
Piceid Octanoate Protects Retinal Cells against Oxidative Damage by Regulating the Sirtuin 1/Poly-ADP-Ribose Polymerase 1 Axis In Vitro and in rd10 Mice

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of 3-O-(6′-O-octanoyl)-β-D-glucopyranoside Resveratrol (Piceid Octanoate, PIC-OCT)
2.2. Pharmacokinetic Study In Silico
2.3. Cell Culture
2.4. Cell Viability
2.5. Measurement SIRT1 Deacetylase Activity
2.6. Measurement of NAD+/NADH Levels
2.7. Immunofluorescence Experiments and Mitochondrial Staining
2.8. Intracellular ROS Level Measurement
2.9. Western Blot (WB)
2.10. Mitochondrial Potential Membrane Assessment
2.11. RNA Isolation, Reverse Transcription, PCR, and Real-Time PCR
2.12. In Vivo Experiments
2.13. Histology of Mouse Retinas
2.14. Statistical Analysis
3. Results
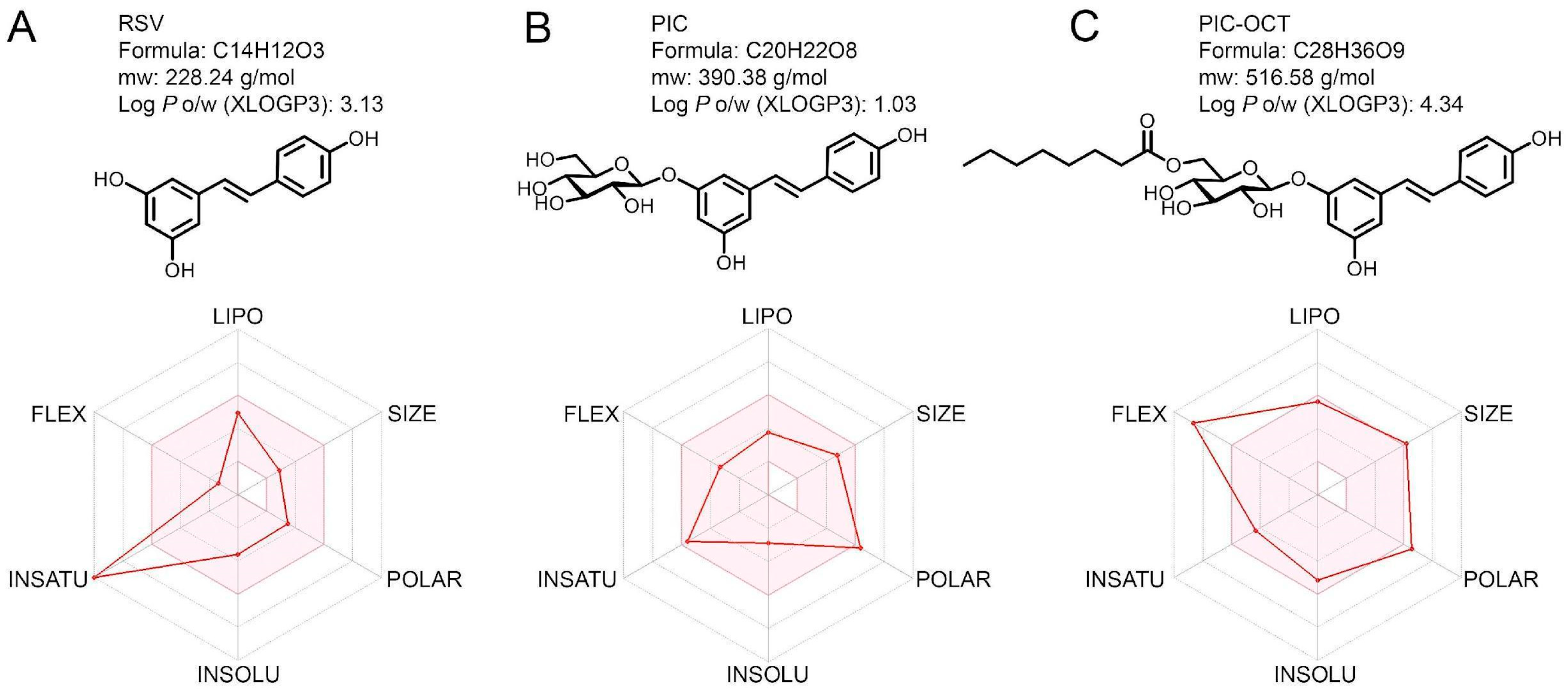
3.1. PIC-OCT Showed High Lipophilicity in an In Silico Pharmacokinetic Profile and Drug-Likeness Evaluation
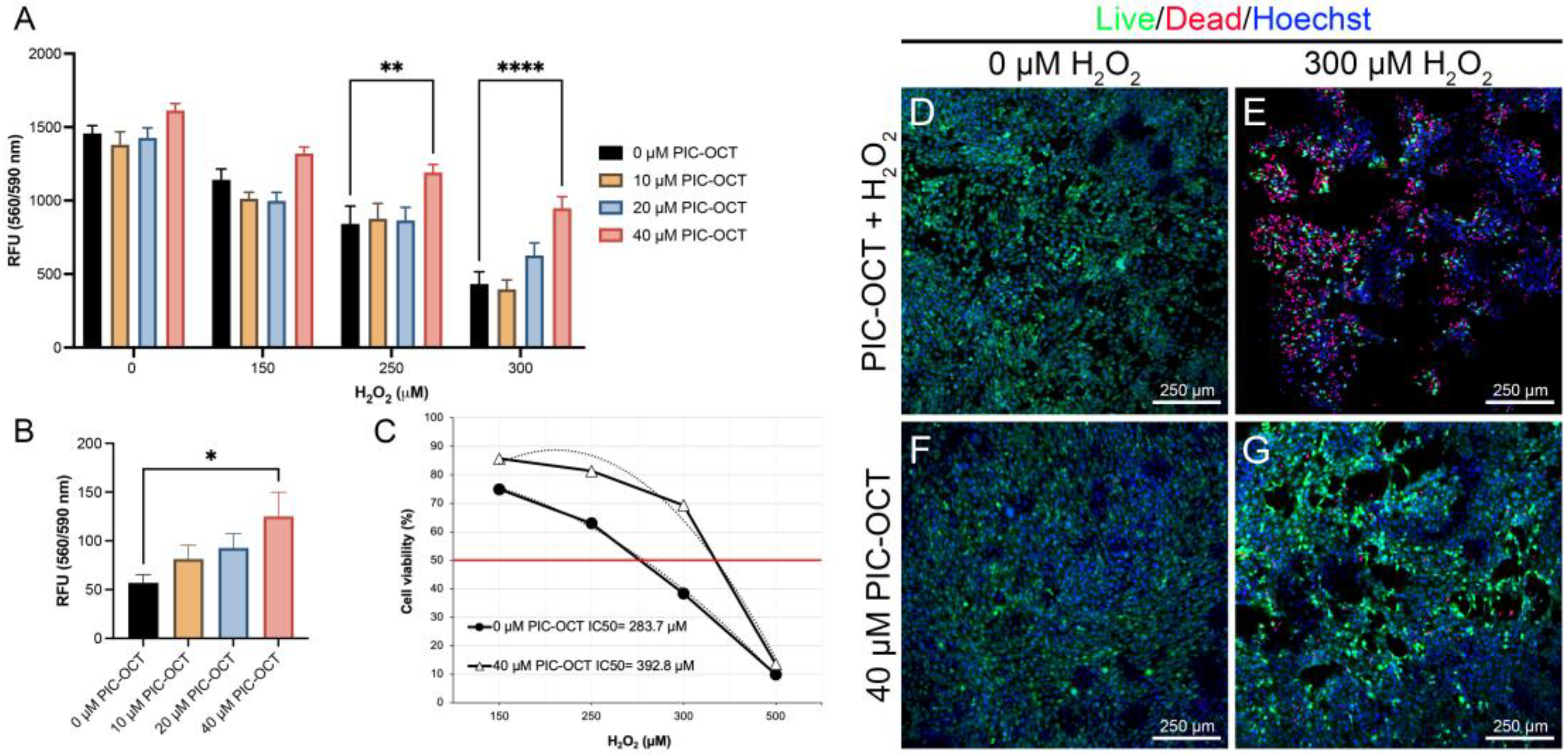
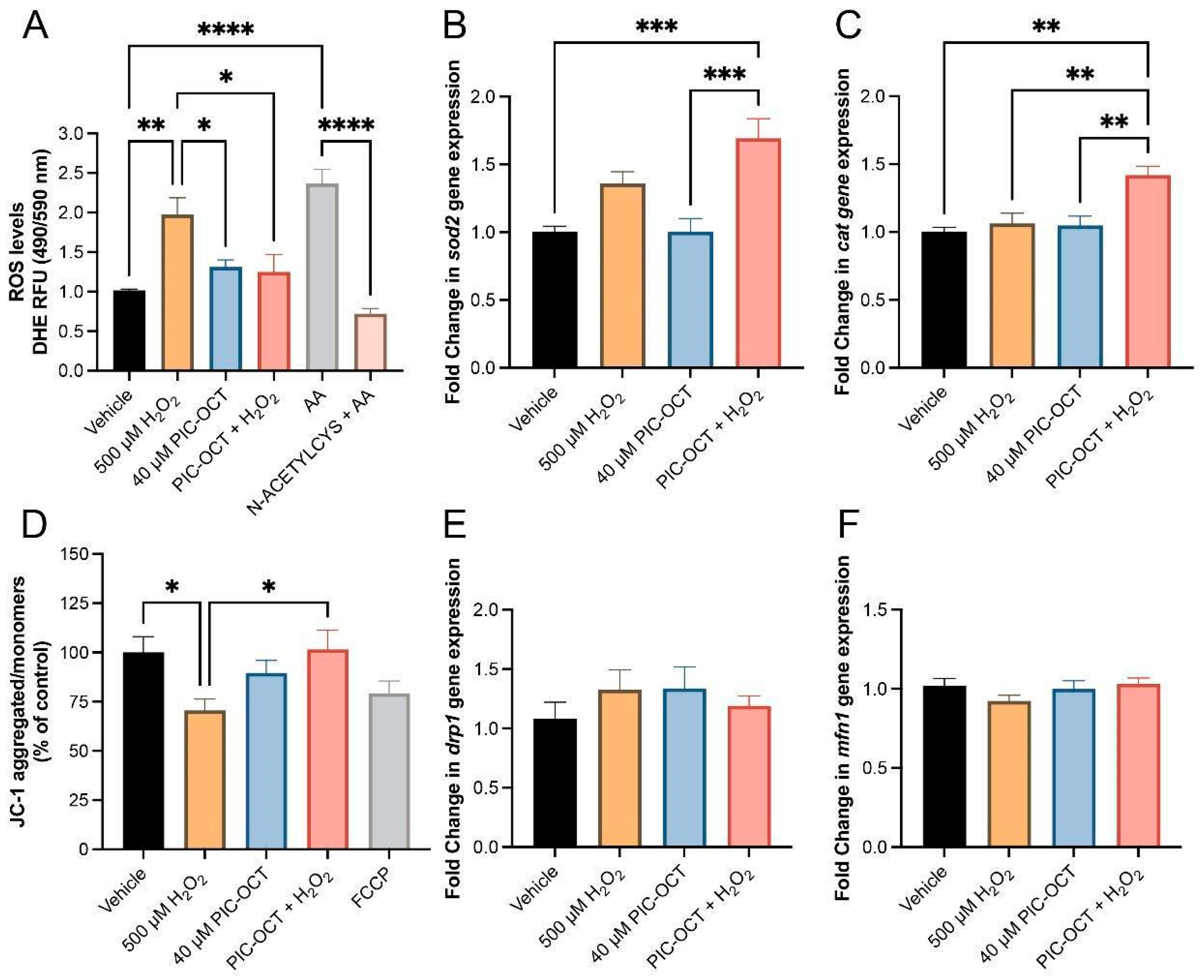
3.2. PIC-OCT Treatment Protects 661W Cells against Oxidative Stress Caused by H2O2 Exposure
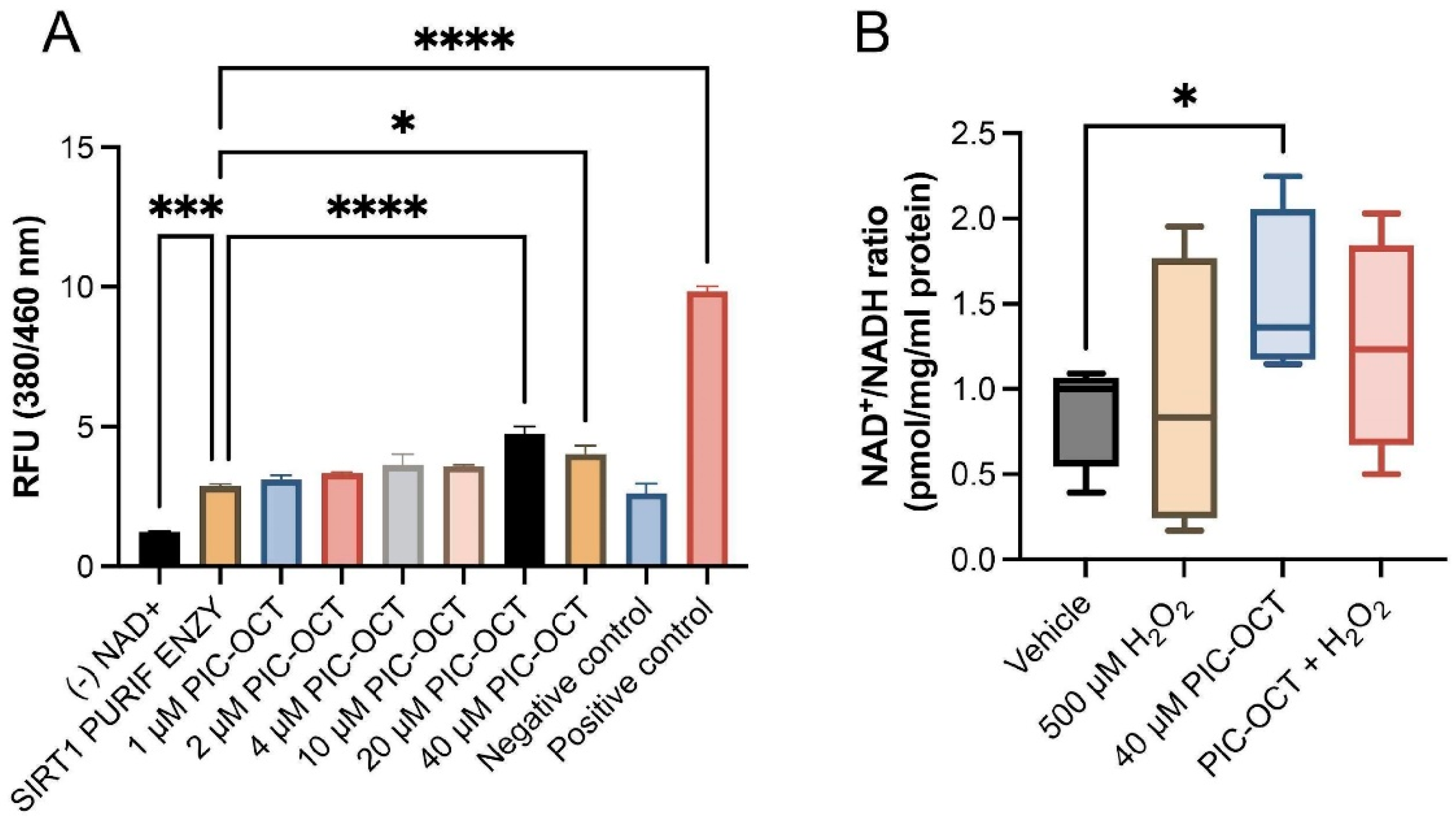
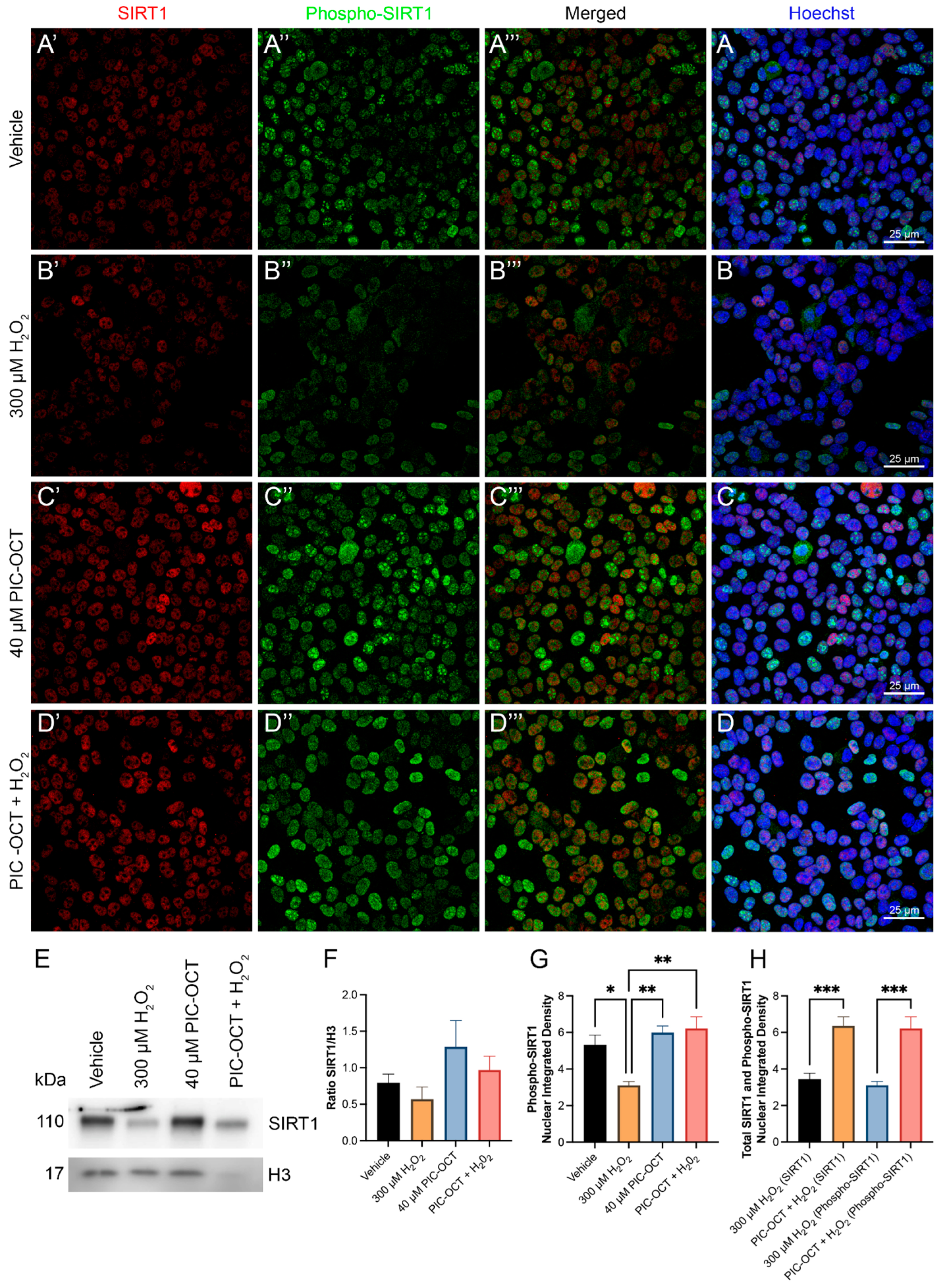
3.3. PIC-OCT Molecule Activates SIRT1, Preserving NAD+/NADH Ratio and Upregulates the Nuclear Expressions of SIRT1 and Phospho-SIRT1 (Ser47)
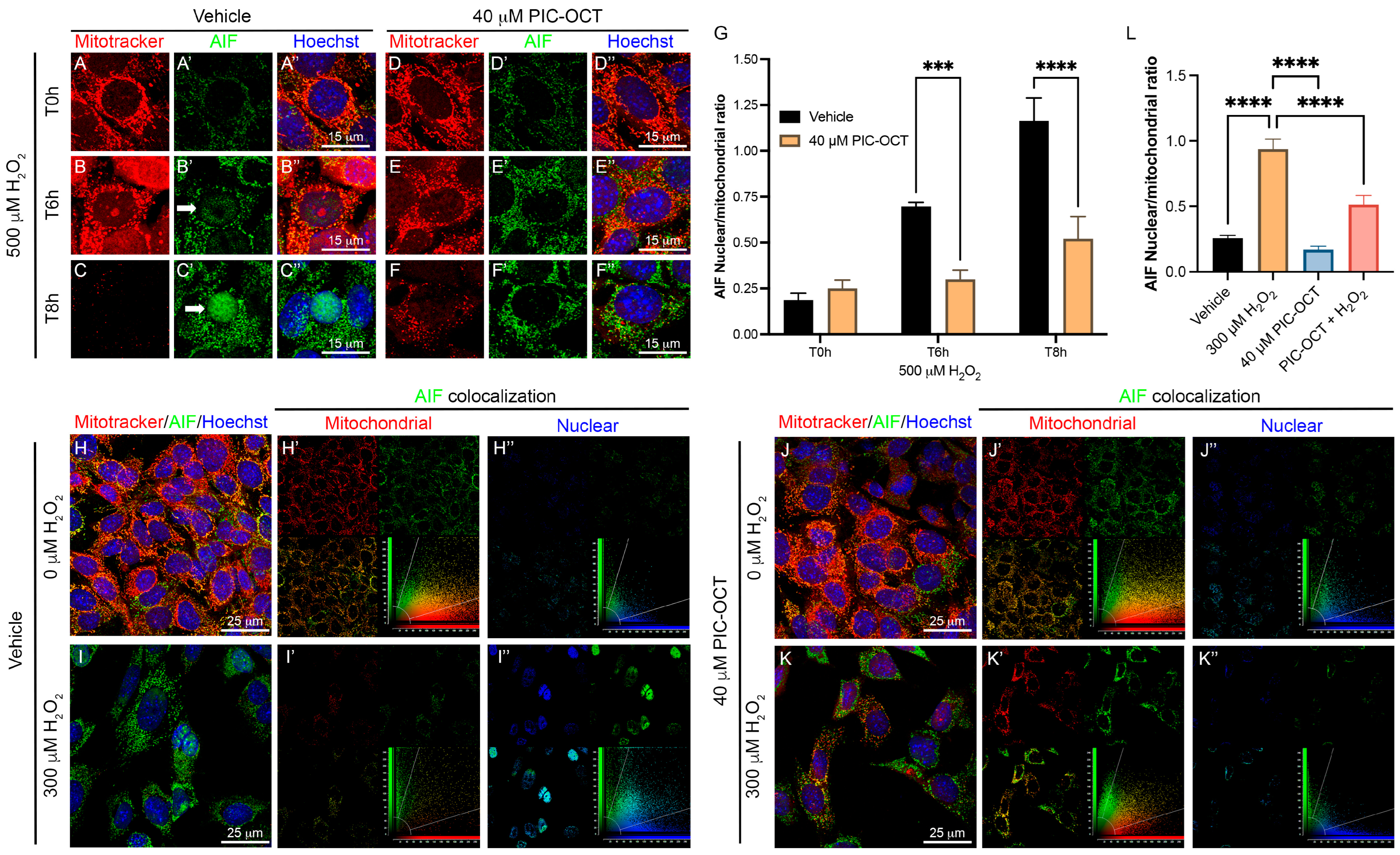
3.4. PIC-OCT Blocked Intracellular ROS Production by Increasing Anti-Oxidant Gene Expression and Preserving the Mitochondrial Membrane Potential
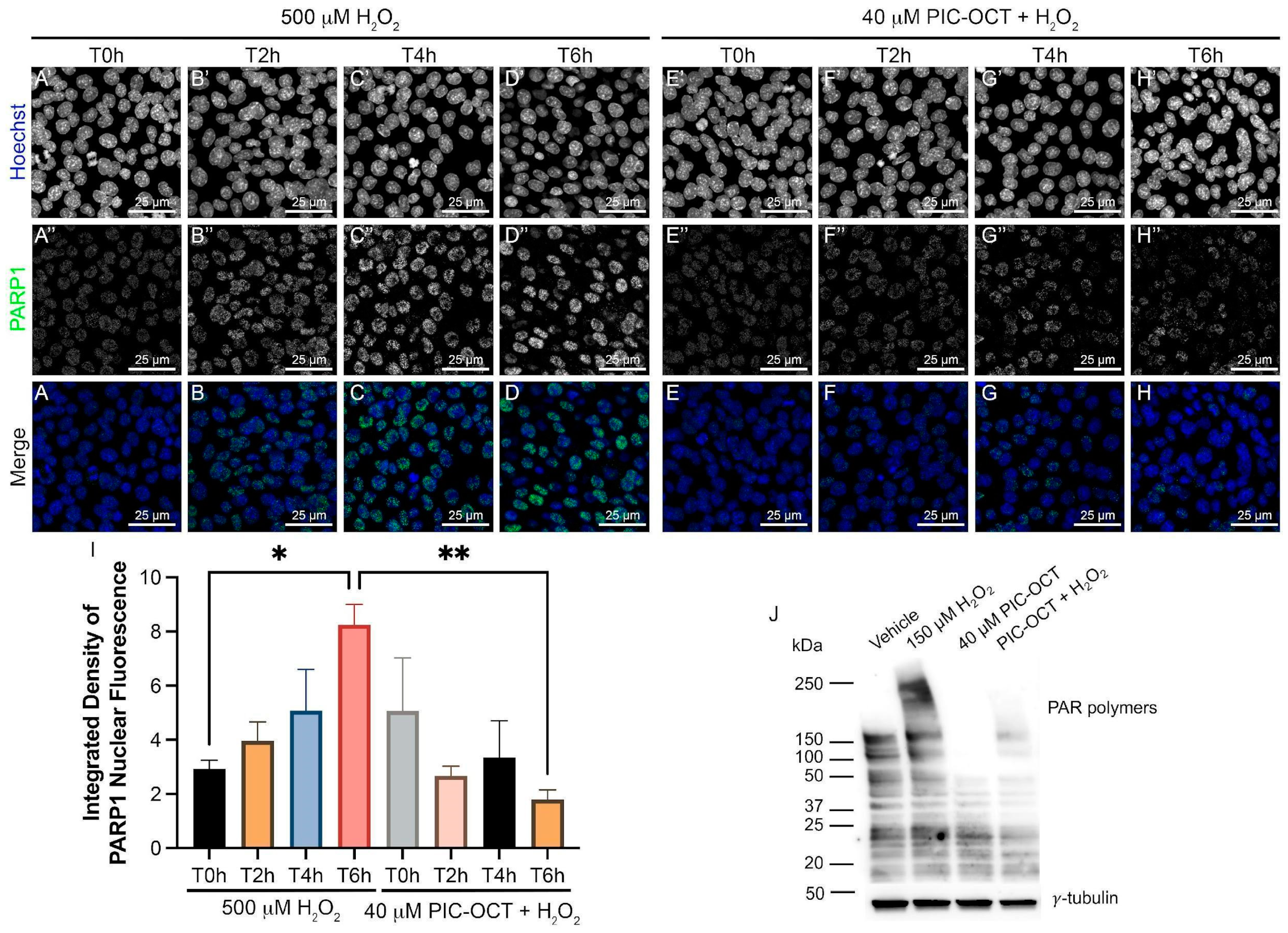
3.5. PIC-OCT Treatment Decreases the Expression of Parthanatos Hallmarks in 661W Cells Exposed to H2O2
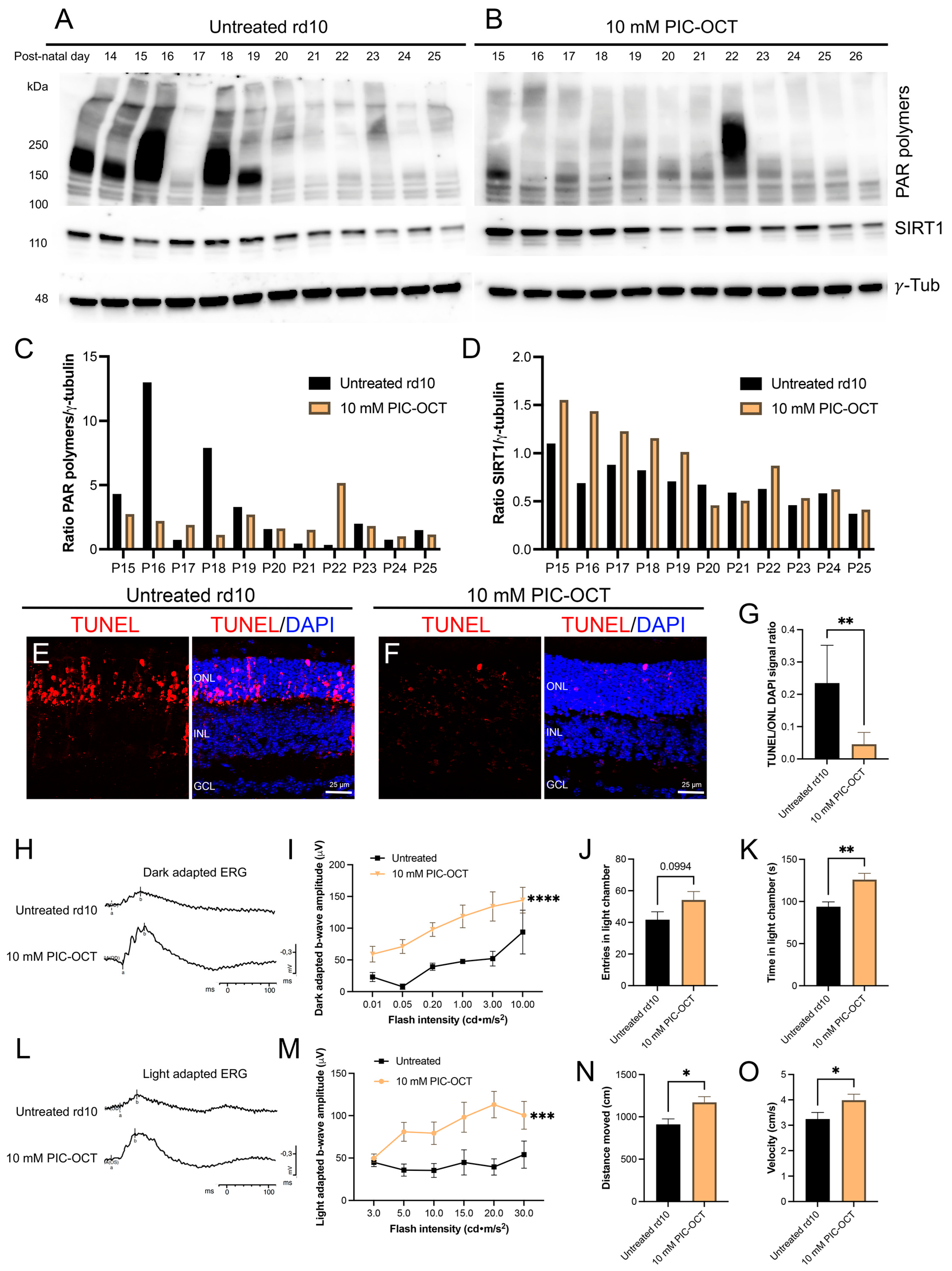
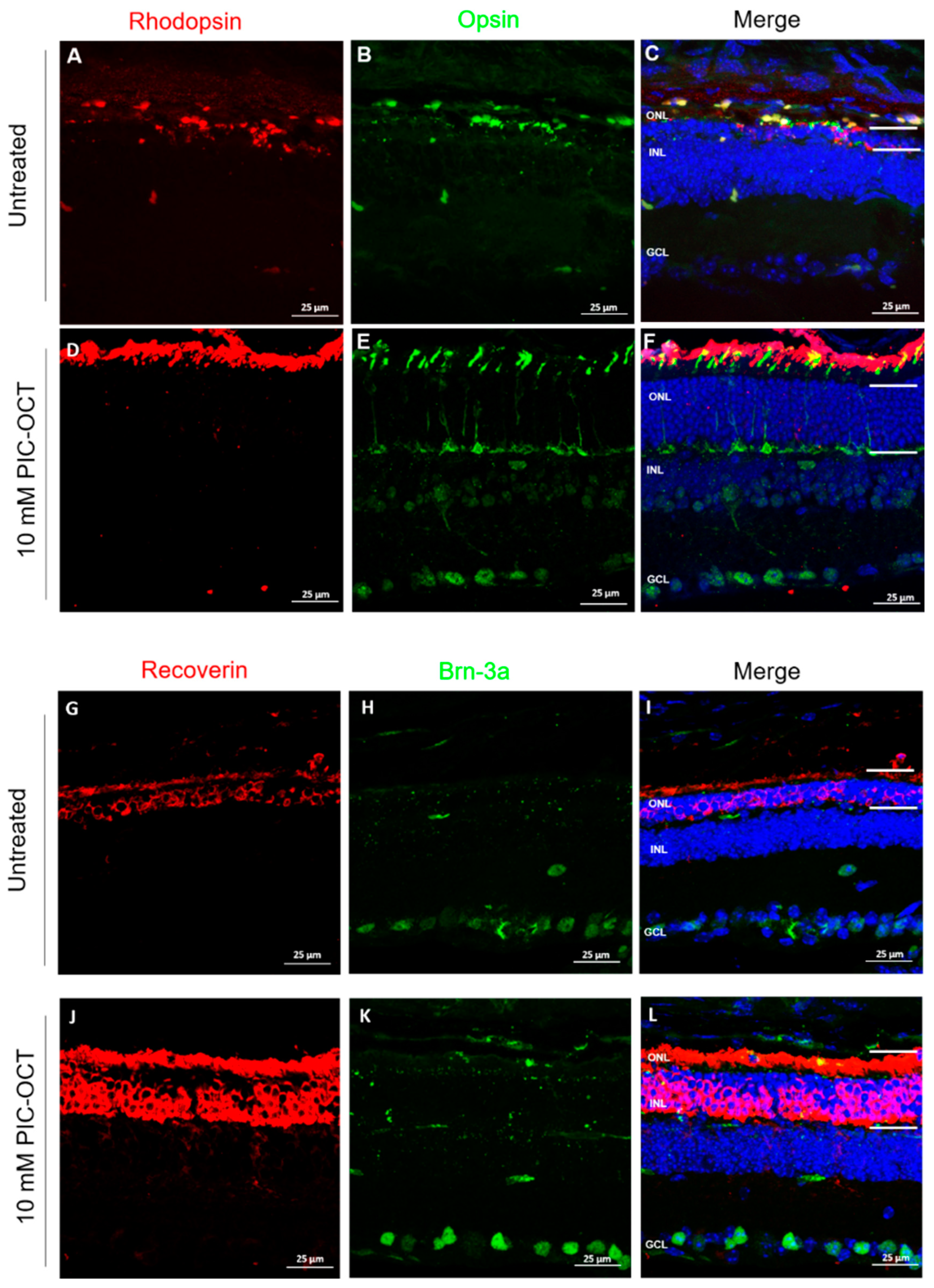
3.6. PIC-OCT Treatment Inhibited the Formation of PAR-Polymers and Preserved Photoreceptors thus Improving Visual Behavior in rd10 Mice
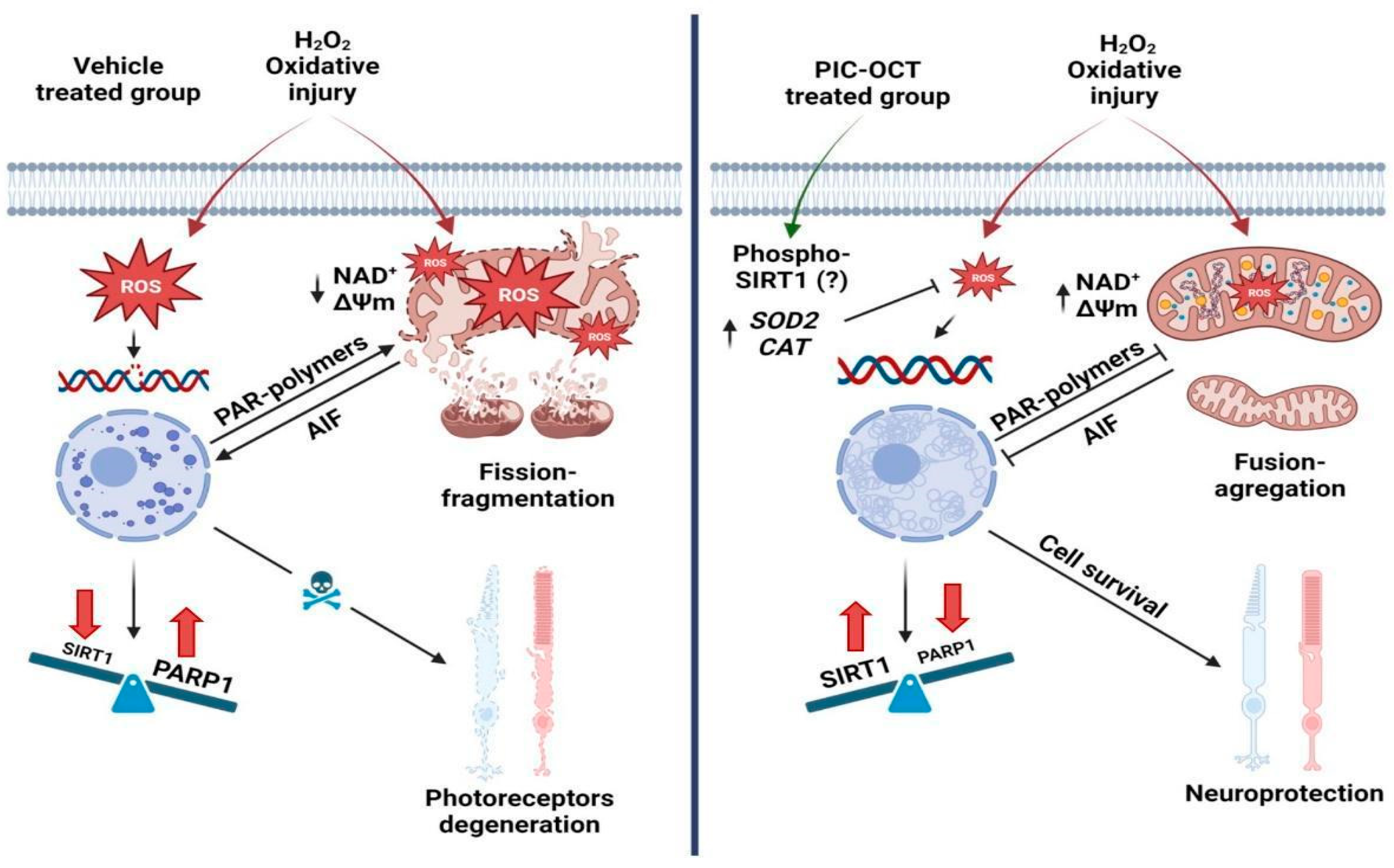
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. [Google Scholar] [CrossRef]
- Liu, M.M.; Zack, D.J. Alternative Splicing and Retinal Degeneration. Clin. Genet. 2013, 84, 142–149. [Google Scholar] [CrossRef]
- Růžičková, Š.; Staněk, D.; Arka, S.; Zi, R.; Stan, D. Mutations in Spliceosomal Proteins and Retina Degeneration. RNA Biol. 2017, 14, 544–552. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-Syndromic Retinitis Pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Yan, J.; Chen, Y.; Zhu, Y.; Paquet-Durand, F. Programmed Non-Apoptotic Cell Death in Hereditary Retinal Degeneration: Crosstalk between CGMP-Dependent Pathways and PARthanatos? Int. J. Mol. Sci. 2021, 22, 10567. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed Necrotic Cell Death of Macrophages: Focus on Pyroptosis, Necroptosis, and Parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-Linked Mechanisms and Therapeutic Opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed]
- Sahaboglu, A.; Tanimoto, N.; Kaur, J.; Sancho-Pelluz, J.; Huber, G.; Fahl, E.; Arango-Gonzalez, B.; Zrenner, E.; Ekström, P.; Löwenheim, H.; et al. PARP1 Gene Knock-out Increases Resistance to Retinal Degeneration without Affecting Retinal Function. PLoS ONE 2010, 5, e15495. [Google Scholar] [CrossRef] [PubMed]
- Arango-Gonzalez, B.; Trifunović, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen-Bienhold, U.; et al. Identification of a Common Non-Apoptotic Cell Death Mechanism in Hereditary Retinal Degeneration. PLoS ONE 2014, 9, e112142. [Google Scholar] [CrossRef] [PubMed]
