
Nucleoredoxin Redox Interactions Are Sensitized by Aging and Potentiated by Chronic Alcohol Consumption in the Mouse Liver

 , , , , , , , , , , and
, , , , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Histopathology and Immunodetection Analyses
2.3. Senescence-Associated β-Galactosidase Activity In Situ (SA-β-gal)
2.4. Western Blot (WB) Analysis
2.5. Determination of Carbonylated Proteins
2.6. Immunoprecipitation (IP) Assay
2.7. Statistical Analysis
3. Results
3.1. Chronic Alcohol Consumption Increases Mice Body Weight and the Relative Liver Weight during Aging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Age | Body Weight (g) | Percentage of Relative Liver Weight | |
|---|---|---|---|---|
| Initial | Final | |||
| C | 7W | 15.28 ± 0.16 | 15.64 ± 0.14 | 5.53 ± 0.12 |
| 12M | 24.93 ± 2.60 | 29.92 ± 1.39 * | 5.65 ± 0.66 | |
| 18M | 32.00 ± 2.08 | 36.00 ± 1.05 * | 4.38 ± 0.28 | |
| ALD | 7W | 17.66 ± 0.37 | 18.44 ± 0.30 | 4.97 ± 0.16 |
| 12M | 26.40 ± 0.58 | 30.34 ± 0.81 * | 6.14 ± 0.31 | |
| 18M | 26.92 ± 0.68 | 30.30 ± 0.47 * | 6.25 ± 0.64 $ | |
3.2. Chronic Alcohol Consumption Modifies Extracellular Matrix Components during Aging
3.3. Cell Proliferation Is Increased by Chronic Alcohol Consumption during Aging
3.4. Chronic Alcohol Consumption Increases Cellular Senescence Markers in the Liver of Aged Mice
3.5. Chronic Alcohol Consumption Promotes the Preferential Localization of NXN Either into or Alongside Senescent Cells in the Liver of Aged Mice
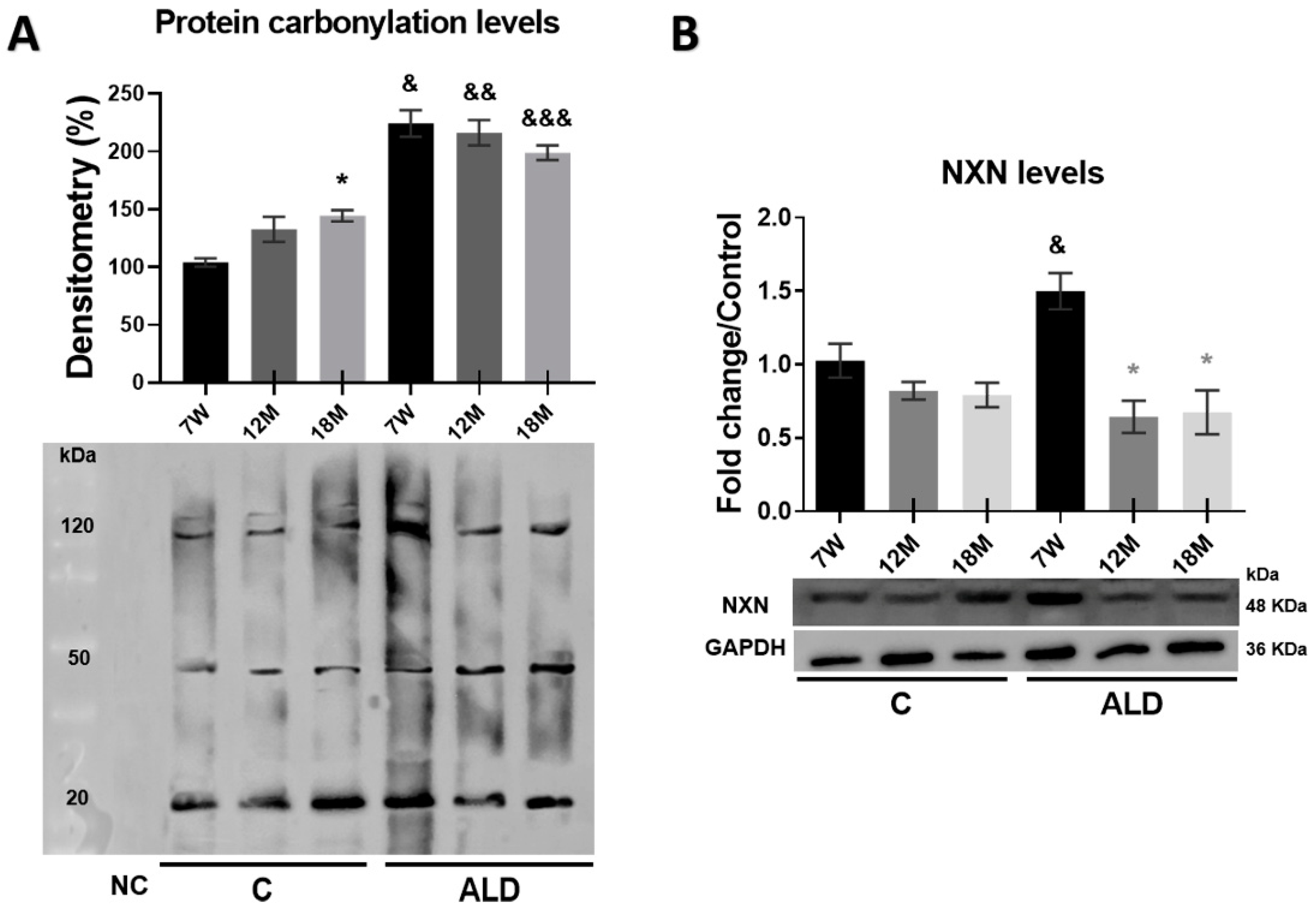
3.6. Carbonylated Proteins Are Increased by Chronic Alcohol Consumption in the Liver of Aged Mice
3.7. Chronic Alcohol Consumption Worsens Aging-Promoted Alteration of NXN-Dependent Interaction Ratios in the Mouse Liver
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flatt, T. A New Definition of Aging? Front. Genet. 2012, 3, 148. [Google Scholar] [CrossRef]
- White, A.M.; Orosz, A.; Powell, P.A.; Koob, G.F. Alcohol and Aging—An Area of Increasing Concern. Alcohol 2023, 107, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Shadyab, A.H.; LaCroix, A.Z. Genetic Factors Associated with Longevity: A Review of Recent Findings. Ageing Res. Rev. 2015, 19, 1–7. [Google Scholar] [CrossRef]
- Aravinthan, A.D.; Alexander, G.J.M. Senescence in Chronic Liver Disease: Is the Future in Aging? J. Hepatol. 2016, 65, 825–834. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Gonzalez, S.; Lu, W.Y.; Raven, A.; Dwyer, B.; Man, T.Y.; O’Duibhir, E.; Lewis, P.J.S.; Campana, L.; Kendall, T.J.; Bird, T.G.; et al. Paracrine Cellular Senescence Exacerbates Biliary Injury and Impairs Regeneration. Nat. Commun. 2018, 9, 1020. [Google Scholar] [CrossRef] [PubMed]
