

The Intersection of Genetic Factors, Aberrant Nutrient Metabolism and Oxidative Stress in the Progression of Cardiometabolic Disease


Abstract
:1. Introduction
2. Oxidative Stress and Cardiometabolic Disease
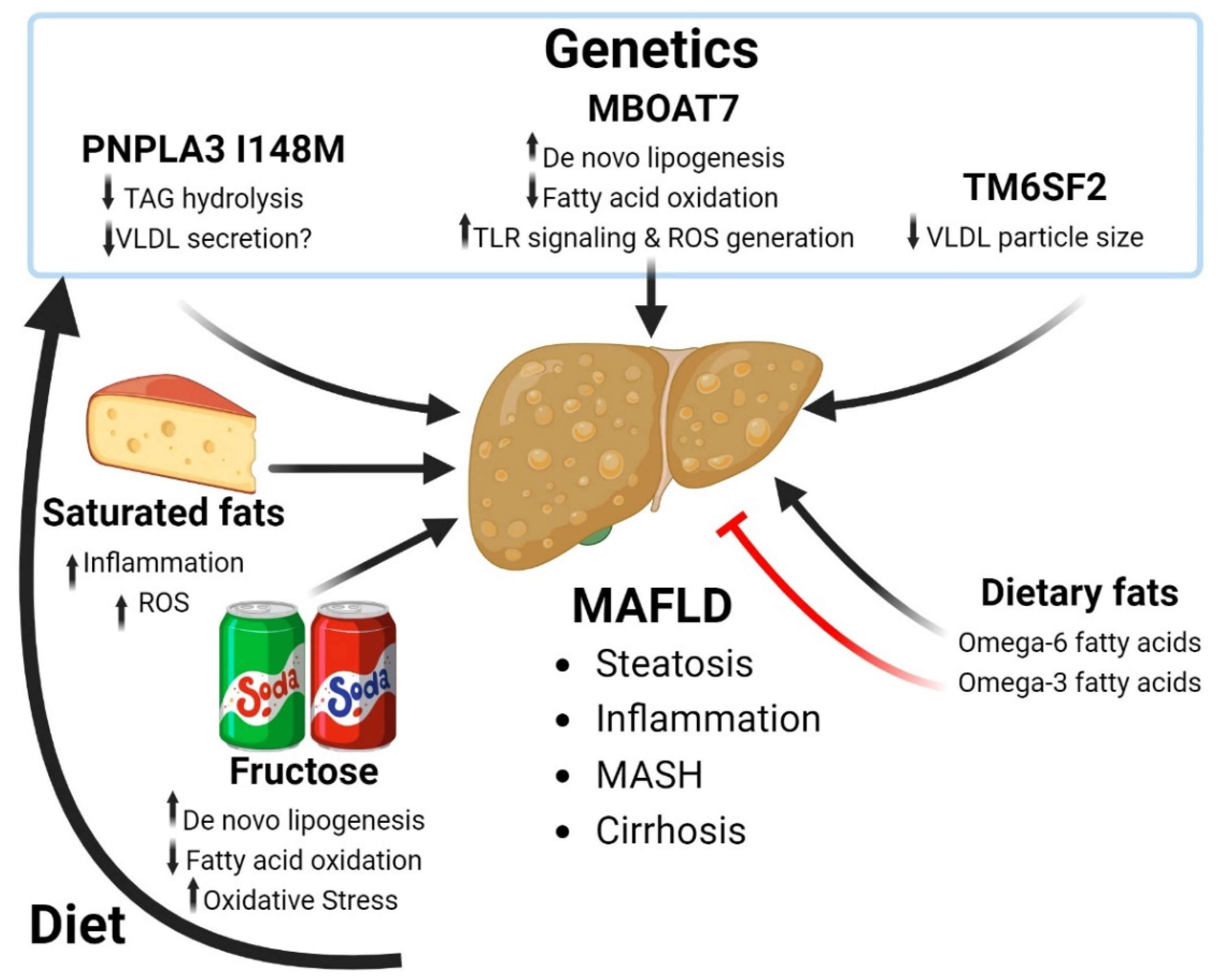
3. Metabolic-Associated Fatty Liver Disease
3.1. Fructose and Progression of MAFLD
3.2. Dietary Fatty Acids and MAFLD
3.3. Genetics of MAFLD