- Viringipurampeer, I.A.; Gregory-Evans, C.Y.; Metcalfe, A.L.; Bashar, E.; Moritz, O.L.; Gregory-Evans, K. Cell Death Pathways in Mutant Rhodopsin Rat Models Identifies Genotype-Specific Targets Controlling Retinal Degeneration. Mol. Neurobiol. 2019, 56, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ Consumption by PARP1 in Response to DNA Damage Triggers Metabolic Shift Critical for Damaged Cell Survival. Mol. Biol. Cell 2019, 30, 2584. [Google Scholar] [CrossRef] [PubMed]
- Strømland, Ø.; Diab, J.; Ferrario, E.; Sverkeli, L.J.; Ziegler, M. The Balance between NAD+ Biosynthesis and Consumption in Ageing. Mech. Ageing Dev. 2021, 199, 111569. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, A.R.; Larrick, J.W. The NAD+/PARP1/SIRT1 Axis in Aging. Rejuvenation Res. 2017, 20, 244–247. [Google Scholar] [CrossRef]
- Sahaboglu, A.; Barth, M.; Secer, E.; Del Amo, E.M.; Urtti, A.; Arsenijevic, Y.; Zrenner, E.; Paquet-Durand, F. Olaparib Significantly Delays Photoreceptor Loss in a Model for Hereditary Retinal Degeneration. Sci. Rep. 2016, 6, 39537. [Google Scholar] [CrossRef]
- Sahaboglu, A.; Miranda, M.; Canjuga, D.; Avci-Adali, M.; Savytska, N.; Secer, E.; Feria-Pliego, J.A.; Kayık, G.; Durdagi, S. Drug Repurposing Studies of PARP Inhibitors as a New Therapy for Inherited Retinal Degeneration. Cell. Mol. Life Sci. 2020, 77, 2199–2216. [Google Scholar] [CrossRef]
- Sahaboglu, A.; Sharif, A.; Feng, L.; Secer, E.; Zrenner, E.; Paquet-Durand, F. Temporal Progression of PARP Activity in the Prph2 Mutant Rd2 Mouse: Neuroprotective Effects of the PARP Inhibitor PJ34. PLoS ONE 2017, 12, e0181374. [Google Scholar] [CrossRef]
- Bixel, K.; Hays, J.L. Olaparib in the Management of Ovarian Cancer. Pharmgenomics Pers. Med. 2015, 8, 127–135. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, S.; Wu, T.; Zhang, H. Comparison of Adverse Reactions Caused by Olaparib for Different Indications. Front. Pharmacol. 2022, 13, 968163. [Google Scholar] [CrossRef] [PubMed]
- Gallyas, F.; Sumegi, B. Mitochondrial Protection by PARP Inhibition. Int. J. Mol. Sci. 2020, 21, 2767. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Song, J.Y.; Fan, B.; Wang, Y.; Pan, Y.R.; Che, L.; Sun, Y.J.; Li, G.Y. Resveratrol Protects Photoreceptors by Blocking Caspase- and PARP-Dependent Cell Death Pathways. Free Radic. Biol. Med. 2018, 129, 569–581. [Google Scholar] [CrossRef]
- Valdés-Sánchez, L.; García-Delgado, A.B.; Montero-Sánchez, A.; de la Cerda, B.; Lucas, R.; Peñalver, P.; Morales, J.C.; Bhattacharya, S.S.; Díaz-Corrales, F.J. The Resveratrol Prodrug JC19 Delays Retinal Degeneration in Rd10 Mice. Adv. Exp. Med. Biol. 2019, 1185, 457–462. [Google Scholar] [CrossRef]
- Arbo, B.D.; André-Miral, C.; Nasre-Nasser, R.G.; Schimith, L.E.; Santos, M.G.; Costa-Silva, D.; Muccillo-Baisch, A.L.; Hort, M.A. Resveratrol Derivatives as Potential Treatments for Alzheimer’s and Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 103. [Google Scholar] [CrossRef]
- Peñalver, P.; Belmonte-Reche, E.; Adán, N.; Caro, M.; Mateos-Martín, M.L.; Delgado, M.; González-Rey, E.; Morales, J.C. Alkylated Resveratrol Prodrugs and Metabolites as Potential Therapeutics for Neurodegenerative Diseases. Eur. J. Med. Chem. 2018, 146, 123–138. [Google Scholar] [CrossRef]