- Kudlova, N.; De Sanctis, J.B.; Hajduch, M. Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs. Int. J. Mol. Sci. 2022, 23, 4168. [Google Scholar] [CrossRef]
- Maldonado, E.; Morales-Pison, S.; Urbina, F.; Solari, A. Aging Hallmarks and the Role of Oxidative Stress. Antioxidants 2023, 12, 651. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy Metabolism in the Liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar]
- Sheedfar, F.; Di Biase, S.; Koonen, D.; Vinciguerra, M. Liver Diseases and Aging: Friends or Foes? Aging Cell 2013, 12, 950–954. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of Liver Diseases in the World. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Dey, A.; Cederbaum, A.I. Alcohol and Oxidative Liver Injury. Hepatology 2006, 43 (Suppl. S1), S63–S74. [Google Scholar] [CrossRef]
- Shearn, C.T.; Orlicky, D.J.; Saba, L.M.; Shearn, A.H.; Petersen, D.R. Increased Hepatocellular Protein Carbonylation in Human End-Stage Alcoholic Cirrhosis. Free Radic. Biol. Med. 2015, 89, 1144–1153. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Choi, Y.; Ha, S.K.; Song, B.J. Cytochrome P450-2e1 Promotes Aging-Related Hepatic Steatosis, Apoptosis and Fibrosis through Increased Nitroxidative Stress. Free Radic. Biol. Med. 2016, 91, 188–202. [Google Scholar] [CrossRef]
- Meier, P.; Seitz, H.K. Age, Alcohol Metabolism and Liver Disease. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Kurooka, H.; Kato, K.; Minoguchi, S.; Takahashi, Y.; Ikeda, J.; Habu, S.; Osawa, N.; Buchberg, A.M.; Moriwaki, K.; Shisa, H.; et al. Cloning and Characterization of the Nucleoredoxin Gene That Encodes a Novel Nuclear Protein Related to Thioredoxin. Genomics 1997, 39, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kneeshaw, S.; Keyani, R.; Delorme-Hinoux, V.; Imrie, L.; Loake, G.J.; Le Bihan, T.; Reichheld, J.P.; Spoel, S.H. Nucleoredoxin Guards against Oxidative Stress by Protecting Antioxidant Enzymes. Proc. Natl. Acad. Sci. USA 2017, 114, 8414–8419. [Google Scholar] [CrossRef] [PubMed]
- Idelfonso-García, O.G.; Alarcón-Sánchez, B.R.; Vásquez-Garzón, V.R.; Baltiérrez-Hoyos, R.; Villa-Treviño, S.; Muriel, P.; Serrano, H.; Pérez-Carreón, J.I.; Arellanes-Robledo, J. Is Nucleoredoxin a Master Regulator of Cellular Redox Homeostasis? Its Implication in Different Pathologies. Antioxidants 2022, 11, 670. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Michiue, T.; Asashima, M.; Miki, H. The Thioredoxin-Related Redox-Regulating Protein Nucleoredoxin Inhibits Wnt-Beta-Catenin Signalling through Dishevelled. Nat. Cell Biol. 2006, 8, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Arellanes-Robledo, J.; Reyes-Gordillo, K.; Shah, R.; Dominguez-Rosales, J.A.; Hernandez-Nazara, Z.H.; Ramirez, F.; Rojkind, M.; Lakshman, M.R. Fibrogenic Actions of Acetaldehyde Are Beta-Catenin Dependent but Wingless Independent: A Critical Role of Nucleoredoxin and Reactive Oxygen Species in Human Hepatic Stellate Cells. Free Radic. Biol. Med. 2013, 65, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Arellanes-Robledo, J.; Reyes-Gordillo, K.; Ibrahim, J.; Leckey, L.; Shah, R.; Lakshman, M.R. Ethanol Targets Nucleoredoxin/Dishevelled Interactions and Stimulates Phosphatidylinositol 4-Phosphate Production In Vivo and In Vitro. Biochem. Pharmacol. 2018, 156, 135–146. [Google Scholar] [CrossRef]
- Alarcon-Sanchez, B.R.; Guerrero-Escalera, D.; Rosas-Madrigal, S.; Aparicio-Bautista, D.I.; Reyes-Gordillo, K.; Lakshman, M.R.; Ortiz-Fernandez, A.; Quezada, H.; Medina-Contreras, O.; Villa-Trevino, S.; et al. Nucleoredoxin Interaction with Flightless-I/Actin Complex Is Differentially Altered in Alcoholic Liver Disease. Basic Clin. Pharmacol. Toxicol. 2020, 127, 389–404. [Google Scholar] [CrossRef]
- Gu, Z.; Tan, W.; Feng, G.; Meng, Y.; Shen, B.; Liu, H.; Cheng, C. Wnt/Beta-Catenin Signaling Mediates the Senescence of Bone Marrow-Mesenchymal Stem Cells from Systemic Lupus Erythematosus Patients through the P53/P21 Pathway. Mol. Cell. Biochem. 2014, 387, 27–37. [Google Scholar] [CrossRef]
- Feng, G.; Zheng, K.; Cao, T.; Zhang, J.; Lian, M.; Huang, D.; Wei, C.; Gu, Z.; Feng, X. Repeated Stimulation by Lps Promotes the Senescence of Dpscs via Tlr4/Myd88-Nf-Kappab-P53/P21 Signaling. Cytotechnology 2018, 70, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Vendemiale, G. The Aging Liver: Redox Biology and Liver Regeneration. Antioxid. Redox Signal. 2021, 35, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Koch, O.R.; Fusco, S.; Ranieri, S.C.; Maulucci, G.; Palozza, P.; Larocca, L.M.; Cravero, A.A.; Farre, S.M.; De Spirito, M.; Galeotti, T.; et al. Role of the Life Span Determinant P66(Shca) in Ethanol-Induced Liver Damage. Lab. Investig. 2008, 88, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin In Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Daniels, S.J.; Nielsen, S.H.; Bager, C.; Rasmussen, D.G.K.; Loomba, R.; Surabattula, R.; Villesen, I.F.; Luo, Y.; Shevell, D.; et al. Collagen Biology and Non-Invasive Biomarkers of Liver Fibrosis. Liver Int. 2020, 40, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Chen, H.; Jiang, L.; Liu, L. Liver Regeneration: Changes in Oxidative Stress, Immune System, Cytokines, and Epigenetic Modifications Associated with Aging. Oxidative Med. Cell. Longev. 2022, 2022, 9018811. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(Adp-Ribose): Novel Functions for an Old Molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Fedorova, M.; Bollineni, R.C.; Hoffmann, R. Protein Carbonylation as a Major Hallmark of Oxidative Damage: Update of Analytical Strategies. Mass Spectrom. Rev. 2014, 33, 79–97. [Google Scholar] [CrossRef]