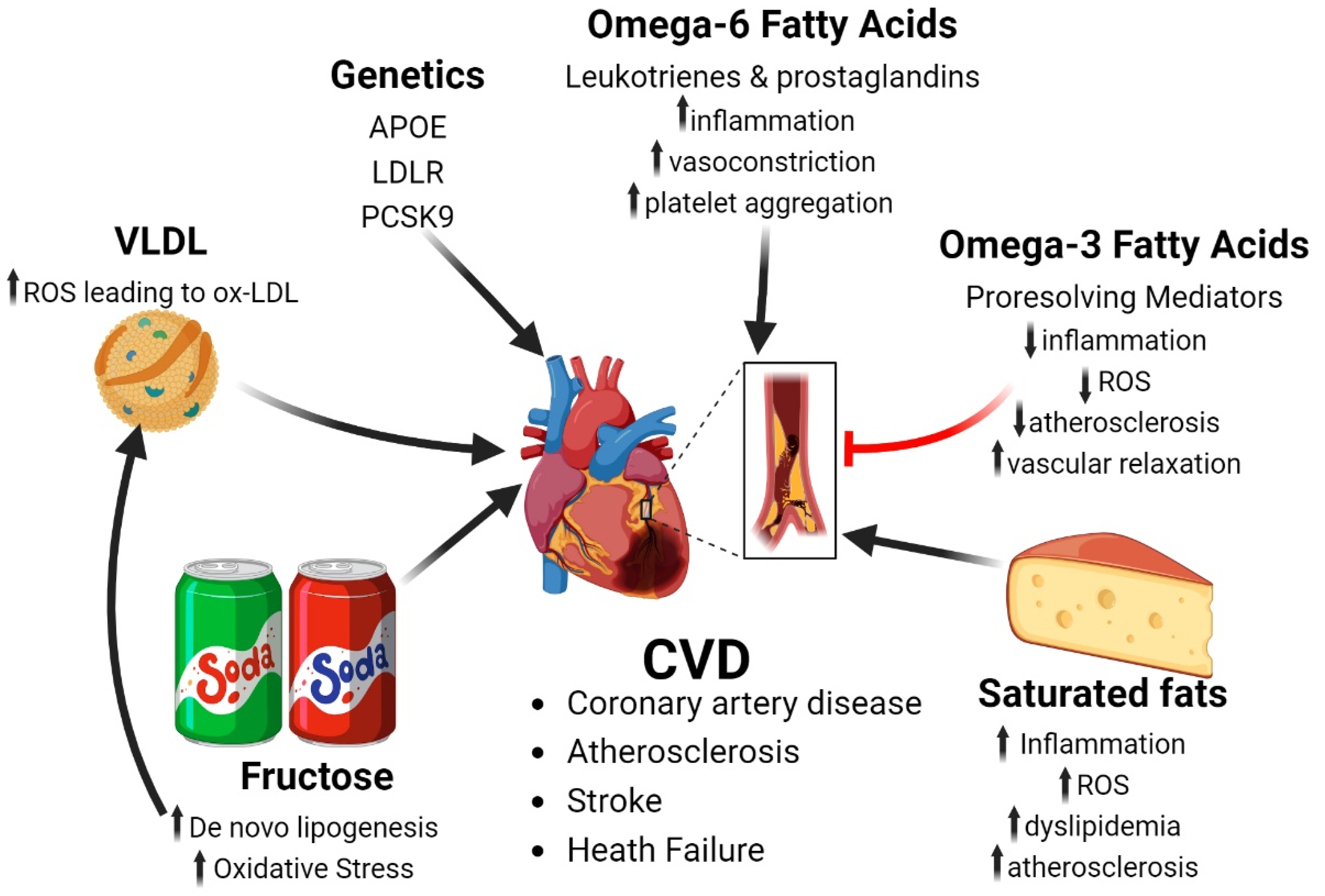
4. Cardiovascular Disease
4.1. Fatty Acids and Risk of Cardiovascular Disease
4.2. Simple Sugars and Cardiovascular Disease
4.3. Genetic Risk Factor of Cardiovascular Disease
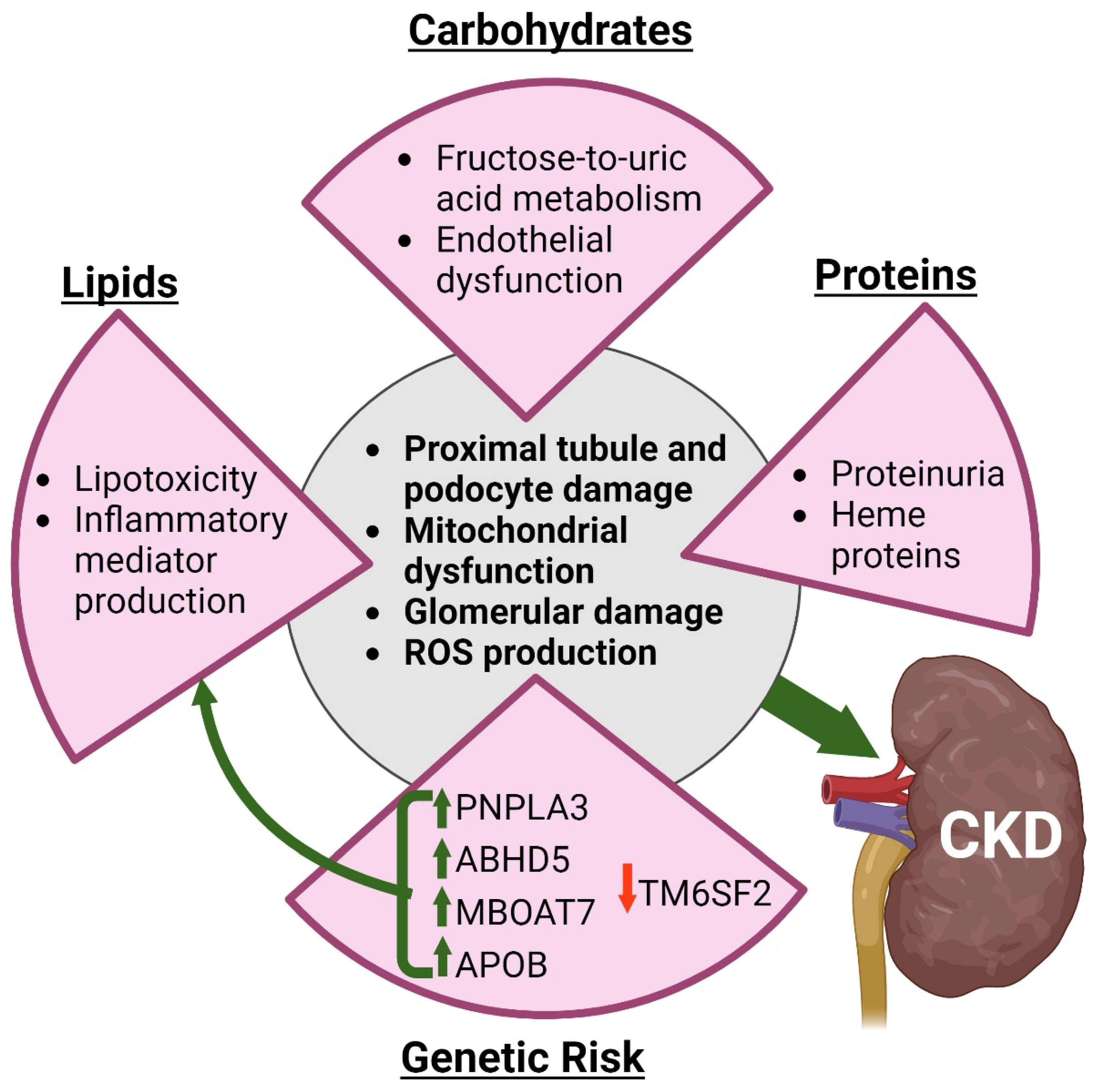
5. Chronic Kidney Disease
5.1. Ectopic Renal Fat Accumulation
5.2. Simple Sugars and Uric Acid
5.3. Protein-Induced Kidney Damage
5.4. Genes Associated with Aberrant Nutrient Metabolism and CKD
6. Intersection of MAFLD and CKD
6.1. Epidemiology of MAFLD and CKD
6.2. Renin-Angiotensin System Activation
6.3. Lipid Dysregulation
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight#:~:text=Worldwide%20obesity%20has%20nearly%20tripled,%2C%20and%2013%25%20were%20obese (accessed on 12 November 2023).