- Bryl, A.; Falkowski, M.; Zorena, K.; Mrugacz, M. The Role of Resveratrol in Eye Diseases-A Review of the Literature. Nutrients 2022, 14, 2974. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Luo, L.J.; Yang, C.J.; Lai, J.Y. Highly Retina-Permeating and Long-Acting Resveratrol/Metformin Nanotherapeutics for Enhanced Treatment of Macular Degeneration. ACS Nano 2023, 17, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, J.; Rashid, K.; Langmann, T. Resveratrol Induces Dynamic Changes to the Microglia Transcriptome, Inhibiting Inflammatory Pathways and Protecting against Microglia-Mediated Photoreceptor Apoptosis. Biochem. Biophys. Res. Commun. 2018, 501, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Fonseca-Kelly, Z.; Callinan, C.; Zuo, L.; Sachdeva, M.M.; Shindler, K.S. SIRT1 Activating Compounds Reduce Oxidative Stress and Prevent Cell Death in Neuronal Cells. Front. Cell. Neurosci. 2012, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Winnik, S.; Auwerx, J.; Sinclair, D.A.; Matter, C.M. Protective Effects of Sirtuins in Cardiovascular Diseases: From Bench to Bedside. Eur. Heart J. 2015, 36, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Giblin, W.; Lombard, D.B. Sirtuins, Healthspan, and Longevity in Mammals. In Handbook of the Biology of Aging, 8th ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 83–132. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Yoshioka, K.; Tatematsu, S.; Hara, Y.; Minakuchi, H.; Washida, N.; Tokuyama, H.; Hayashi, K.; Itoh, H. Sirt1 Protects against Oxidative Stress-Induced Renal Tubular Cell Apoptosis by the Bidirectional Regulation of Catalase Expression. Biochem. Biophys. Res. Commun. 2008, 372, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.B.; Apte, R.S. NAD+ and Sirtuins in Retinal Degenerative Diseases: A Look at Future Therapies. Prog. Retin. Eye Res. 2018, 67, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Derout, J.; Shao, X.M.; Bankole, E.; Hasan, K.M.; Mtume, N.; Liu, Y.; Sinha-Hikim, A.P.; Friedman, T.C. Hepatic DNA Damage Induced by Electronic Cigarette Exposure Is Associated With the Modulation of NAD+/PARP1/SIRT1 Axis. Front. Endocrinol. 2019, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.T.; Joe, Y. Antagonistic Crosstalk between SIRT1, PARP-1, and-2 in the Regulation of Chronic Inflammation Associated with Aging and Metabolic Diseases. Integr. Med. Res. 2014, 3, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Luna, A.; Aladjem, M.I.; Kohn, K.W. SIRT1/PARP1 Crosstalk: Connecting DNA Damage and Metabolism. Genome Integr. 2013, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of Human SIRT1 Activation by Resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Guarente, L. Sirtuins at a Glance. J. Cell Sci. 2011, 124, 833–838. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Pharmacological Approaches for Modulating Sirtuins. In Introductory Review on Sirtuins in Biology, Aging, and Disease; Academic Press: Cambridge, MA, USA, 2018; pp. 71–81. [Google Scholar] [CrossRef]
- Donmez, G.; Outeiro, T.F. SIRT1 and SIRT2: Emerging Targets in Neurodegeneration. EMBO Mol. Med. 2013, 5, 344. [Google Scholar] [CrossRef]
- Singh, P.; Hanson, P.S.; Morris, C.M. SIRT1 Ameliorates Oxidative Stress Induced Neural Cell Death and Is Down-Regulated in Parkinson’s Disease. BMC Neurosci. 2017, 18, 46. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. SIRT1 in Neurodevelopment and Brain Senescence. Neuron 2014, 81, 471–483. [Google Scholar] [CrossRef]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-Pinzon, M.A. Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front. Endocrinol. 2018, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E.; Walle, U.K. High Absorption but Very Low Bioavailability of Oral Resveratrol in Humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Pentek, T.; Newenhouse, E.; O’Brien, B.; Singh Chauhan, A.; Chauhan, A.S.; Singh Chauhan, A. Development of a Topical Resveratrol Formulation for Commercial Applications Using Dendrimer Nanotechnology. Molecules 2017, 22, 137. [Google Scholar] [CrossRef]