- Arellanes-Robledo, J.; Ibrahim, J.; Reyes-Gordillo, K.; Shah, R.; Leckey, L.; Lakshman, M.R. Flightless-I Is a Potential Biomarker for the Early Detection of Alcoholic Liver Disease. Biochem. Pharmacol. 2021, 183, 114323. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.H.; Kisseleva, T.; Brenner, D.A. Aging and Liver Disease. Curr. Opin. Gastroenterol. 2015, 31, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, X.; Wang, B.; Zhou, H.; Dang, S.; Shi, Y.; Hao, L.; Luo, Q.; Jin, M.; Zhou, Q.; et al. Aging-Associated Oxidative Stress Inhibits Liver Progenitor Cell Activation in Mice. Aging 2017, 9, 1359–1374. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Alcoholic Liver Disease in the Elderly. Clin. Geriatr. Med. 2007, 23, 905–921, viii. [Google Scholar] [CrossRef] [PubMed]
- Fujino, G.; Noguchi, T.; Takeda, K.; Ichijo, H. Thioredoxin and Protein Kinases in Redox Signaling. Semin. Cancer Biol. 2006, 16, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. 2017, 38, 147–161. [Google Scholar]
- Adrover, J.M.; Nicolas-Avila, J.A.; Hidalgo, A. Aging: A Temporal Dimension for Neutrophils. Trends Immunol. 2016, 37, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Bologna-Molina, R.; Mosqueda-Taylor, A.; Molina-Frechero, N.; Mori-Estevez, A.D.; Sanchez-Acuna, G. Comparison of the Value of Pcna and Ki-67 as Markers of Cell Proliferation in Ameloblastic Tumors. Med. Oral Patol. Oral Cir. Bucal 2013, 18, e174–e179. [Google Scholar] [CrossRef]
- Pibiri, M. Liver Regeneration in Aged Mice: New Insights. Aging 2018, 10, 1801–1824. [Google Scholar] [CrossRef]
- Mendenhall, C.L.; Rouster, S.D.; Roselle, G.A.; Grossman, C.J.; Ghosn, S.; Gartside, P. Impact of Chronic Alcoholism on the Aging Rat: Changes in Nutrition, Liver Composition, and Mortality. Alcohol Clin. Exp. Res. 1993, 17, 847–853. [Google Scholar] [CrossRef]
- Rao, R. Endotoxemia and Gut Barrier Dysfunction in Alcoholic Liver Disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef]
- Piskunova, T.S.; Yurova, M.N.; Ovsyannikov, A.I.; Semenchenko, A.V.; Zabezhinski, M.A.; Popovich, I.G.; Wang, Z.Q.; Anisimov, V.N. Deficiency in Poly(Adp-Ribose) Polymerase-1 (Parp-1) Accelerates Aging and Spontaneous Carcinogenesis in Mice. Curr. Gerontol. Geriatr. Res. 2008, 2008, 754190. [Google Scholar] [CrossRef]