- Ward, Z.J.; Bleich, S.N.; Cradock, A.L.; Barrett, J.L.; Giles, C.M.; Flax, C.; Long, M.W.; Gortmaker, S.L. Projected U.S. State-Level Prevalence of Adult Obesity and Severe Obesity. N. Engl. J. Med. 2019, 381, 2440–2450. [Google Scholar] [CrossRef]
- Control, C.f.D. The High Obesity Program (HOP 2023). Available online: https://www.cdc.gov/nccdphp/dnpao/state-local-programs/fundingopp/2023/hop.html (accessed on 8 November 2023).
- Virtue, S.; Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome—An allostatic perspective. Biochim. Biophys. Acta 2010, 1801, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 2010, 1801, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Britton, K.A.; Fox, C.S. Ectopic fat depots and cardiovascular disease. Circulation 2011, 124, e837–e841. [Google Scholar] [CrossRef] [PubMed]
- Klop, B.; Elte, J.W.; Cabezas, M.C. Dyslipidemia in obesity: Mechanisms and potential targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef] [PubMed]
- Pai, J.K. Inflammatory Markers and the Risk of Coronary Heart Disease in Men and Women. N. Engl. J. Med. 2004, 351, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D. Influence of Body Fat Distribution on Free Fatty Acid Metabolism in Obesity. J. Clin. Investig. 1989, 83, 1168–1173. [Google Scholar] [CrossRef]
- Fain, J. Release of Interleukins and Other Inflammatory Cytokines by Hunman Adipose Tissue is Enhanced in Obesity and Primarily due to the Nonfat Cells. Vitam. Horm. 2006, 74C, 443–477. [Google Scholar]
- Calabro, P.; Willerson, J.T.; Yeh, E.T. Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation 2003, 108, 1930–1932. [Google Scholar] [CrossRef]
- Emerging Risk Factors, C.; Kaptoge, S.; Di Angelantonio, E.; Pennells, L.; Wood, A.M.; White, I.R.; Gao, P.; Walker, M.; Thompson, A.; Sarwar, N.; et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N. Engl. J. Med. 2012, 367, 1310–1320. [Google Scholar] [CrossRef]
- Badimon, L.; Pena, E.; Arderiu, G.; Padro, T.; Slevin, M.; Vilahur, G.; Chiva-Blanch, G. C-Reactive Protein in Atherothrombosis and Angiogenesis. Front. Immunol. 2018, 9, 430. [Google Scholar] [CrossRef] [PubMed]
- Hauck, A.K.; Huang, Y.; Hertzel, A.V.; Bernlohr, D.A. Adipose oxidative stress and protein carbonylation. J. Biol. Chem. 2019, 294, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Murdolo, G.; Piroddi, M.; Luchetti, F.; Tortoioli, C.; Canonico, B.; Zerbinati, C.; Galli, F.; Iuliano, L. Oxidative stress and lipid peroxidation by-products at the crossroad between adipose organ dysregulation and obesity-linked insulin resistance. Biochimie 2013, 95, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Delbosc, S.; Paizanis, E.; Magous, R.; Araiz, C.; Dimo, T.; Cristol, J.P.; Cros, G.; Azay, J. Involvement of oxidative stress and NADPH oxidase activation in the development of cardiovascular complications in a model of insulin resistance, the fructose-fed rat. Atherosclerosis 2005, 179, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Albert-Garay, J.S.; Riesgo-Escovar, J.R.; Salceda, R. High glucose concentrations induce oxidative stress by inhibiting Nrf2 expression in rat Muller retinal cells in vitro. Sci. Rep. 2022, 12, 1261. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.B.; Gunn, P.J.; Fielding, B.A. The role of dietary sugars and de novo lipogenesis in non-alcoholic fatty liver disease. Nutrients 2014, 6, 5679–5703. [Google Scholar] [CrossRef] [PubMed]
