
Exosomal Dynamics and Brain Redox Imbalance: Implications in Alzheimer’s Disease Pathology and Diagnosis
 , , , and
, , , and {kind=link}
{kind=link}
Abstract
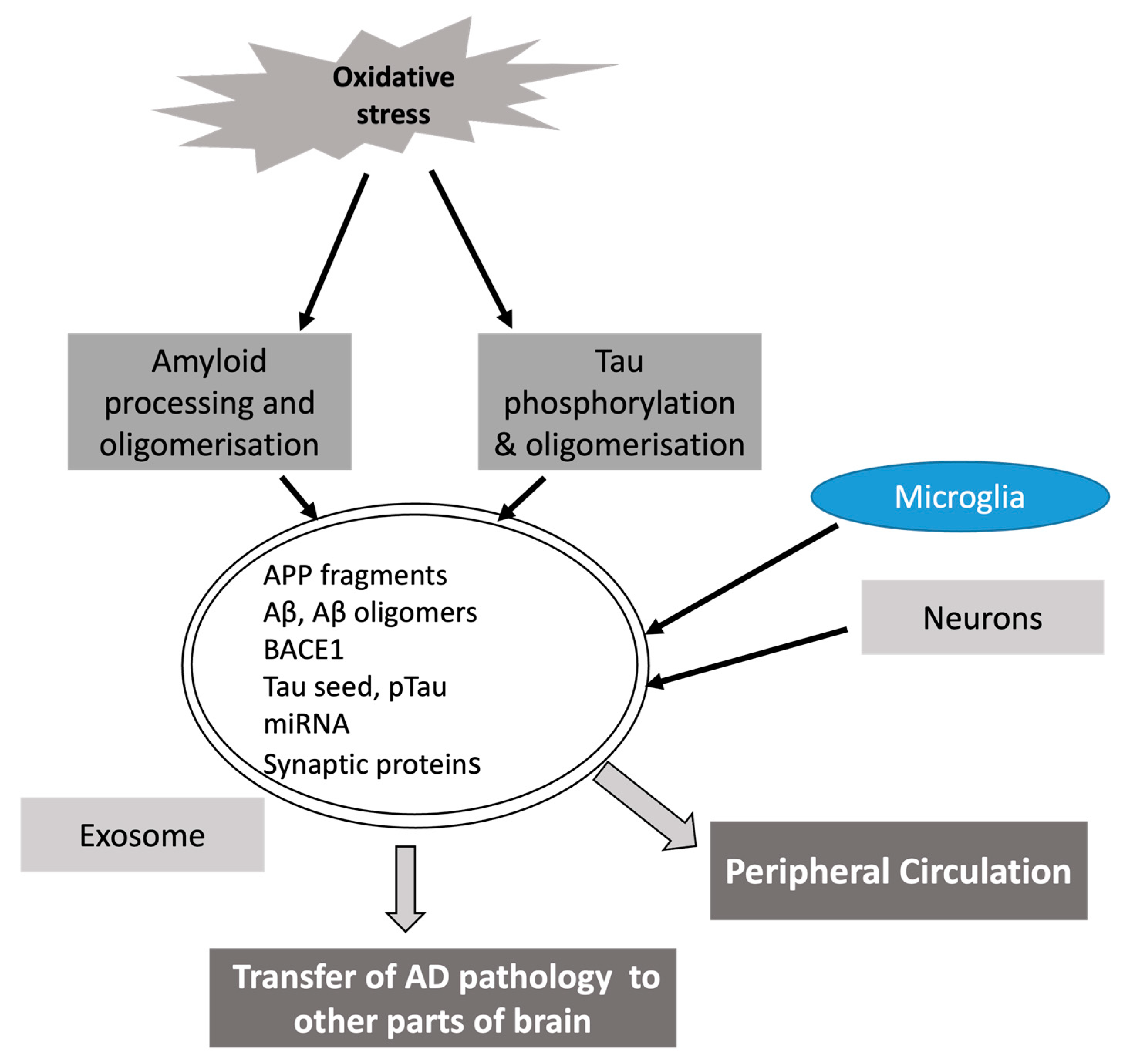
:1. Introduction
2. Oxidative Stress and Alzheimer’s Disease
2.1. Accumulation of Amyloid Beta Due to Oxidative Stress
2.2. Induction of Oxidative Stress Mediated by Amyloid Beta
2.3. Role of Tau Phosphorylation in Oxidative Stress
2.4. Mitochondria Dysfunction, Oxidative Stress and AD
2.5. Metal Ion Homeostasis and Oxidative Stress in AD
2.6. Abnormal Glucose Metabolism, Oxidative Stress and AD
3. Exosome Dynamics and Functional Significance
3.1. Biogenesis, Release and Transport of Exosomes
3.2. Oxidative Stress Induces Changes in Exosomes
4. Exosomes in AD
4.1. Proteins of Exosomal Cargo in AD
4.2. Exosomal miRNAs and AD
5. The Crosstalk between Exosomal miRNAs and Oxidative Stress in AD
6. Exosomes as a Therapeutic Cargo against Oxidative Stress in AD
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, X.X.; Tian, Y.; Wang, Z.T.; Ma, Y.H.; Tan, L.; Yu, J.T. The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. J. Prev. Alzheimers Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Qiu, C.; Kivipelto, M.; Von Strauss, E. Epidemiology of Alzheimer’s Disease: Occurrence, Determinants, and Strategies toward Intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.g.; Zhu, X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Rojas-Gutierrez, E.; Muñoz-Arenas, G.; Treviño, S.; Espinosa, B.; Chavez, R.; Rojas, K.; Flores, G.; Díaz, A.; Guevara, J. Alzheimer’s Disease and Metabolic Syndrome: A Link from Oxidative Stress and Inflammation to Neurodegeneration. Synapse 2017, 71, e21990. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Kanaan, N.M.; Yan, S.S. Du Mitochondrial Oxidative Stress Contributes to the Pathological Aggregation and Accumulation of Tau Oligomers in Alzheimer’s Disease. Hum. Mol. Genet. 2022, 31, 2498–2507. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The Diagnosis of Dementia Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Chapleau, M.; Iaccarino, L.; Soleimani-Meigooni, D.; Rabinovici, G.D. The Role of Amyloid PET in Imaging Neurodegenerative Disorders: A Review. J. Nucl. Med. 2022, 63, 13S–19S. [Google Scholar] [CrossRef]
- Kaur, U.; Reddy, J.; Tiwari, A.; Chakrabarti, S.; Chakrabarti, S.S. Lecanemab: More Questions Than Answers! Clin. Drug Investig. 2024, 44, 1–10. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Anastasiou, A.I.; Zachariou, V.; Pelidou, S.H. Reasons for Failed Trials of Disease-Modifying Treatments for Alzheimer Disease and Their Contribution in Recent Research. Biomedicines 2019, 7, 97. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef]
- Wang, Y.; Xia, X. Editorial: The Role of Exosomes in Neuroinflammation and Neurodegeneration. Front. Cell Neurosci. 2022, 16, 1109885. [Google Scholar] [CrossRef]
- Soares Martins, T.; Trindade, D.; Vaz, M.; Campelo, I.; Almeida, M.; Trigo, G.; da Cruz e Silva, O.A.B.; Henriques, A.G. Diagnostic and Therapeutic Potential of Exosomes in Alzheimer’s Disease. J. Neurochem. 2021, 156, 162–181. [Google Scholar] [CrossRef]
- Wang, T.; Jian, Z.; Baskys, A.; Yang, J.; Li, J.; Guo, H.; Hei, Y.; Xian, P.; He, Z.; Li, Z.; et al. MSC-Derived Exosomes Protect against Oxidative Stress-Induced Skin Injury via Adaptive Regulation of the NRF2 Defense System. Biomaterials 2020, 257, 120264. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Sinha, M.; Thakurta, I.; Banerjee, P.; Chattopadhyay, M. Oxidative Stress and Amyloid Beta Toxicity in Alzheimer’s Disease: Intervention in a Complex Relationship by Antioxidants. CMC 2013, 20, 4648–4664. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, M.; Dranka, B.; Kumar, S.N.; Myers, C.R.; Bennett, B.; Garces, A.M.; Dias Duarte Machado, L.G.; Thiebaut, D.; Ouari, O.; et al. Detection of Mitochondria-Generated Reactive Oxygen Species in Cells Using Multiple Probes and Methods:. Potentials, Pitfalls, and the Future. J. Biol. Chem. 2018, 293, 10363–10380. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Guéraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P.M.; Huc, L.; Jouanin, I.; Siems, W.; Uchida, K. Chemistry and Biochemistry of Lipid Peroxidation Products. Free Radic. Res. 2010, 44, 1098–1124. [Google Scholar] [CrossRef]
- Kourie, J.I. Interaction of Reactive Oxygen Species with Ion Transport Mechanisms. Am. J. Physiol.-Cell Physiol. 1998, 275, C1–C24. [Google Scholar] [CrossRef]
- Stark, G. Functional Consequences of Oxidative Membrane Damage. J. Membr. Biol. 2005, 205, 1–16. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative Stress and the Pathogenesis of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef]
- Cioffi, F.; Adam, R.H.I.; Bansal, R.; Broersen, K. A Review of Oxidative Stress Products and Related Genes in Early Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 83, 977–1001. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; De Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated Levels of 3-Nitrotyrosine in Brain from Subjects with Amnestic Mild Cognitive Impairment: Implications for the Role of Nitration in the Progression of Alzheimer’s Disease. Brain Res. 2007, 1148, 243–248. [Google Scholar] [CrossRef]
- Montine, T.J.; Markesbery, W.R.; Zackert, W.; Sanchez, S.C.; Roberts, L.J.; Morrow, J.D. The Magnitude of Brain Lipid Peroxidation Correlates with the Extent of Degeneration but Not with Density of Neuritic Plaques or Neurofibrillary Tangles or with APOE Genotype in Alzheimer’s Disease Patients. Am. J. Pathol. 1999, 155, 863–868. [Google Scholar] [CrossRef]