- Nunes, T.; Almeida, L.; Rocha, J.F.; Falcão, A.; Fernandes-Lopes, C.; Loureiro, A.I.; Wright, L.; Vaz-Da-Silva, M.; Soares-Da-Silva, P. Pharmacokinetics of Trans-Resveratrol Following Repeated Administration in Healthy Elderly and Young Subjects. J. Clin. Pharmacol. 2009, 49, 1477–1482. [Google Scholar] [CrossRef]
- Larrosa, M.; Tomé-Carneiro, J.; Yáñez-Gascón, M.J.; Alcántara, D.; Selma, M.V.; Beltrán, D.; García-Conesa, M.T.; Urbán, C.; Lucas, R.; Tomás-Barberán, F.; et al. Preventive Oral Treatment with Resveratrol Pro-Prodrugs Drastically Reduce Colon Inflammation in Rodents. J. Med. Chem. 2010, 53, 7365–7376. [Google Scholar] [CrossRef]
- Diaz-Corrales, F.J.; Morales-Sánchez, J.C.; Valdés-Sánchez, L.; Lucas-Rodríguez, R.; Rodriguez-Martinez, D.; Peñalver, P.; Diez-Lloret, A.; Bhatia, V.; la Cerda, B.D.; Rodriguez-Bocanegra, E.; et al. Sirt1 Activators Induced Neuroprotection of Photoreceptors in Rd10 Mice. Proceedings of the Annual Meeting of the Association for Research in Vision and Ophthalmology (ARVO), Seattle, Washington, USA, 1–5 May 2016. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4388. [Google Scholar]
- Tan, T.E.; Fenner, B.J.; Barathi, V.A.; Tun, S.B.B.; Wey, Y.S.; Tsai, A.S.H.; Su, X.; Lee, S.Y.; Cheung, C.M.G.; Wong, T.Y.; et al. Gene-Based Therapeutics for Acquired Retinal Disease: Opportunities and Progress. Front. Genet. 2021, 12, 795010. [Google Scholar] [CrossRef] [PubMed]
- Astuti, G.D.N.; Bertelsen, M.; Preising, M.N.; Ajmal, M.; Lorenz, B.; Faradz, S.M.H.; Qamar, R.; Collin, R.W.J.; Rosenberg, T.; Cremers, F.P.M. Comprehensive Genotyping Reveals RPE65 as the Most Frequently Mutated Gene in Leber Congenital Amaurosis in Denmark. Eur. J. Hum. Genet. 2016, 24, 1071. [Google Scholar] [CrossRef] [PubMed]
- Drag, S.; Dotiwala, F.; Upadhyay, A.K. Gene Therapy for Retinal Degenerative Diseases: Progress, Challenges, and Future Directions. Investig. Ophthalmol. Vis. Sci. 2023, 64, 39. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Russell, S.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Drack, A.V.; Simonelli, F.; Leroy, B.P.; Reape, K.Z.; High, K.A.; et al. Durability of Voretigene Neparvovec for Biallelic RPE65-Mediated Inherited Retinal Disease: Phase 3 Results at 3 and 4 Years. Ophthalmology 2021, 128, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Timmers, A.M.; Newmark, J.A.; Turunen, H.T.; Farivar, T.; Liu, J.; Song, C.; Ye, G.J.; Pennock, S.; Gaskin, C.; Knop, D.R.; et al. Ocular Inflammatory Response to Intravitreal Injection of Adeno-Associated Virus Vector: Relative Contribution of Genome and Capsid. Hum. Gene Ther. 2020, 31, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Edelshtain, V.; Peled, A.; Tzameret, A.; Chen, S.P.; Ziv, H.; Derazne, E.; Harats, D.; Greenberg, R.; Harari, A.; Shaish, A.; et al. Long-Term Treatment with 9-Cis-β-Carotene Rich Alga Dunaliella Bardawil Ameliorates Photoreceptor Degeneration in a Mouse Model of Retinoid Cycle Defect. Algal Res. 2019, 43, 101607. [Google Scholar] [CrossRef]
- Swinkels, D.; Baes, M. The Essential Role of Docosahexaenoic Acid and Its Derivatives for Retinal Integrity. Pharmacol. Ther. 2023, 247, 108440. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.H.-L.; Lam, H.I.; Choi, K.Y.; Li, S.Z.C.; Lakshmanan, Y.; Yu, W.Y.; Chang, R.C.C.; Lai, J.S.M.; So, K. fai Delay of Cone Degeneration in Retinitis Pigmentosa Using a 12-Month Treatment with Lycium Barbarum Supplement. J. Ethnopharmacol. 2019, 236, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Sayyad, Z.; Sirohi, K.; Radha, V.; Swarup, G. 661W Is a Retinal Ganglion Precursor-like Cell Line in Which Glaucoma-Associated Optineurin Mutants Induce Cell Death Selectively. Sci. Rep. 2017, 7, 16855. [Google Scholar] [CrossRef]