- Dong, R.; Wang, X.; Wang, L.; Wang, C.; Huang, K.; Fu, T.; Liu, K.; Wu, J.; Sun, H.; Meng, Q. Yangonin Inhibits Ethanol-Induced Hepatocyte Senescence Via Mir-194/Fxr Axis. Eur. J. Pharmacol. 2021, 890, 173653. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ge, T.; Shao, Y.; Cui, W.; Li, Z.; Xu, W.; Bao, X. Znf281 Drives Hepatocyte Senescence in Alcoholic Liver Disease by Reducing Hk2-Stabilized Pink1/Parkin-Mediated Mitophagy. Cell Prolif. 2023, 56, e13378. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nunez, S.; Chicas, A.; Hearn, S.A.; Myers, M.P.; Lowe, S.W. A Novel Role for High-Mobility Group a Proteins in Cellular Senescence and Heterochromatin Formation. Cell 2006, 126, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing Human Cells and Ageing Mice Accumulate DNA Lesions with Unrepairable Double-Strand Breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef]
- Calder, P.C.; Bosco, N.; Bourdet-Sicard, R.; Capuron, L.; Delzenne, N.; Dore, J.; Franceschi, C.; Lehtinen, M.J.; Recker, T.; Salvioli, S.; et al. Health Relevance of the Modification of Low Grade Inflammation in Ageing (Inflammageing) and the Role of Nutrition. Ageing Res. Rev. 2017, 40, 95–119. [Google Scholar] [CrossRef] [PubMed]
- Maeso-Diaz, R.; Ortega-Ribera, M.; Fernandez-Iglesias, A.; Hide, D.; Munoz, L.; Hessheimer, A.J.; Vila, S.; Frances, R.; Fondevila, C.; Albillos, A.; et al. Effects of Aging on Liver Microcirculatory Function and Sinusoidal Phenotype. Aging Cell 2018, 17, e12829. [Google Scholar] [CrossRef] [PubMed]
- Nechemia-Arbely, Y.; Shriki, A.; Denz, U.; Drucker, C.; Scheller, J.; Raub, J.; Pappo, O.; Rose-John, S.; Galun, E.; Axelrod, J.H. Early Hepatocyte DNA Synthetic Response Posthepatectomy Is Modulated by Il-6 Trans-Signaling and Pi3k/Akt Activation. J. Hepatol. 2011, 54, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.; Idelchik, M.; Melendez, J.A. Redox Control of Senescence and Age-Related Disease. Redox Biol. 2017, 11, 91–102. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein Carbonylation in Human Diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Shearn, C.T.; Pulliam, C.F.; Pedersen, K.; Meredith, K.; Mercer, K.E.; Saba, L.M.; Orlicky, D.J.; Ronis, M.J.; Petersen, D.R. Knockout of the Gsta4 Gene in Male Mice Leads to an Altered Pattern of Hepatic Protein Carbonylation and Enhanced Inflammation Following Chronic Consumption of an Ethanol Diet. Alcohol Clin. Exp. Res. 2018, 42, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Deguine, J.; Barton, G.M. Myd88: A Central Player in Innate Immune Signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Ruzehaji, N.; Mills, S.J.; Melville, E.; Arkell, R.; Fitridge, R.; Cowin, A.J. The Influence of Flightless I on Toll-Like-Receptor-Mediated Inflammation in a Murine Model of Diabetic Wound Healing. BioMed Res. Int. 2013, 2013, 389792. [Google Scholar] [CrossRef]