- Aksenov, M.Y.; Aksenova, M.V.; Butterfield, D.A.; Geddes, J.W.; Markesbery, W.R. Protein Oxidation in the Brain in Alzheimer’s Disease. Neuroscience 2001, 103, 373–383. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative Damage Is the Earliest Event in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain Iron Is Associated with Accelerated Cognitive Decline in People with Alzheimer Pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef]
- Ayton, S.; Portbury, S.; Kalinowski, P.; Agarwal, P.; Diouf, I.; Schneider, J.A.; Morris, M.C.; Bush, A.I. Regional Brain Iron Associated with Deterioration in Alzheimer’s Disease: A Large Cohort Study and Theoretical Significance. Alzheimer’s Dement. 2021, 17, 1244–1256. [Google Scholar] [CrossRef]
- Damulina, A.; Pirpamer, L.; Soellradl, M.; Sackl, M.; Tinauer, C.; Hofer, E.; Enzinger, C.; Gesierich, B.; Duering, M.; Ropele, S.; et al. Cross-Sectional and Longitudinal Assessment of Brain Iron Level in Alzheimer Disease Using 3-T MRI. Radiology 2020, 296, 619–626. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. Oxidative Stress in the Progression of Alzheimer Disease in the Frontal Cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence Supporting Oxidative Stress in a Moderately Affected Area of the Brain in Alzheimer’s Disease. Sci. Rep. 2018, 8, 11553. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Di Domenico, F.; Swomley, A.M.; Head, E.; Perluigi, M. Redox Proteomics Analysis to Decipher the Neurobiology of Alzheimer-like Neurodegeneration: Overlaps in Down’s Syndrome and Alzheimer’s Disease Brain. Biochem. J. 2014, 463, 177–189. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Redox Proteomics and Amyloid Β-peptide: Insights into Alzheimer Disease. J. Neurochem. 2019, 151, 459–487. [Google Scholar] [CrossRef]
- Bello-Medina, P.C.; González-Franco, D.A.; Vargas-Rodríguez, I.; Díaz-Cintra, S. Oxidative Stress, the Immune Response, Synaptic Plasticity, and Cognition in Transgenic Models of Alzheimer Disease. Neurología 2022, 37, 682–690. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Picciano, M.; La Francois, J.; Duff, K. Fibrillar β-Amyloid Evokes Oxidative Damage in a Transgenic Mouse Model of Alzheimer’s Disease. Neuroscience 2001, 104, 609–613. [Google Scholar] [CrossRef]
- Schrag, M.; Mueller, C.; Oyoyo, U.; Smith, M.A.; Kirsch, W.M. Iron, Zinc and Copper in the Alzheimer’s Disease Brain: A Quantitative Meta-Analysis. Some Insight on the Influence of Citation Bias on Scientific Opinion. Prog. Neurobiol. 2011, 94, 296–306. [Google Scholar] [CrossRef]
- Zabel, M.; Nackenoff, A.; Kirsch, W.M.; Harrison, F.E.; Perry, G.; Schrag, M. Markers of Oxidative Damage to Lipids, Nucleic Acids and Proteins and Antioxidant Enzymes Activities in Alzheimer’s Disease Brain: A Meta-Analysis in Human Pathological Specimens. Free Radic. Biol. Med. 2018, 115, 351–360. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; Gutiérrez, D.A.; Barranco-Almohalla, A.; Crepin, G.; Tajes, M.; Ill-Raga, G.; Guix, F.X.; Menéndez, S.; Arumí-Uría, M.; Vicente, R.; et al. Amyloid Beta-Peptide Increases BACE1 Translation through the Phosphorylation of the Eukaryotic Initiation Factor-2 α. Oxid. Med. Cell Longev. 2020, 2020, 2739459. [Google Scholar] [CrossRef]
- Chandran, S.; Binninger, D. Role of Oxidative Stress, Methionine Oxidation and Methionine Sulfoxide Reductases (MSR) in Alzheimer’s Disease. Antioxidants 2023, 13, 21. [Google Scholar] [CrossRef]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, Oxidative Stress and Mitochondrial Dysfunction: Cross Talk in Alzheimer’s Disease and Parkinson’s Disease. Drug Des. Dev Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An Iron-Responsive Element Type II in the 5’-Untranslated Region of the Alzheimer’s Amyloid Precursor Protein Transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef]
- Storck, S.E.; Hartz, A.M.S.; Pietrzik, C.U. The Blood-Brain Barrier in Alzheimer’s Disease. Handb. Exp. Pharmacol. 2022, 273, 247–266. [Google Scholar] [CrossRef]
- Swomley, A.M.; Förster, S.; Keeney, J.T.; Triplett, J.; Zhang, Z.; Sultana, R.; Butterfield, D.A. Abeta, Oxidative Stress in Alzheimer Disease: Evidence Based on Proteomics Studies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1248–1257. [Google Scholar] [CrossRef]
- Harris, M.E.; Hensley, K.; Butterfield, D.A.; Leedle, R.A.; Carney, J.M. Direct Evidence of Oxidative Injury Produced by the Alzheimer’s β-Amyloid Peptide (1–40) in Cultured Hippocampal Neurons. Exp. Neurol. 1995, 131, 193–202. [Google Scholar] [CrossRef]
- Ledezma, C.; Coria-Lucero, C.; Delsouc, M.B.; Casais, M.; Della Vedova, C.; Ramirez, D.; Devia, C.M.; Delgado, S.M.; Navigatore-Fonzo, L.; Anzulovich, A.C. Effect of an Intracerebroventricular Injection of Aggregated Beta-Amyloid (1–42) on Daily Rhythms of Oxidative Stress Parameters in the Prefrontal Cortex. Neuroscience 2021, 458, 99–107. [Google Scholar] [CrossRef]
- Rehman, I.U.; Ahmad, R.; Khan, I.; Lee, H.J.; Park, J.; Ullah, R.; Choi, M.J.; Kang, H.Y.; Kim, M.O. Nicotinamide Ameliorates Amyloid Beta-Induced Oxidative Stress-Mediated Neuroinflammation and Neurodegeneration in Adult Mouse Brain. Biomedicines 2021, 9, 408. [Google Scholar] [CrossRef]
- Oku, Y.; Murakami, K.; Irie, K.; Hoseki, J.; Sakai, Y. Synthesized Aβ42 Caused Intracellular Oxidative Damage, Leading to Cell Death, via Lysosome Rupture. Cell Struct. Funct. 2017, 42, 71–79. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Yoo, B.; McElheny, D.; Tay, W.; Ishii, Y. Capturing a Reactive State of Amyloid Aggregates: NMR-Based Characterization of Copper-Bound Alzheimer Disease Amyloid β-Fibrils in a Redox Cycle. J. Biol. Chem. 2014, 289, 9998–10010. [Google Scholar] [CrossRef]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The Redox Chemistry of the Alzheimer’s Disease Amyloid β Peptide. Biochim. Biophys. Acta (BBA)-Biomembr. 2007, 1768, 1976–1990. [Google Scholar] [CrossRef]
