van Tassell, B.W.; Farkas, D.; Marchetti, C.; Voelkel, N.F.; Abbate, A. Interleukin-1beta blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp. Physiol. 2013, 98, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Trankle, C.R.; Buckley, L.F.; Lipinski, M.J.; Appleton, D.; Kadariya, D.; Canada, J.M.; Carbone, S.; Roberts, C.S.; Abouzaki, N.; et al. Interleukin-1 Blockade Inhibits the Acute Inflammatory Response in Patients with ST-Segment-Elevation Myocardial Infarction. J. Am. Heart Assoc. 2020, 9, e014941. [Google Scholar] [CrossRef]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef]
- Abbate, A.; Kontos, M.C.; Grizzard, J.D.; Biondi-Zoccai, G.G.; Van Tassell, B.W.; Robati, R.; Roach, L.M.; Arena, R.A.; Roberts, C.S.; Varma, A.; et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am. J. Cardiol. 2010, 105, 1371–1377.e1. [Google Scholar] [CrossRef]
- Abbate, A.; Kontos, M.C.; Abouzaki, N.A.; Melchior, R.D.; Thomas, C.; Van Tassell, B.W.; Oddi, C.; Carbone, S.; Trankle, C.R.; Roberts, C.S.; et al. Comparative safety of interleukin-1 blockade with anakinra in patients with ST-segment elevation acute myocardial infarction (from the VCU-ART and VCU-ART2 pilot studies). Am. J. Cardiol. 2015, 115, 288–292. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; Group, C.T. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy with Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly; regulation; signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Van Tassell, B.W.; Mezzaroma, E.; Del Buono, M.G.; Prestamburgo, A.; Potere, N.; Abbate, A. The NLRP3 Inflammasome Inhibitor, OLT1177 (Dapansutrile), Reduces Infarct Size and Preserves Contractile Function After Ischemia Reperfusion Injury in the Mouse. J. Cardiovasc. Pharmacol. 2019, 73, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Wohlford, G.F.; Van Tassell, B.W.; Billingsley, H.E.; Kadariya, D.; Canada, J.M.; Carbone, S.; Mihalick, V.L.; Bonaventura, A.; Vecchie, A.; Chiabrando, J.G.; et al. Phase 1B, Randomized, Double-Blinded, Dose Escalation, Single-Center, Repeat Dose Safety and Pharmacodynamics Study of the Oral NLRP3 Inhibitor Dapansutrile in Subjects With NYHA II-III Systolic Heart Failure. J. Cardiovasc. Pharmacol. 2020, 77, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Molad, Y.; Reibman, J.; Balakhane, E.; Levin, R.I.; Weissmann, G. Colchicine alters the quantitative and qualitative display of selectins on endothelial cells and neutrophils. J. Clin. Investig. 1995, 96, 994–1002. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Deftereos, S.; Giannopoulos, G.; Angelidis, C.; Alexopoulos, N.; Filippatos, G.; Papoutsidakis, N.; Sianos, G.; Goudevenos, J.; Alexopoulos, D.; Pyrgakis, V.; et al. Anti-Inflammatory Treatment with Colchicine in Acute Myocardial Infarction: A Pilot Study. Circulation 2015, 132, 1395–1403. [Google Scholar] [CrossRef]
- Markousis-Mavrogenis, G.; Tromp, J.; Ouwerkerk, W.; Devalaraja, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.S.; van der Harst, P.; Lang, C.C.; et al. The clinical significance of interleukin-6 in heart failure: Results from the BIOSTAT-CHF study. Eur. J. Heart Fail. 2019, 21, 965–973. [Google Scholar] [CrossRef]