- Hayashi, T.; Funato, Y.; Terabayashi, T.; Morinaka, A.; Sakamoto, R.; Ichise, H.; Fukuda, H.; Yoshida, N.; Miki, H. Nucleoredoxin Negatively Regulates Toll-Like Receptor 4 Signaling via Recruitment of Flightless-I to Myeloid Differentiation Primary Response Gene (88). J. Biol. Chem. 2010, 285, 18586–18593. [Google Scholar] [CrossRef]
- Tran, B.N.; Valek, L.; Wilken-Schmitz, A.; Fuhrmann, D.C.; Namgaladze, D.; Wittig, I.; Tegeder, I. Reduced Exploratory Behavior in Neuronal Nucleoredoxin Knockout Mice. Redox Biol. 2021, 45, 102054. [Google Scholar] [CrossRef]
- Yalcin, A.; Telang, S.; Clem, B.; Chesney, J. Regulation of Glucose Metabolism by 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatases in Cancer. Exp. Mol. Pathol. 2009, 86, 174–179. [Google Scholar] [CrossRef]
- Funato, Y.; Hayashi, T.; Irino, Y.; Takenawa, T.; Miki, H. Nucleoredoxin Regulates Glucose Metabolism via Phosphofructokinase 1. Biochem. Biophys. Res. Commun. 2013, 440, 737–742. [Google Scholar] [CrossRef]
- Bellentani, S.; Scaglioni, F.; Ciccia, S.; Bedogni, G.; Tiribelli, C. Hcv, Hbv and Alcohol—The Dionysos Study. Dig. Dis. 2010, 28, 799–801. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idelfonso-García, O.G.; Alarcón-Sánchez, B.R.; Guerrero-Escalera, D.; López-Hernández, N.A.; Pérez-Hernández, J.L.; Pacheco-Rivera, R.; Serrano-Luna, J.; Resendis-Antonio, O.; Muciño-Olmos, E.A.; Aparicio-Bautista, D.I.; et al. Nucleoredoxin Redox Interactions Are Sensitized by Aging and Potentiated by Chronic Alcohol Consumption in the Mouse Liver. Antioxidants 2024, 13, 257. https://doi.org/10.3390/antiox13030257
Idelfonso-García OG, Alarcón-Sánchez BR, Guerrero-Escalera D, López-Hernández NA, Pérez-Hernández JL, Pacheco-Rivera R, Serrano-Luna J, Resendis-Antonio O, Muciño-Olmos EA, Aparicio-Bautista DI, et al. Nucleoredoxin Redox Interactions Are Sensitized by Aging and Potentiated by Chronic Alcohol Consumption in the Mouse Liver. Antioxidants. 2024; 13(3):257. https://doi.org/10.3390/antiox13030257
Chicago/Turabian StyleIdelfonso-García, Osiris Germán, Brisa Rodope Alarcón-Sánchez, Dafne Guerrero-Escalera, Norma Arely López-Hernández, José Luis Pérez-Hernández, Ruth Pacheco-Rivera, Jesús Serrano-Luna, Osbaldo Resendis-Antonio, Erick Andrés Muciño-Olmos, Diana Ivette Aparicio-Bautista, and et al. 2024. "Nucleoredoxin Redox Interactions Are Sensitized by Aging and Potentiated by Chronic Alcohol Consumption in the Mouse Liver" Antioxidants 13, no. 3: 257. https://doi.org/10.3390/antiox13030257