- Atwood, C.S.; Obrenovich, M.E.; Liu, T.; Chan, H.; Perry, G.; Smith, M.A.; Martins, R.N. Amyloid-β: A Chameleon Walking in Two Worlds: A Review of the Trophic and Toxic Properties of Amyloid-β. Brain Res. Rev. 2003, 43, 1–16. [Google Scholar] [CrossRef]
- Sinha, M.; Bhowmick, P.; Banerjee, A.; Chakrabarti, S. Antioxidant Role of Amyloid β Protein in Cell-Free and Biological Systems: Implication for the Pathogenesis of Alzheimerdisease. Free Radic. Biol. Med. 2013, 56, 184–192. [Google Scholar] [CrossRef]
- Mitra, S.; Prasad, P.; Chakraborty, S. A Unified View of Assessing the Pro-Oxidant versus Antioxidant Nature of Amyloid Beta Conformers. Chembiochem 2018, 19, 2360–2371. [Google Scholar] [CrossRef]
- Carrillo-Mora, P.; Luna, R.; Colín-Barenque, L. Amyloid Beta: Multiple Mechanisms of Toxicity and Only Some Protective Effects? Oxid. Med. Cell Longev. 2014, 2014, 795375. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Amyloid-β-Induced Reactive Oxygen Species Production and Priming Are Differentially Regulated by Ion Channels in Microglia. J. Cell Physiol. 2011, 226, 3295–3302. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Febbraio, M. CD36, a Scavenger Receptor Involved in Immunity, Metabolism, Angiogenesis, and Behavior. Sci. Signal 2009, 2, re3. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, X.; Qian, L.; Chen, S.H.; Zhou, H.; Wilson, B.; Miller, D.S.; Hong, J.S. Microglial MAC1 Receptor and PI3K Are Essential in Mediating β-Amyloid Peptide-Induced Microglial Activation and Subsequent Neurotoxicity. J. Neuroinflam. 2011, 8, 3. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef]
- Gong, C.-X.; Iqbal, K. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Braithwaite, S.P.; Stock, J.B.; Lombroso, P.J.; Nairn, A.C. Protein Phosphatases and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2012, 106, 343–379. [Google Scholar] [CrossRef]
- Fan, X.; Xia, L.; Zhou, Z.; Qiu, Y.; Zhao, C.; Yin, X.; Qian, W. Tau Acts in Concert With Kinase/Phosphatase Underlying Synaptic Dysfunction. Front. Aging Neurosci. 2022, 14, 908881. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Lee, H.-G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic Oxidative Stress Causes Increased Tau Phosphorylation in M17 Neuroblastoma Cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef]
- Bartolome, F.; Carro, E.; Alquezar, C. Oxidative Stress in Tauopathies: From Cause to Therapy. Antioxidants 2022, 11, 1421. [Google Scholar] [CrossRef]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A Current View on Tau Protein Phosphorylation in Alzheimer’s Disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef]
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The Ambiguous Relationship of Oxidative Stress, Tau Hyperphosphorylation, and Autophagy Dysfunction in Alzheimer’s Disease. Oxid. Med. Cell Longev. 2015, 2015, 352723. [Google Scholar] [CrossRef]
- Cabezas-Opazo, F.A.; Vergara-Pulgar, K.; Pérez, M.J.; Jara, C.; Osorio-Fuentealba, C.; Quintanilla, R.A. Mitochondrial Dysfunction Contributes to the Pathogenesis of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2015, 2015, 509654. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 57. [Google Scholar] [CrossRef]
- Jurcău, M.C.; Andronie-Cioara, F.L.; Jurcău, A.; Marcu, F.; Ţiț, D.M.; Pașcalău, N.; Nistor-Cseppentö, D.C. The Link between Oxidative Stress, Mitochondrial Dysfunction and Neuroinflammation in the Pathophysiology of Alzheimer’s Disease: Therapeutic Implications and Future Perspectives. Antioxidants 2022, 11, 2167. [Google Scholar] [CrossRef]
- Morsy, A.; Trippier, P.C. Amyloid-Binding Alcohol Dehydrogenase (ABAD) Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2019, 62, 4252–4264. [Google Scholar] [CrossRef]
- Wilkins, H.M. Interactions between Amyloid, Amyloid Precursor Protein, and Mitochondria. Biochem. Soc. Trans. 2023, 51, 173–182. [Google Scholar] [CrossRef]
- Fišar, Z. Linking the Amyloid, Tau, and Mitochondrial Hypotheses of Alzheimer’s Disease and Identifying Promising Drug Targets. Biomolecules 2022, 12, 1676. [Google Scholar] [CrossRef]
- Vassallo, N. Amyloid Pores in Mitochondrial Membranes. Neural Regen. Res. 2021, 16, 2225–2226. [Google Scholar] [CrossRef]
- Pavlov, P.F.; Petersen, C.H.; Glaser, E.; Ankarcrona, M. Mitochondrial Accumulation of APP and Abeta: Significance for Alzheimer Disease Pathogenesis. J. Cell. Mol. Med. 2009, 13, 4137–4145. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Simonyi, A.; He, Y.; Sheng, W.; Sun, A.Y.; Wood, W.G.; Weisman, G.A.; Sun, G.Y. Targeting NADPH Oxidase and Phospholipases A2 in Alzheimer’s Disease. Mol. Neurobiol. 2010, 41, 73–86. [Google Scholar] [CrossRef]