- George, M.J.; Jasmin, N.H.; Cummings, V.T.; Richard-Loendt, A.; Launchbury, F.; Woollard, K.; Turner-Stokes, T.; Diaz, A.I.G.; Lythgoe, M.; Stuckey, D.J.; et al. Selective Interleukin-6 Trans-Signaling Blockade Is More Effective Than Panantagonism in Reperfused Myocardial Infarction. JACC Basic. Transl. Sci. 2021, 6, 431–443. [Google Scholar] [CrossRef]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Broch, K.; Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Tollefsen, I.M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damas, J.K.; et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients With Acute ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Huse, C.; Anstensrud, A.K.; Michelsen, A.E.; Ueland, T.; Broch, K.; Woxholt, S.; Yang, K.; Sharma, K.; Tollefsen, I.M.; Bendz, B.; et al. Interleukin-6 inhibition in ST-elevation myocardial infarction: Immune cell profile in the randomised ASSAIL-MI trial. EBioMedicine 2022, 80, 104013. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling, Nature reviews. Cardiology 2014, 11, 255–265. [Google Scholar]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar] [CrossRef]
- Garcia, R.A.; Lupisella, J.A.; Ito, B.R.; Hsu, M.Y.; Fernando, G.; Carson, N.L.; Allocco, J.J.; Ryan, C.S.; Zhang, R.; Wang, Z.; et al. Selective FPR2 Agonism Promotes a Proresolution Macrophage Phenotype and Improves Cardiac Structure-Function Post Myocardial Infarction. JACC Basic. Transl. Sci. 2021, 6, 676–689. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I/R Model | Cell/Molecular Target | Animal Model/Intervention | Major Findings | Proposed Mechanism | Ref |
|---|---|---|---|---|---|
| Ischemia: 45 min Reperfusion: 1, 3, 5, and 7 days | Characterize overall immune response in the heart | Mice Flow cytometry | Reperfusion accelerated immune cell infiltration versus non-reperfused MI | Speculate early resolution of inflammatory response in the reperfused heart | [96] |
| Ischemia: 30 min Reperfusion: 24 h | TLR2 signaling | Mice TLR2-/- global KO Administration of OPN-301 (TLR2 inhibitor) | ↓ infarct size ↓ myeloid infiltration ↓ inflammation ↓ cardiomyocyte apoptosis ↑ cardiac function | Attenuated p38-MAPK and JNK signaling | [23] |
| Ischemia: 45 min Reperfusion: 3 days | TLR3 signaling | Mice TLR3-/- global KO | ↓ infarct size ↓ cardiomyocyte apoptosis ↓ myeloid infiltration ↑ cardiac function | Attenuated NF-κB and TNFα signaling. Reduced BAX/Bak signaling. | [28] |
| Ischemia: 30 min Reperfusion: 24 h 7 and 28 days | TLR4 signaling | Mice TLR4-/- global KO Administration of TAK-242-NP (TLR4 inhibitor) | ↓ infarct size ↓ myeloid infiltration ↓ inflammation ↓ pathological remodeling ↑ cardiac function | TLR4 inhibition at reperfusion suppressed CCR2-mediated inflammatory cell recruitment | [21] |
| Ischemia: 1 h Reperfusion: 24 h | TLR4 signaling | Mice TLR4 deficient strains | ↓ infarct size ↓ inflammation | Attenuated neutrophil infiltration, reduced ROS, and reduced C3 complement | [18] |
| Ischemia: 30 min Reperfusion: 1 h (ex vivo in mice) | RAGE signaling | Mice: RAGE-/- global KO Rats: Administration of soluble RAGE (sRAGE) decoys | ↓ cardiac injury ↓ cGMP, nitrite/nitrate levels in myocardium ↑ Energy metabolism | Attenuated iNOS signaling, possibly due to decreased glycolysis and peroxynitrite formation | [25] |