- Ogita, M.; Ogita, A.; Usuki, Y.; Fujita, K.I.; Tanaka, T. Antimycin A-Induced Cell Death Depends on AIF Translocation through NO Production and PARP Activation and Is Not Involved in ROS Generation, Cytochrome c Release and Caspase-3 Activation in HL-60 Cells. J. Antibiot. 2009, 62, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS Inhibitor N-Acetyl-l-Cysteine Antagonizes the Activity of Proteasome Inhibitors. Biochem. J. 2013, 454, 201. [Google Scholar] [CrossRef] [PubMed]
- Pensado, A.; Diaz-Corrales, F.J.; De la Cerda, B.; Valdés-Sánchez, L.; del Boz, A.A.; Rodriguez-Martinez, D.; García-Delgado, A.B.; Seijo, B.; Bhattacharya, S.S.; Sanchez, A. Span Poly-L-Arginine Nanoparticles Are Efficient Non-Viral Vectors for PRPF31 Gene Delivery: An Approach of Gene Therapy to Treat Retinitis Pigmentosa. Nanomedicine 2016, 12, 2251–2260. [Google Scholar] [CrossRef]
- Valdés-Sánchez, L.; De La Cerda, B.; Diaz-Corrales, F.J.; Massalini, S.; Chakarova, C.F.; Wright, A.F.; Bhattacharya, S.S. ATR Localizes to the Photoreceptor Connecting Cilium and Deficiency Leads to Severe Photoreceptor Degeneration in Mice. Hum. Mol. Genet. 2013, 22, 1507–1515. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, Z.; Gao, L.; Sun, D.; Hu, X.; Xue, L.; Dai, J.; Zeng, Y.X.; Chen, S.; Pan, B.; et al. Curcumin Delays Retinal Degeneration by Regulating Microglia Activation in the Retina of Rd1 Mice. Cell. Physiol. Biochem. 2017, 44, 479–493. [Google Scholar] [CrossRef]
- Chini, C.C.S.S.; Tarrag, M.G.; Chini, E.N.; Tarragó, M.G.; Chini, E.N. NAD and the Aging Process: Role in Life, Death and Everything in Between. Mol. Cell. Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef]
- Nasrin, N.; Kaushik, V.K.; Fortier, E.; Wall, D.; Pearson, K.J.; de Cabo, R.; Bordone, L. JNK1 Phosphorylates SIRT1 and Promotes Its 1286 Enzymatic Activity. PLoS ONE 2009, 4, 8414. [Google Scholar] [CrossRef]
- Xu, J.J.; Cui, J.; Lin, Q.; Chen, X.Y.; Zhang, J.; Gao, E.H.; Wei, B.; Zhao, W. Protection of the Enhanced Nrf2 Deacetylation and Its Downstream Transcriptional Activity by SIRT1 in Myocardial Ischemia/Reperfusion Injury. Int. J. Cardiol. 2021, 342, 82–93. [Google Scholar] [CrossRef]
- Carroll, J.; Dubra, A.; Gardner, J.C.; Mizrahi-Meissonnier, L.; Cooper, R.F.; Dubis, A.M.; Nordgren, R.; Genead, M.; Connor, T.B.; Stepien, K.E.; et al. The Effect of Cone Opsin Mutations on Retinal Structure and the Integrity of the Photoreceptor Mosaic. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8006. [Google Scholar] [CrossRef] [PubMed]
- Barone, I.; Novelli, E.; Strettoi, E. Long-Term Preservation of Cone Photoreceptors and Visual Acuity in Rd10 Mutant Mice Exposed to Continuous Environmental Enrichment. Mol. Vis. 2014, 20, 1545. [Google Scholar]
- Cha, S.; Ahn, J.; Jeong, Y.; Lee, Y.H.; Kim, H.K.; Lee, D.; Yoo, Y.; Goo, Y.S. Stage-Dependent Changes of Visual Function and Electrical Response of the Retina in the Rd10 Mouse Model. Front. Cell Neurosci. 2022, 16, 926096. [Google Scholar] [CrossRef]
- Gambini, J.; Inglés, M.; Olaso, G.; Lopez-Grueso, R.; Bonet-Costa, V.; Gimeno-Mallench, L.; Mas-Bargues, C.; Abdelaziz, K.M.; Gomez-Cabrera, M.C.; Vina, J.; et al. Properties of Resveratrol: In Vitro and In Vivo Studies about Metabolism, Bioavailability, and Biological Effects in Animal Models and Humans. Oxid. Med. Cell. Longev. 2015, 2015, 837042. [Google Scholar] [CrossRef] [PubMed]