- Yauger, Y.J.; Bermudez, S.; Moritz, K.E.; Glaser, E.; Stoica, B.; Byrnes, K.R. Iron Accentuated Reactive Oxygen Species Release by NADPH Oxidase in Activated Microglia Contributes to Oxidative Stress in Vitro. J. Neuroinflam. 2019, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron Homeostasis and Oxidative Stress: An Intimate Relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Spotorno, N.; Acosta-Cabronero, J.; Stomrud, E.; Lampinen, B.; Strandberg, O.T.; Van Westen, D.; Hansson, O. Relationship between Cortical Iron and Tau Aggregation in Alzheimer’s Disease. Brain 2020, 143, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Moreira, P.I.; Liu, G.; Dobson, J.; Perry, G.; Smith, M.A.; Zhu, X. Iron: The Redox-Active Center of Oxidative Stress in Alzheimer Disease. Neurochem. Res. 2007, 32, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Svobodová, H.; Kosnáč, D.; Balázsiová, Z.; Tanila, H.; Miettinen, P.O.; Sierra, A.; Vitovič, P.; Wagner, A.; Polák, Š.; Kopáni, M. Elevated Age-Related Cortical Iron, Ferritin and Amyloid Plaques in APPswe/PS1ΔE9 Transgenic Mouse Model of Alzheimer’s Disease. Physiol. Res. 2019, 68, S445–S451. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Ventriglia, M.; Simonelli, I.; Bonvicini, C.; Costa, A.; Perini, G.; Binetti, G.; Benussi, L.; Ghidoni, R.; Koch, G.; et al. Copper Imbalance in Alzheimer’s Disease: Meta-Analysis of Serum, Plasma, and Brain Specimens, and Replication Study Evaluating ATP7B Gene Variants. Biomolecules 2021, 11, 960. [Google Scholar] [CrossRef]
- Agarwal, P.; Dhana, K.; Schneider, J.A.; Ayton, S.; Wang, Y.; Agrawal, S.; Bennett, D.A.; Barnes, L.L.; Leurgans, S.E.; Bush, A.I.; et al. Association of Brain Copper with Alzheimer’s Disease Neuropathology: A Community-based Neuropathologic Study: Human Neuropathology/Clinicopathologic Correlations. Alzheimer’s Dement. 2020, 16, e045980. [Google Scholar] [CrossRef]
- Noda, Y.; Asada, M.; Kubota, M.; Maesako, M.; Watanabe, K.; Uemura, M.; Kihara, T.; Shimohama, S.; Takahashi, R.; Kinoshita, A.; et al. Copper Enhances APP Dimerization and Promotes Aβ Production. Neurosci. Lett. 2013, 547, 10–15. [Google Scholar] [CrossRef]
- Chen, Y.T.; Chen, W.Y.; Huang, X.T.; Xu, Y.C.; Zhang, H.Y. Iron Dysregulates APP Processing Accompanying with SAPPα Cellular Retention and β-Secretase Inhibition in Rat Cortical Neurons. Acta Pharmacol. Sin. 2018, 39, 177–183. [Google Scholar] [CrossRef]
- Zheng, W.; Xin, N.; Chi, Z.-H.; Zhao, B.-L.; Zhang, J.; Li, J.-Y.; Wang, Z.-Y. Divalent Metal Transporter 1 Is Involved in Amyloid Precursor Protein Processing and Abeta Generation. FASEB J. 2009, 23, 4207–4217. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative Stress, Dysfunctional Glucose Metabolism and Alzheimer Disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Wang, F.; Yu, H. Systemic Alterations of Tricarboxylic Acid Cycle Enzymes in Alzheimer’s Disease. Front. Neurosci. 2023, 17, 1206688. [Google Scholar] [CrossRef]
- Zündorf, G.; Reiser, G. Calcium Dysregulation and Homeostasis of Neural Calcium in the Molecular Mechanisms of Neurodegenerative Diseases Provide Multiple Targets for Neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- McDaid, J.; Mustaly-Kalimi, S.; Stutzmann, G.E. Ca2+ Dyshomeostasis Disrupts Neuronal and Synaptic Function in Alzheimer’s Disease. Cells 2020, 9, 2655. [Google Scholar] [CrossRef]
- Piccirillo, S.; Preziuso, A.; Amoroso, S.; Serfilippi, T.; Miceli, F.; Magi, S.; Lariccia, V. A New K+channel-Independent Mechanism Is Involved in the Antioxidant Effect of XE-991 in an in Vitro Model of Glucose Metabolism Impairment: Implications for Alzheimer’s Disease. Cell Death Discov. 2022, 8, 391. [Google Scholar] [CrossRef]
- Chandan, G.; Ganguly, U.; Pal, S.; Singh, S.; Saini, R.V.; Chakrabarti, S.S.; Saini, A.K.; Chakrabarti, S. GLUT Inhibitor WZB117 Induces Cytotoxicity with Increased Production of Amyloid-Beta Peptide in SH-SY5Y Cells Preventable by Beta-Hydroxybutyrate: Implications in Alzheimer’s Disease. Pharmacol. Rep. 2023, 75, 482–489. [Google Scholar] [CrossRef]
- Perluigi, M.; Di Domenico, F.; Barone, E.; Butterfield, D.A. MTOR in Alzheimer Disease and Its Earlier Stages: Links to Oxidative Damage in the Progression of This Dementing Disorder. Free Radic. Biol. Med. 2021, 169, 382–396. [Google Scholar] [CrossRef]
- D’cunha, N.M.; Sergi, D.; Lane, M.M.; Naumovski, N.; Gamage, E.; Rajendran, A.; Kouvari, M.; Gauci, S.; Dissanayka, T.; Marx, W.; et al. The Effects of Dietary Advanced Glycation End-Products on Neurocognitive and Mental Disorders. Nutrients 2022, 14, 2421. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Mosquera-Heredia, M.I.; Morales, L.C.; Vidal, O.M.; Barceló, E.; Silvera-Redondo, C.; Vélez, J.I.; Garavito-Galofre, P. Exosomes: Potential Disease Biomarkers and New Therapeutic Targets. Biomedicines 2021, 9, 1061. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The Exosome Journey: From Biogenesis to Uptake and Intracellular Signalling. Cell Commun. Signal 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, Biologic Function and Clinical Potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Melo, S.A. Non-Coding RNAs in Exosomes: New Players in Cancer Biology. Curr. Genom. 2015, 16, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. IJMS 2020, 21, 4407. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, S.; Israel, S.; Nagy, C.; Turecki, G. The Emerging Role of Exosomes in Mental Disorders. Transl. Psychiatry 2019, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Console, L.; Scalise, M.; Indiveri, C. Exosomes in Inflammation and Role as Biomarkers. Clin. Chim. Acta 2019, 488, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Hade, M.D.; Suire, C.N.; Suo, Z. Mesenchymal Stem Cell-Derived Exosomes: Applications in Regenerative Medicine. Cells 2021, 10, 1959. [Google Scholar] [CrossRef] [PubMed]
- Tienda-Vázquez, M.A.; Hanel, J.M.; Márquez-Arteaga, E.M.; Salgado-Álvarez, A.P.; Scheckhuber, C.Q.; Alanis-Gómez, J.R.; Espinoza-Silva, J.I.; Ramos-Kuri, M.; Hernández-Rosas, F.; Melchor-Martínez, E.M.; et al. Exosomes: A Promising Strategy for Repair, Regeneration and Treatment of Skin Disorders. Cells 2023, 12, 1625. [Google Scholar] [CrossRef] [PubMed]