| Ischemia: 30 min Reperfusion: 2 weeks | RAGE signaling | Mice Administration of soluble RAGE (sRAGE) recombinant protein | ↑ cardiac function ↑ angiogenesis ↓ pathological remodeling ↓ endothelial apoptosis in myocardium | Increased angiogenesis via STAT3-mediated activation of VEGFR2 in myocardial endothelial cells | [162] |
| Ischemia: 30 min Reperfusion: 1 and 24 h 4 weeks | Complement cascade | Mice C5aR-/- global KO | ↓ infarct size ↓ leukocyte infiltration ↓ cardiomyocyte apoptosis ↑ cardiac function | Decreased neutrophil and T cell infiltration and related inflammation | [163] |
| Ischemia: 30 min Reperfusion: 4 h | Complement cascade | Rats Use of 18A, 16C (C5 neutralizing antibodies) | ↓ infarct size ↓ cardiac injury ↓ myeloperoxidase ↓ cardiomyocyte apoptosis | Attenuated neutrophil infiltration and preserved C3b-related immunoprotection | [164] |
| Ischemia: 30 min Reperfusion: 24 h | NF-κB pathway | Mice p50-/- global KO | ↓ infarct size ↑ inflammation ↓ neutrophil infiltration | Suppressed adhesion of leukocytes | [165] |
| Ischemia: 30 min Reperfusion: 1 h (ex vivo) 24 h | NF-κB pathway | Mice p65 cardiac KO | ↓ infarct size ↓ cardiomyocyte apoptosis ↑ cardiac function | Sustained intracellular calcium cycling, possibly through alterations in PLN | [166] |
| Ischemia: 30 min Reperfusion: 2 h | NF-κB pathway | Mice Administration of Bay 65-1942 (IKKβ inhibitor) | ↓ infarct size ↓ cardiac injury ↓ inflammation ↑ cardiac function | Suppression of TNFα and IL-6 | [167] |
| Ischemia: 30 min Reperfusion: 2 h | TNFα signaling | Mice TNFα-/- global KO TNFα neutralizing antibodies | ↓ arrhythmia ↓ infarct size ↓ inflammation ↑ cardiac function | Attenuated NF-κB activation Reduced neutrophil infiltration | [168] |
| Ischemia: 30 min Reperfusion: 24 and 48 h | NLRP3 Inflammasome | Mice ASC-/- global KO Caspase-1-/- global KO | ↓ infarct size ↓ pathological remodeling ↓ myeloid infiltration ↓ inflammation ↑ cardiac function | Activation of the inflammasome in fibroblasts facilitates leukocyte infiltration. | [169] |
| Ischemia: 30, 75 min Reperfusion: 1, 3, 6, and 24 h | NLRP3 Inflammasome | Mice Administration of NLRP3 inhibitor (NLRP3inh) | ↓ infarct size ↓ caspase-1 activity | Early inhibition of NLRP3 after reperfusion suppressed pyroptotic cell death | [170] |
| Ischemia: 30 min Reperfusion: 3, 24, and 48 h | NLRP3 Inflammasome | Mice Administration of NLRP3 siRNA or BAY 11-7028 (inflammasome inhibitor) | ↓ myeloid infiltration ↓ cardiomyocyte apoptosis ↓ infarct size ↑ cardiac function | Suppression of ROS-induced inflammasome activation in the microvasculature, but not necessarily cardiomyocytes | [171] |
| Ischemia: 30 min Reperfusion: 24 and 48 h | NLRP3 Inflammasome | Mice Administration of 16673-34-0 (NLRP3 inflammasome inhibitor) | ↓ infarct size ↓ cardiac injury | Inhibition of NLRP3 inflammasome is cardioprotective | [172] |
| Ischemia: 30 min Reperfusion: 3 and 24 h | Gasdermin D (GSDMD), pyroptosis | Mice GSDMD-/- global KO | ↓ infarct size ↓ cardiac injury ↓ cardiomyocyte death | I/R-induced oxidative stress activates and cleaves GSDMD and kills cardiomyocytes through pyroptosis | [173] |
| Ischemia: 30 min Reperfusion: 1, 3, 6, and 12 h 1, 3, and 7 days | S100a9 alarmins | Mice S100a9 transgenic S100a9 global KO Administration of S100a9 neutralizing antibodies | S100a9 TG: ↑ infarct size ↑ fibrosis ↓ cardiac function S100a9 KO or Abs: ↓ infarct size ↓ fibrosis ↑ cardiac function | Altered ETC complex I expression and activity in cardiomyocytes modulates I/R injury | [174] |