- Regev-Shoshani, G.; Shoseyov, O.; Bilkis, I.; Kerem, Z. Glycosylation of Resveratrol Protects It from Enzymic Oxidation. Biochem. J. 2003, 374, 157. [Google Scholar] [CrossRef]
- Su, D.; Cheng, Y.; Liu, M.; Liu, D.; Cui, H.; Zhang, B.; Zhou, S.; Yang, T.; Mei, Q. Comparision of Piceid and Resveratrol in Antioxidation and Antiproliferation Activities In Vitro. PLoS ONE 2013, 8, e54505. [Google Scholar] [CrossRef]
- Burkon, A.; Somoza, V. Quantification of Free and Protein-Bound Trans-Resveratrol Metabolites and Identification of Trans-Resveratrol-C/O-Conjugated Diglucuronides-Two Novel Resveratrol Metabolites in Human Plasma. Mol. Nutr. Food Res. 2008, 52, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Falomir, E.; Lucas, R.; Peñalver, P.; Martí-Centelles, R.; Dupont, A.; Zafra-Gómez, A.; Carda, M.; Morales, J.C. Cytotoxic, Antiangiogenic and Antitelomerase Activity of Glucosyl- and Acyl- Resveratrol Prodrugs and Resveratrol Sulfate Metabolites. Chembiochem 2016, 17, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.H.; Hwang, Y.; Kim, E. PARP1 Impedes SIRT1-Mediated Autophagy during Degeneration of the Retinal Pigment Epithelium under Oxidative Stress. Mol. Cells 2020, 43, 632. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, N.; Zhao, H.H.; Zhang, X.; Kawano, H.; Liu, L.; Zhao, L.; Li, H.P. Interactions between Sirt1 and MAPKs Regulate Astrocyte Activation Induced by Brain Injury in Vitro and in Vivo. J. Neuroinflamm. 2017, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Simeone, P.; Toraldo, D.; Del Boccio, P.; Vergaro, V.; Leporatti, S.; Pieragostino, D.; Tinelli, A.; De Domenico, S.; Alberti, S.; et al. Resveratrol Downregulates Akt/GSK and ERK Signalling Pathways in OVCAR-3 Ovarian Cancer Cells. Mol. Biosyst. 2012, 8, 1078–1087. [Google Scholar] [CrossRef]
- Olivares-González, L.; Velasco, S.; Millán, J.M.; Rodrigo, R. Intravitreal Administration of Adalimumab Delays Retinal Degeneration in Rd10 Mice. FASEB J. 2020, 34, 13839–13861. [Google Scholar] [CrossRef]
- Bournival, J.; Patrik, A.E.; Ae, Q.; Martinoli, M.-G. Protective Effects of Resveratrol and Quercetin Against MPP +-Induced Oxidative Stress Act by Modulating Markers of Apoptotic Death in Dopaminergic Neurons. Cell. Mol. Neurobiol. 2009, 29, 1169–1180. [Google Scholar] [CrossRef]
- Kumar, S.J.; Eroglu, E.; Stokes, J.A.; Scissum-Gunn, K.; Saldanha, S.N.; Singh, U.P.; Manne, U.; Ponnazhagan, S.; Mishra, M.K. Resveratrol Induces Mitochondria-Mediated, Caspase-Independent Apoptosis in Murine Prostate Cancer Cells. Oncotarget 2017, 8, 20895–20908. [Google Scholar] [CrossRef]
- Yuan, Z.; Du, W.; He, X.; Zhang, D.; He, W. Tribulus Terrestris Ameliorates Oxidative Stress-Induced ARPE-19 Cell Injury through the PI3K/Akt-Nrf2 Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 7962393. [Google Scholar] [CrossRef]
- Xu, X.R.; Yu, H.T.; Yang, Y.; Hang, L.; Yang, X.W.; Ding, S.H. Quercetin Phospholipid Complex Significantly Protects against Oxidative Injury in ARPE-19 Cells Associated with Activation of Nrf2 Pathway. Eur. J. Pharmacol. 2016, 770, 1–8. [Google Scholar] [CrossRef]
- Alhasani, R.H.; Biswas, L.; Tohari, A.M.; Zhou, X.; Reilly, J.; He, J.F.; Shu, X. Gypenosides Protect Retinal Pigment Epithelium Cells from Oxidative Stress. Food Chem. Toxicol. 2018, 112, 76–85. [Google Scholar] [CrossRef]
- Xu, X.; Li, M.; Chen, W.; Yu, H.; Yang, Y.; Hang, L. Apigenin Attenuates Oxidative Injury in ARPE-19 Cells Thorough Activation of Nrf2 Pathway. Oxid. Med. Cell. Longev. 2016, 2016, 4378461. [Google Scholar] [CrossRef]
- Ren, Z.; He, H.; Zuo, Z.; Xu, Z.; Wei, Z.; Deng, J. The Role of Different SIRT1-Mediated Signaling Pathways in Toxic Injury. Cell. Mol. Biol. Lett. 2019, 24, 36. [Google Scholar] [CrossRef] [PubMed]