- Gangadaran, P.; Madhyastha, H.; Madhyastha, R.; Rajendran, R.L.; Nakajima, Y.; Watanabe, N.; Velikkakath, A.K.G.; Hong, C.M.; Gopi, R.V.; Muthukalianan, G.K.; et al. The Emerging Role of Exosomes in Innate Immunity, Diagnosis and Therapy. Front. Immunol. 2023, 13, 1085057. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.M.; Carneiro, F.; Machado, J.C.; Melo, S.A. Exosomes and Immune Response in Cancer: Friends or Foes? Front. Immunol. 2018, 9, 730. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.C.; Wang, H. Pathogenic and Therapeutic Role of Exosomes in Neurodegenerative Disorders. Neural Regen. Res. 2024, 19, 75–79. [Google Scholar] [CrossRef]
- Chivet, M.; Hemming, F.; Pernet-Gallay, K.; Fraboulet, S.; Sadoul, R. Emerging Role of Neuronal Exosomes in the Central Nervous System. Front. Physiol. 2012, 3, 145. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Y.; Qin, Y.; Zhao, S.; Zheng, J.C. Exosome: A Novel Neurotransmission Modulator or Non-Canonical Neurotransmitter? Ageing Res. Rev. 2022, 74, 101558. [Google Scholar] [CrossRef]
- Jiang, L.; Dong, H.; Cao, H.; Ji, X.; Luan, S.; Liu, J. Exosomes in Pathogenesis, Diagnosis, and Treatment of Alzheimer’s Disease. Med. Sci. Monit. 2019, 25, 3329–3335. [Google Scholar] [CrossRef]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimers Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef]
- Yarana, C.; St. Clair, D.K. Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses. Antioxidants 2017, 6, 75. [Google Scholar] [CrossRef]
- Benedikter, B.J.; Weseler, A.R.; Wouters, E.F.M.; Savelkoul, P.H.M.; Rohde, G.G.U.; Stassen, F.R.M. Redox-Dependent Thiol Modifications: Implications for the Release of Extracellular Vesicles. Cell Mol. Life Sci. 2018, 75, 2321–2337. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, H.; Xu, W.; Wang, B.; Wu, H.; Tao, Y.; Zhang, B.; Wang, M.; Mao, F.; Yan, Y.; et al. Exosomes Released by Human Umbilical Cord Mesenchymal Stem Cells Protect against Cisplatin-Induced Renal Oxidative Stress and Apoptosis in Vivo and in Vitro. Stem Cell Res. Ther. 2013, 4, 34. [Google Scholar] [CrossRef]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.E.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal Stem Cell-Derived Exosomes Increase ATP Levels, Decrease Oxidative Stress and Activate PI3K/Akt Pathway to Enhance Myocardial Viability and Prevent Adverse Remodeling after Myocardial Ischemia/Reperfusion Injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Tancini, B.; Buratta, S.; Sagini, K.; Costanzi, E.; Delo, F.; Urbanelli, L.; Emiliani, C. Insight into the Role of Extracellular Vesicles in Lysosomal Storage Disorders. Genes 2019, 10, 510. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Øverbye, A.; Brech, A.; Torgersen, M.L.; Jakobsen, I.S.; Sandvig, K.; Llorente, A. PIKfyve Inhibition Increases Exosome Release and Induces Secretory Autophagy. Cell Mol. Life Sci. 2016, 73, 4717–4737. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Sánchez, D.; Furlán, M.; Colombo, M.I. Induction of Autophagy Promotes Fusion of Multivesicular Bodies with Autophagic Vacuoles in K562 Cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Kim, J.Y.; Lee, J.C.; Moon, M.H. Investigation of Lipidomic Perturbations in Oxidatively Stressed Subcellular Organelles and Exosomes by Asymmetrical Flow Field-Flow Fractionation and Nanoflow Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry. Anal. Chim. Acta 2019, 1073, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Nagaeva, O.; Kargl, D.; Baranov, V.; Mincheva-Nilsson, L. Thermal- and Oxidative Stress Causes Enhanced Release of NKG2D Ligand-Bearing Immunosuppressive Exosomes in Leukemia/Lymphoma T and B Cells. PLoS ONE 2011, 6, e16899. [Google Scholar] [CrossRef]
- Wang, R.; Li, J.; Zhang, X.; Zhang, X.; Zhang, X.; Zhu, Y.; Chen, C.; Liu, Z.; Wu, X.; Wang, D.; et al. Extracellular Vesicles Promote Epithelial-to-Mesenchymal Transition of Lens Epithelial Cells under Oxidative Stress. Exp. Cell Res. 2021, 398, 112362. [Google Scholar] [CrossRef]
- Chiaradia, E.; Tancini, B.; Emiliani, C.; Delo, F.; Pellegrino, R.M.; Tognoloni, A.; Urbanelli, L.; Buratta, S. Extracellular Vesicles under Oxidative Stress Conditions: Biological Properties and Physiological Roles. Cells 2021, 10, 1763. [Google Scholar] [CrossRef]
- Li, G.; Huang, D.; Li, N.; Ritter, J.K.; Li, P.L. Regulation of TRPML1 Channel Activity and Inflammatory Exosome Release by Endogenously Produced Reactive Oxygen Species in Mouse Podocytes. Redox Biol. 2021, 43, 102013. [Google Scholar] [CrossRef] [PubMed]
- Biasutto, L.; Chiechi, A.; Couch, R.; Liotta, L.A.; Espina, V. Retinal Pigment Epithelium (RPE) Exosomes Contain Signaling Phosphoproteins Affected by Oxidative Stress. Exp. Cell Res. 2013, 319, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Manèek-Keber, M.; Frank-Bertoncelj, M.; Hafner-Bratkoviè, I.; Smole, A.; Zorko, M.; Pirher, N.; Hayer, S.; Kralj-Igliè, V.; Rozman, B.; Ilc, N.; et al. Toll-like Receptor 4 Senses Oxidative Stress Mediated by the Oxidation of Phospholipids in Extracellular Vesicles. Sci. Signal 2015, 8, ra60. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J. Advancing Medicine for Alzheimer’s Disease: A Plasma Neural Exosome Platform. FASEB J. 2020, 34, 13079–13084. [Google Scholar] [CrossRef]