| Ischemia: 45 min Reperfusion: 3 h, 45 days | Neutrophil extracellular traps (NETs) | Rats Administration of DNase I +/- plasminogen activator | ↓ infarct size ↓ pathological remodeling ↓ no reflow ↑ cardiac function | Cleavage/reduction in NETs with DNAse I treatment, decreased MPO activity, and reduced thrombosis afforded cardioprotection | [175] |
| Ischemia: 45 min Reperfusion: 4 h, 3, 7, 14, and 28 days | Macrophage, MerTK | Mice MerTK myeloid KO | ↑ infarct size ↓ cardiac function | Cleavage of MerTK on resident macrophages during I/R enhances injury and suppresses repair | [103] |
| Ischemia: 45 min Reperfusion: 24 h | Dectin-1 | Mice Dectin 1-/- global KO | ↓ infarct size ↓ immune cell infiltration ↑ cardiac function | Dectin-1 positively regulates NF-κB signaling, inflammatory cytokines, and neutrophil recruitment | [176] |
| Ischemia: 40 min Reperfusion: 60 min | Plasmacytoid dendritic cells (pDC) | Mice Administration of pDC antigen-1 antibody depletion | ↓ infarct size ↓ cardiac injury | Suppressed secretion of Type 1 interferons | [120] |
| Ischemia: 30 min Reperfusion: 180 min | Dendritic cells, HMGB1 | Rats Administration of HMGB1 neutralizing antibodies | ↓ infarct size ↓ cardiac injury ↑ cardiac function | Suppression of inflammatory dendritic cell recruitment and cardiomyocyte apoptosis | [123] |
| Ischemia: 30 min Reperfusion: 3, 7, and 14 days | Tregs, IL-2 signaling | Mice Use of PC61 (CD25) neutralizing antibodies | ↑ infarct size ↑ cardiac injury ↑ cardiac remodeling ↓ cardiac function | IL-2C Treg suppression enhances inflammation and injury | [177] |
| Ischemia: 45 min Reperfusion: 15 min, 60 min, 24 h | T cells CD4+ and CD8+ subtypes | Mice Administration of CD4 or CD8 neutralizing antibodies | CD4 depletion: ↓ infarct size ↓ leukocyte recruitment CD8 depletion: No change in infarct | CD4 T cells regulate IFNγ, and inflammatory cell recruitment | [135] |
| Ischemia: 90 min, closed chest Reperfusion: 2 weeks | B cell/ Total depletion | Mice Administration of CD20 neutralizing antibodies or Pirfenidone | Survival advantage after I/R, but no significant difference in cardiac function | Pirfenidone-mediated reduction in B cell infiltration and inflammation may afford benefit | [152] |
| Ischemia: 1 h Reperfusion: 24 h | B cell/ IgM activation | Mice Cr2-/- global KO RAG1-/- global KO | ↓ infarct size ↓ neutrophil recruitment ↑ cardiac function | I/R-induced autoactivation of B cells is mediated by IgM and contributes to I/R injury | [178] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francisco, J.; Del Re, D.P. Inflammation in Myocardial Ischemia/Reperfusion Injury: Underlying Mechanisms and Therapeutic Potential. Antioxidants 2023, 12, 1944. https://doi.org/10.3390/antiox12111944
Francisco J, Del Re DP. Inflammation in Myocardial Ischemia/Reperfusion Injury: Underlying Mechanisms and Therapeutic Potential. Antioxidants. 2023; 12(11):1944. https://doi.org/10.3390/antiox12111944
Chicago/Turabian StyleFrancisco, Jamie, and Dominic P. Del Re. 2023. "Inflammation in Myocardial Ischemia/Reperfusion Injury: Underlying Mechanisms and Therapeutic Potential" Antioxidants 12, no. 11: 1944. https://doi.org/10.3390/antiox12111944