- Adijanto, J.; Du, J.; Moffat, C.; Seifert, E.L.; Hurley, J.B.; Philp, N.J. The Retinal Pigment Epithelium Utilizes Fatty Acids for Ketogenesis: Implications for Metabolic Coupling with the Outer Retina. J. Biol. Chem. 2014, 289, 20570. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Amorim, J.A.; Moustafa, G.A.; Lee, J.J.; Yu, Z.; Ishihara, K.; Iesato, Y.; Barbisan, P.; Ueta, T.; Togka, K.A.; et al. Neuroprotective Effects and Mechanisms of Action of Nicotinamide Mononucleotide (NMN) in a Photoreceptor Degenerative Model of Retinal Detachment. Aging 2020, 12, 24504. [Google Scholar] [CrossRef]
- Cao, W.; Hong, Y.; Chen, H.; Wu, F.; Wei, X.; Ying, W. SIRT2 Mediates NADH-Induced Increases in Nrf2, GCL, and Glutathione by Modulating Akt Phosphorylation in PC12 Cells. FEBS Lett. 2016, 590, 2241–2255. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Song, Y.; Kincaid, B.; Bossy, B.; Bossy-Wetzel, E. Mutant SOD1G93A Triggers Mitochondrial Fragmentation in Spinal Cord Motor Neurons: Neuroprotection by SIRT3 and PGC-1α. Neurobiol. Dis. 2013, 51, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Fisher, C.R.; Kowluru, R.A. Mitochondrial Defects Drive Degenerative Retinal Diseases Cell-Specific Mitochondrial Defects Cause Distinct Diseases. Trends Mol. Med. 2019, 26, 105–118. [Google Scholar] [CrossRef]
- Pagano, G.; Pallardó, F.V.; Lyakhovich, A.; Tiano, L.; Trifuoggi, M. Mitigating the Pro-Oxidant State and Melanogenesis of Retinitis Pigmentosa: By Counteracting Mitochondrial Dysfunction. Cell. Mol. Life Sci. 2021, 78, 7491–7503. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of Mitochondrial Dysfunction and Their Impact on Age-Related Macular Degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Zhou, B.; Fang, L.; Dong, Y.; Yang, J.; Chen, X.; Zhang, N.; Zhu, Y.; Huang, T. Mitochondrial Quality Control Protects Photoreceptors against Oxidative Stress in the H2O2-Induced Models of Retinal Degeneration Diseases. Cell Death Dis. 2021, 12, 413. [Google Scholar] [CrossRef]
- Cilleros-Holgado, P.; Gómez-Fernández, D.; Piñero-Pérez, R.; Reche-López, D.; Álvarez-Córdoba, M.; Munuera-Cabeza, M.; Talaverón-Rey, M.; Povea-Cabello, S.; Suárez-Carrillo, A.; Romero-González, A.; et al. MtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. Int. J. Mol. Sci. 2023, 24, 1482. [Google Scholar] [CrossRef] [PubMed]
- Lefevere, E.; Toft-Kehler, A.K.; Vohra, R.; Kolko, M.; Moons, L.; Van Hove, I. Mitochondrial Dysfunction Underlying Outer Retinal Diseases. Mitochondrion 2017, 36, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Ya-An Chang, J.; Yu, F.; Shi, L.; Ko, M.L.; Ko, G.Y.-P. Melatonin Affects Mitochondrial Fission/Fusion Dynamics in the Diabetic Retina. J. Diabetes Res. 2019, 2019, 8463125. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, D.H.; Yang, S.G.; Kim, D.Y. Improved Effect of a Mitochondria-Targeted Antioxidant on Hydrogen Peroxide-Induced Oxidative Stress in Human Retinal Pigment Epithelium Cells. BMC Pharmacol. Toxicol. 2021, 22, 7. [Google Scholar] [CrossRef] [PubMed]
- Jaliffa, C.; Ameqrane, I.; Dansault, A.; Leemput, J.; Vieira, V.; Lacassagne, E.; Provost, A.; Bigot, K.; Masson, C.; Menasche, M.; et al. Sirt1 Involvement in Rd10 Mouse Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3562–3572. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.; Tomarev, S. Evaluating Retinal Ganglion Cell Loss and Dysfunction. Exp. Eye Res. 2016, 151, 96. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, Y.; Oishi, A.; Miyata, M.; Oishi, M.; Hasegawa, T.; Numa, S.; Ikeda, H.O.; Tsujikawa, A. Wavelength of Light and Photophobia in Inherited Retinal Dystrophy. Sci. Rep. 2020, 10, 14798. [Google Scholar] [CrossRef] [PubMed]