- Lim, C.Z.J.; Zhang, Y.; Chen, Y.; Zhao, H.; Stephenson, M.C.; Ho, N.R.Y.; Chen, Y.; Chung, J.; Reilhac, A.; Loh, T.P.; et al. Subtyping of Circulating Exosome-Bound Amyloid β Reflects Brain Plaque Deposition. Nat. Commun. 2019, 10, 1144. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Qiu, Q.; Zhang, H.; Chu, L.; Du, Y.; Zhang, J.; Zhou, C.; Liang, F.; Shi, S.; Wang, S.; et al. Concordance between the Assessment of Aβ42, T-Tau, and P-T181-Tau in Peripheral Blood Neuronal-Derived Exosomes and Cerebrospinal Fluid. Alzheimers Dement. 2019, 15, 1071–1080. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of Preclinical Alzheimer’s Disease by a Profile of Pathogenic Proteins in Neurally Derived Blood Exosomes: A Case-Control Study. Alzheimers Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of Conversion from Mild Cognitive Impairment to Dementia with Neuronally Derived Blood Exosome Protein Profile. Alzheimers Dement. 2016, 3, 63–72. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Usuki, S.; Sakai, S.; Hanamatsu, H.; Mioka, T.; Kimura, N.; Okada, M.; Tahara, H.; Furukawa, J.I.; et al. A Potential Function for Neuronal Exosomes: Sequestering Intracerebral Amyloid-β Peptide. FEBS Lett. 2015, 589, 84–88. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome Reduction in Vivo Is Associated with Lower Amyloid Plaque Load in the 5XFAD Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Hamdane, M.; Loyens, A.; Gelé, P.; Drobeck, H.; Bégard, S.; Galas, M.C.; Delacourte, A.; Beauvillain, J.C.; Buée, L.; et al. Alkalizing Drugs Induce Accumulation of Amyloid Precursor Protein By-Products in Luminal Vesicles of Multivesicular Bodies. J. Biol. Chem. 2007, 282, 18197–18205. [Google Scholar] [CrossRef]
- Laulagnier, K.; Javalet, C.; Hemming, F.J.; Chivet, M.; Lachenal, G.; Blot, B.; Chatellard, C.; Sadoul, R. Amyloid Precursor Protein Products Concentrate in a Subset of Exosomes Specifically Endocytosed by Neurons. Cell Mol. Life Sci. 2018, 75, 757–773. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s Disease Beta-Amyloid Peptides Are Released in Association with Exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s Disease Pathology Propagation by Exosomes Containing Toxic Amyloid-Beta Oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Schneider, J.A.; Tangney, C.; Tremblay-Mercier, J.; Fortier, M.; Bennett, D.A.; Morris, M.C. Plasma and Brain Fatty Acid Profiles in Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimers Dis. 2012, 29, 691–697. [Google Scholar] [CrossRef]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Götz, J. Extracellular Vesicles Isolated from the Brains of RTg4510 Mice Seed Tau Protein Aggregation in a Threshold-Dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef] [PubMed]
- Winston, C.N.; Aulston, B.; Rockenstein, E.M.; Adame, A.; Prikhodko, O.; Dave, K.N.; Mishra, P.; Rissman, R.A.; Yuan, S.H. Neuronal Exosome-Derived Human Tau Is Toxic to Recipient Mouse Neurons in Vivo. J. Alzheimers Dis. 2019, 67, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of Microglia and Inhibition of Exosome Synthesis Halt Tau Propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Zhu, B.; Liu, Y.; Hwang, S.; Archuleta, K.; Huang, H.; Campos, A.; Murad, R.; Piña-Crespo, J.; Xu, H.; Huang, T.Y. Trem2 Deletion Enhances Tau Dispersion and Pathology through Microglia Exosomes. Mol. Neurodegener. 2022, 17, 58. [Google Scholar] [CrossRef]
- Crotti, A.; Sait, H.R.; McAvoy, K.M.; Estrada, K.; Ergun, A.; Szak, S.; Marsh, G.; Jandreski, L.; Peterson, M.; Reynolds, T.L.; et al. BIN1 Favors the Spreading of Tau via Extracellular Vesicles. Sci. Rep. 2019, 9, 9477. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Stewart, T.; Hong, Z.; Guo, Z.; Aro, P.; Soltys, D.; Pan, C.; Peskind, E.R.; Zabetian, C.P.; Shaw, L.M.; et al. Blood Extracellular Vesicles Carrying Synaptic Function- and Brain-related Proteins as Potential Biomarkers for Alzheimer’s Disease. Alzheimer’s Dement. 2023, 19, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Sun, Y.; Zhen, H.; Guo, M.; Ye, J.; Liu, Z.; Yang, Y.; Zhang, X. Differential Expression of Plasma Exo-miRNA in Neurodegenerative Diseases by Next-Generation Sequencing. Front. Neurosci. 2020, 14, 438. [Google Scholar] [CrossRef]
- Li, T.-R.; Wang, X.-N.; Sheng, C.; Li, Y.-X.; Li, F.Z.-T.; Sun, Y.; Han, Y. Extracellular Vesicles as an Emerging Tool for the Early Detection of Alzheimer’s Disease. Mech. Ageing Dev. 2019, 184, 111175. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased Synaptic Proteins in Neuronal Exosomes of Frontotemporal Dementia and Alzheimer’s Disease. FASEB J. 2016, 30, 4141–4148. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered Lysosomal Proteins in Neural-Derived Plasma Exosomes in Preclinical Alzheimer Disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Carlson, O.D.; Mustapic, M.; Kapogiannis, D. Low Neural Exosomal Levels of Cellular Survival Factors in Alzheimer’s Disease. Ann. Clin. Transl. Neurol. 2015, 2, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, S.; Jedrychowski, M.P.; Yanamandra, K.; Ikezu, S.; Gygi, S.P.; Ikezu, T. Proteomic Profiling of Extracellular Vesicles Derived from Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Pilot Study. Cells 2020, 9, 1959. [Google Scholar] [CrossRef]
- Muraoka, S.; Jedrychowski, M.P.; Tatebe, H.; DeLeo, A.M.; Ikezu, S.; Tokuda, T.; Gygi, S.P.; Stern, R.A.; Ikezu, T. Proteomic Profiling of Extracellular Vesicles Isolated From Cerebrospinal Fluid of Former National Football League Players at Risk for Chronic Traumatic Encephalopathy. Front. Neurosci. 2019, 13, 10559. [Google Scholar] [CrossRef]
- Gerber, H.; Mosser, S.; Boury-Jamot, B.; Stumpe, M.; Piersigilli, A.; Goepfert, C.; Dengjel, J.; Albrecht, U.; Magara, F.; Fraering, P.C. The APMAP Interactome Reveals New Modulators of APP Processing and Beta-Amyloid Production That Are Altered in Alzheimer’s Disease. Acta Neuropathol. Commun. 2019, 7, 13. [Google Scholar] [CrossRef]
- Hensen, S.M.M.; Heldens, L.; Van Enckevort, C.M.W.; Van Genesen, S.T.; Pruijn, G.J.M.; Lubsen, N.H. Activation of the Antioxidant Response in Methionine Deprived Human Cells Results in an HSF1-Independent Increase in HSPA1A MRNA Levels. Biochimie 2013, 95, 1245–1251. [Google Scholar] [CrossRef]
- Ren, G.; Ma, Z.; Hui, M.; Kudo, L.C.; Hui, K.S.; Karsten, S.L. Cu, Zn-Superoxide Dismutase 1 (SOD1) Is a Novel Target of Puromycin-Sensitive Aminopeptidase (PSA/NPEPPS): PSA/NPEPPS Is a Possible Modifier of Amyotrophic Lateral Sclerosis. Mol. Neurodegener. 2011, 6, 29. [Google Scholar] [CrossRef]
- Cheng, L.; Doecke, J.D.; Sharples, R.A.; Villemagne, V.L.; Fowler, C.J.; Rembach, A.; Martins, R.N.; Rowe, C.C.; Macaulay, S.L.; Masters, C.L.; et al. Prognostic Serum MiRNA Biomarkers Associated with Alzheimer’s Disease Shows Concordance with Neuropsychological and Neuroimaging Assessment. Mol. Psychiatry 2015, 20, 1188–1196. [Google Scholar] [CrossRef]
- Li, W.; Zheng, Y. MicroRNAs in Extracellular Vesicles of Alzheimer’s Disease. Cells 2023, 12, 1378. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma Exosomal MiRNAs in Persons with and without Alzheimer Disease: Altered Expression and Prospects for Biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.X.; Liu, H.; Zhang, L.S.; Lv, W.; Hu, X.Y. Altered MicroRNA Profiles in Cerebrospinal Fluid Exosome in Parkinson Disease and Alzheimer Disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef]
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J.; et al. MicroRNA Expression Levels Are Altered in the Cerebrospinal Fluid of Patients with Young-Onset Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8826–8841. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Vázquez-Higuera, J.L.; Pozueta, A.; Lage, C.; Kazimierczak, M.; Bravo, M.; Calero, M.; González, A.; Rodríguez, E.; Lleó, A.; et al. MicroRNA Profile in Patients with Alzheimer’s Disease: Analysis of MiR-9-5p and MiR-598 in Raw and Exosome Enriched Cerebrospinal Fluid Samples. J. Alzheimers Dis. 2017, 57, 483–491. [Google Scholar] [CrossRef]
- Barbagallo, C.; Mostile, G.; Baglieri, G.; Giunta, F.; Luca, A.; Raciti, L.; Zappia, M.; Purrello, M.; Ragusa, M.; Nicoletti, A. Specific Signatures of Serum miRNAs as Potential Biomarkers to Discriminate Clinically Similar Neurodegenerative and Vascular-Related Diseases. Cell Mol. Neurobiol. 2020, 40, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Valero, A.; Campdelacreu, J.; Vilas, D.; Ispierto, L.; Reñé, R.; Álvarez, R.; Armengol, M.P.; Borràs, F.E.; Beyer, K. Exploratory Study on microRNA Profiles from Plasma-Derived Extracellular Vesicles in Alzheimer’s Disease and Dementia with Lewy Bodies. Transl. Neurodegener. 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.G.; Song, J.; Zhang, Y.Q.; Wang, P.C. MicroRNA-193b Is a Regulator of Amyloid Precursor Protein in the Blood and Cerebrospinal Fluid Derived Exosomal MicroRNA-193b Is a Biomarker of Alzheimer’s Disease. Mol. Med. Rep. 2014, 10, 2395–2400. [Google Scholar] [CrossRef]
- Wang, L.L.; Min, L.; Guo, Q.D.; Zhang, J.X.; Jiang, H.L.; Shao, S.; Xing, J.G.; Yin, L.L.; Liu, J.H.; Liu, R.; et al. Profiling MicroRNA from Brain by Microarray in a Transgenic Mouse Model of Alzheimer’s Disease. Biomed. Res. Int. 2017, 2017, 8030369. [Google Scholar] [CrossRef]
- Fu, Y.; Hu, X.; Zheng, C.; Sun, G.; Xu, J.; Luo, S.; Cao, P. Intrahippocampal MiR-342-3p Inhibition Reduces β-Amyloid Plaques and Ameliorates Learning and Memory in Alzheimer’s Disease. Metab. Brain Dis. 2019, 34, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yu, J.T.; Tan, M.S.; Liu, Q.Y.; Wang, H.F.; Zhang, W.; Jiang, T.; Tan, L. Genome-Wide Serum MicroRNA Expression Profiling Identifies Serum Biomarkers for Alzheimer’s Disease. J. Alzheimers Dis. 2014, 40, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Elsherbini, A.; Zhu, Z.; Quadri, Z.; Crivelli, S.M.; Ren, X.; Vekaria, H.J.; Tripathi, P.; Zhang, L.; Zhi, W.; Bieberich, E. Novel Isolation Method Reveals Sex-Specific Composition and Neurotoxicity of Small Extracellular Vesicles in a Mouse Model of Alzheimer’s Disease. Cells 2023, 12, 1623. [Google Scholar] [CrossRef] [PubMed]
- Elsherbini, A.; Kirov, A.S.; Dinkins, M.B.; Wang, G.; Qin, H.; Zhu, Z.; Tripathi, P.; Crivelli, S.M.; Bieberich, E. Association of Aβ with Ceramide-Enriched Astrosomes Mediates Aβ Neurotoxicity. Acta Neuropathol. Commun. 2020, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, Y.; Gao, Q.; Ping, D.; Wang, Y.; Wu, W.; Lin, X.; Fang, Y.; Zhang, J.; Shao, A. The Role of Exosomal microRNAs and Oxidative Stress in Neurodegenerative Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 3232869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 Family: A Potential Tumor Suppressor and Therapeutic Candidate in Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef] [PubMed]
- Kalfert, D.; Ludvikova, M.; Pesta, M.; Ludvik, J.; Dostalova, L.; Kholová, I. Multifunctional Roles of MiR-34a in Cancer: A Review with the Emphasis on Head and Neck Squamous Cell Carcinoma and Thyroid Cancer with Clinical Implications. Diagnostics 2020, 10, 563. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.; Chertkow, H.; Schipper, H.M.; Yuan, Z.; Shetty, V.; Jenkins, S.; Jones, T.; Wang, E. Increased MicroRNA-34c Abundance in Alzheimer’s Disease Circulating Blood Plasma. Front. Mol. Neurosci. 2014, 7, 2. [Google Scholar] [CrossRef]
- Sarkar, S.; Jun, S.; Rellick, S.; Quintana, D.D.; Cavendish, J.Z.; Simpkins, J.W. Expression of MicroRNA-34a in Alzheimer’s Disease Brain Targets Genes Linked to Synaptic Plasticity, Energy Metabolism, and Resting State Network Activity. Brain Res. 2016, 1646, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Engler-Chiurazzi, E.B.; Cavendish, J.Z.; Povroznik, J.M.; Russell, A.E.; Quintana, D.D.; Mathers, P.H.; Simpkins, J.W. Over-Expression of MiR-34a Induces Rapid Cognitive Impairment and Alzheimer’s Disease-like Pathology. Brain Res. 2019, 1721, 146327. [Google Scholar] [CrossRef] [PubMed]