- Bourin, M.; Hascoët, M. The Mouse Light/Dark Box Test. Eur. J. Pharmacol. 2003, 463, 55–65. [Google Scholar] [CrossRef]
- Crawley, J.; Goodwin, F.K. Preliminary Report of a Simple Animal Behavior Model for the Anxiolytic Effects of Benzodiazepines. Pharmacol. Biochem. Behav. 1980, 13, 167–170. [Google Scholar] [CrossRef]
- Milla-Navarro, S.; Pazo-González, M.; Germain, F.; de la Villa, P. Phenotype Characterization of a Mice Genetic Model of Absolute Blindness. Int. J. Mol. Sci. 2022, 23, 8152. [Google Scholar] [CrossRef]
- Wei, R.M.; Zhang, Y.M.; Feng, Y.Z.; Zhang, K.X.; Zhang, J.Y.; Chen, J.; Luo, B.L.; Li, X.Y.; Chen, G.H. Resveratrol Ameliorates Maternal Separation-Induced Anxiety- and Depression-like Behaviors and Reduces Sirt1-NF-KB Signaling-Mediated Neuroinflammation. Front. Behav. Neurosci. 2023, 17, 1172091. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, F.; Novelli, E.; Strettoi, E. Retinal Ganglion Cells Survive and Maintain Normal Dendritic Morphology in a Mouse Model of Inherited Photoreceptor Degeneration. J. Neurosci. 2008, 28, 14282–14292. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Alibrandi, S.; Id, C.S.; Rinaldi, C.; Dascola, A.; Id, A.C.; D’angelo, R.; Sidoti, A. The Impact of Modifier Genes on Cone-Rod Dystrophy Heterogeneity: An Explorative Familial Pilot Study and a Hypothesis on Neurotransmission Impairment. PLoS ONE 2022, 17, e0278857. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.; Ding, X.Q.; Saadi, A.; Agarwal, N.; Naash, M.I.; Al-Ubaidi, M.R. Expression of Cone-Photoreceptor–Specific Antigens in a Cell Line Derived from Retinal Tumors in Transgenic Mice. Investig. Ophthalmol. Vis. Sci. 2004, 45, 764–768. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TaqMan® Assay | Function | Gene Symbol |
|---|---|---|
| Assay ID: Mm00679214_g1 | Mitochondrial fission | Dnm1/Drp1 |
| Assay ID: Mm00612599_m1 | Mitochondrial fusion | Mfn1 |
| Assay ID: Mm01313000_m1 | Superoxide dismutase 2, mitochondrial | Sod2 |
| Assay ID: Mm00437992_m1 | Peroxidase | Cat |
| Assay ID: Mm99999915_g1 | Dehydrogenase housekeeping | Gapdh |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moshtaghion, S.M.; Caballano-Infantes, E.; Plaza Reyes, Á.; Valdés-Sánchez, L.; Fernández, P.G.; de la Cerda, B.; Riga, M.S.; Álvarez-Dolado, M.; Peñalver, P.; Morales, J.C.; et al. Piceid Octanoate Protects Retinal Cells against Oxidative Damage by Regulating the Sirtuin 1/Poly-ADP-Ribose Polymerase 1 Axis In Vitro and in rd10 Mice. Antioxidants 2024, 13, 201. https://doi.org/10.3390/antiox13020201
Moshtaghion SM, Caballano-Infantes E, Plaza Reyes Á, Valdés-Sánchez L, Fernández PG, de la Cerda B, Riga MS, Álvarez-Dolado M, Peñalver P, Morales JC, et al. Piceid Octanoate Protects Retinal Cells against Oxidative Damage by Regulating the Sirtuin 1/Poly-ADP-Ribose Polymerase 1 Axis In Vitro and in rd10 Mice. Antioxidants. 2024; 13(2):201. https://doi.org/10.3390/antiox13020201
Chicago/Turabian StyleMoshtaghion, Seyed Mohamadmehdi, Estefanía Caballano-Infantes, Álvaro Plaza Reyes, Lourdes Valdés-Sánchez, Patricia Gallego Fernández, Berta de la Cerda, Maurizio S. Riga, Manuel Álvarez-Dolado, Pablo Peñalver, Juan C. Morales, and et al. 2024. "Piceid Octanoate Protects Retinal Cells against Oxidative Damage by Regulating the Sirtuin 1/Poly-ADP-Ribose Polymerase 1 Axis In Vitro and in rd10 Mice" Antioxidants 13, no. 2: 201. https://doi.org/10.3390/antiox13020201