- Freitas, R.; McDonald, K.; Wang, H.; Sarkar, S.; Corbin, D.; Bix, G.J.; Simpkins, J.W.; Engler-Chiurazzi, E. Inducible microRNA-34a Overexpression Impairs Cognition and Promotes Alzheimer’s Pathology. Alzheimer’s Dement. 2022, 18, e064580. [Google Scholar] [CrossRef]
- Jian, C.; Lu, M.; Zhang, Z.; Liu, L.; Li, X.; Huang, F.; Xu, N.; Qin, L.; Zhang, Q.; Zou, D. MiR-34a Knockout Attenuates Cognitive Deficits in APP/PS1 Mice through Inhibition of the Amyloidogenic Processing of APP. Life Sci. 2017, 182, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Kou, X.; Li, J.; Liu, X.; Chang, J.; Zhao, Q.; Jia, S.; Fan, J.; Chen, N. Swimming Attenuates d -Galactose-Induced Brain Aging via Suppressing miR-34a-Mediated Autophagy Impairment and Abnormal Mitochondrial Dynamics. J. Appl. Physiol. 2017, 122, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lin, Y.; Liu, Y. MiR-34a Increases Inflammation and Oxidative Stress Levels in Patients with Necrotizing Enterocolitis by Downregulating SIRT1 Expression. Mol. Med. Rep. 2021, 24, 664. [Google Scholar] [CrossRef]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in Plasma and Cerebrospinal Fluid as Potential Markers for Alzheimer’s Disease. JAD 2014, 39, 253–259. [Google Scholar] [CrossRef]
- Jin, Y.; Tu, Q.; Liu, M. MicroRNA-125b Regulates Alzheimer’s Disease through SphK1 Regulation. Mol. Med. Rep. 2018, 18, 2373–2380. [Google Scholar] [CrossRef]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. MicroRNA-125b Induces Tau Hyperphosphorylation and Cognitive Deficits in Alzheimer’s Disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef]
- Shen, Y.; Shen, Z.; Guo, L.; Zhang, Q.; Wang, Z.; Miao, L.; Wang, M.; Wu, J.; Guo, W.; Zhu, Y. MiR-125b-5p Is Involved in Oxygen and Glucose Deprivation Injury in PC-12 Cells via CBS/H2S Pathway. Nitric Oxide 2018, 78, 11–21. [Google Scholar] [CrossRef]
- Li, P.; Xu, Y.; Wang, B.; Huang, J.; Li, Q. MiR-34a-5p and MiR-125b-5p Attenuate Aβ-Induced Neurotoxicity through Targeting BACE1. J. Neurol. Sci. 2020, 413, 116793. [Google Scholar] [CrossRef]
- Ji, J.; Qin, Y.; Ren, J.; Lu, C.; Wang, R.; Dai, X.; Zhou, R.; Huang, Z.; Xu, M.; Chen, M.; et al. Mitochondria-Related MiR-141-3p Contributes to Mitochondrial Dysfunction in HFD-Induced Obesity by Inhibiting PTEN. Sci. Rep. 2015, 5, 16262. [Google Scholar] [CrossRef]
- Qin, Q.; Cui, L.; Zhou, Z.; Zhang, Z.; Wang, Y.; Zhou, C. Inhibition of microRNA-141-3p Reduces Hypoxia-Induced Apoptosis in H9c2 Rat Cardiomyocytes by Activating the RP105-Dependent PI3K/AKT Signaling Pathway. Med. Sci. Monit. 2019, 25, 7016–7025. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, R.; Chen, Y.; Wang, M.; Du, J. Crosstalk between Oxidative Stress and Exosomes. Oxid. Med. Cell Longev. 2022, 2022, 3553617. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Ren, X.S.; Tong, Y.; Qiu, Y.; Ye, C.; Wu, N.; Xiong, X.Q.; Wang, J.J.; Han, Y.; Zhou, Y.B.; Zhang, F.; et al. MiR155-5p in Adventitial Fibroblasts-Derived Extracellular Vesicles Inhibits Vascular Smooth Muscle Cell Proliferation via Suppressing Angiotensin-Converting Enzyme Expression. J. Extracell. Vesicles 2019, 9, 1698795. [Google Scholar] [CrossRef]
- Yao, W.; Tai, L.W.; Liu, Y.; Hei, Z.; Li, H. Oxidative Stress and Inflammation Interaction in Ischemia Reperfusion Injury: Role of Programmed Cell Death. Oxid. Med. Cell Longev. 2019, 2019, 6780816. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Qian, C.; Hou, S.; Lu, J.; Zou, Q.; Li, H.; Huang, B. Exosomal MiRNA-320a Is Released from HAMSCs and Regulates SIRT4 to Prevent Reactive Oxygen Species Generation in POI. Mol. Ther. Nucleic Acids 2020, 21, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Kuang, X.; Cai, S.; Wang, X.; Du, D.; Wang, J.; Wang, Y.; Chen, Y.; Bihl, J.; Chen, Y.; et al. MiR-132-3p Priming Enhances the Effects of Mesenchymal Stromal Cell-Derived Exosomes on Ameliorating Brain Ischemic Injury. Stem Cell Res. Ther. 2020, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Gatti, M.; Zavatti, M.; Beretti, F.; Giuliani, D.; Vandini, E.; Ottani, A.; Bertucci, E.; Maraldi, T. Oxidative Stress in Alzheimer’s Disease: In Vitro Therapeutic Effect of Amniotic Fluid Stem Cells Extracellular Vesicles. Oxid. Med. Cell Longev. 2020, 2020, 2785343. [Google Scholar] [CrossRef] [PubMed]
- de Godoy, M.A.; Saraiva, L.M.; de Carvalho, L.R.P.; Vasconcelos-dos-Santos, A.; Beiral, H.J.V.; Ramos, A.B.; de Paula Silva, L.R.; Leal, R.B.; Monteiro, V.H.S.; Braga, C.V.; et al. Mesenchymal Stem Cells and Cell-Derived Extracellular Vesicles Protect Hippocampal Neurons from Oxidative Stress and Synapse Damage Induced by Amyloid-β Oligomers. J. Biol. Chem. 2018, 293, 1957–1975. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bir, A.; Ghosh, A.; Chauhan, A.; Saha, S.; Saini, A.K.; Bisaglia, M.; Chakrabarti, S. Exosomal Dynamics and Brain Redox Imbalance: Implications in Alzheimer’s Disease Pathology and Diagnosis. Antioxidants 2024, 13, 316. https://doi.org/10.3390/antiox13030316
Bir A, Ghosh A, Chauhan A, Saha S, Saini AK, Bisaglia M, Chakrabarti S. Exosomal Dynamics and Brain Redox Imbalance: Implications in Alzheimer’s Disease Pathology and Diagnosis. Antioxidants. 2024; 13(3):316. https://doi.org/10.3390/antiox13030316
Chicago/Turabian StyleBir, Aritri, Arindam Ghosh, Aman Chauhan, Sarama Saha, Adesh K. Saini, Marco Bisaglia, and Sasanka Chakrabarti. 2024. "Exosomal Dynamics and Brain Redox Imbalance: Implications in Alzheimer’s Disease Pathology and Diagnosis" Antioxidants 13, no. 3: 316. https://doi.org/10.3390/antiox13030316