- Heianza, Y.; Qi, L. Gene-Diet Interaction and Precision Nutrition in Obesity. Int. J. Mol. Sci. 2017, 18, 787. [Google Scholar] [CrossRef]
- Drozdz, D.; Alvarez-Pitti, J.; Wojcik, M.; Borghi, C.; Gabbianelli, R.; Mazur, A.; Herceg-Cavrak, V.; Lopez-Valcarcel, B.G.; Brzezinski, M.; Lurbe, E.; et al. Obesity and Cardiometabolic Risk Factors: From Childhood to Adulthood. Nutrients 2021, 13, 4176. [Google Scholar] [CrossRef]
- Heianza, Y.; Qi, L. Impact of Genes and Environment on Obesity and Cardiovascular Disease. Endocrinology 2019, 160, 81–100. [Google Scholar] [CrossRef]
- Marcadenti, A. Diet, Cardiometabolic Factors and Type-2 Diabetes Mellitus: The Role of Genetics. Curr. Diabetes Rev. 2016, 12, 322–330. [Google Scholar] [CrossRef]
- Lombardi, R.; Iuculano, F.; Pallini, G.; Fargion, S.; Fracanzani, A.L. Nutrients, Genetic Factors, and Their Interaction in Non-Alcoholic Fatty Liver Disease and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 8761. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Francisqueti, F.V.; Chiaverini, L.C.; Santos, K.C.; Minatel, I.O.; Ronchi, C.B.; Ferron, A.J.; Ferreira, A.L.; Correa, C.R. The role of oxidative stress on the pathophysiology of metabolic syndrome. Rev Assoc Med Bras 2017, 63, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Akash, M.S.H. Mechanism of Generation of Oxidative Stress and Pathophysiology of Type 2 Diabetes Mellitus: How Are They Interlinked? J. Cell Biochem. 2017, 118, 3577–3585. [Google Scholar] [CrossRef]
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quinones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef]
- Swiatkiewicz, I.; Wroblewski, M.; Nuszkiewicz, J.; Sutkowy, P.; Wroblewska, J.; Wozniak, A. The Role of Oxidative Stress Enhanced by Adiposity in Cardiometabolic Diseases. Int. J. Mol. Sci. 2023, 24, 6382. [Google Scholar] [CrossRef]
- Palinski, W.; Rosenfeld, M.E.; Yla-Herttuala, S.; Gurtner, G.C.; Socher, S.S.; Butler, S.W.; Parthasarathy, S.; Carew, T.E.; Steinberg, D.; Witztum, J.L. Low density lipoprotein undergoes oxidative modification in vivo. Proc. Natl. Acad. Sci. USA 1989, 86, 1372–1376. [Google Scholar] [CrossRef]
- Haberland, M.E.; Mottino, G.; Le, M.; Frank, J.S. Sequestration of aggregated LDL by macrophages studied with freeze-etch electron microscopy. J. Lipid Res. 2001, 42, 605–619. [Google Scholar] [CrossRef]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm. 2013, 2013, 152786. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Mathews, M.T.; Berk, B.C. PARP-1 inhibition prevents oxidative and nitrosative stress-induced endothelial cell death via transactivation of the VEGF receptor 2. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernandez-Fernandez, C.; Mourino-Bayolo, D. Mitochondrial beta-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef]
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 266–271. [Google Scholar] [CrossRef]
- Han, H.J.; Lee, Y.J.; Park, S.H.; Lee, J.H.; Taub, M. High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am. J. Physiol. Renal Physiol. 2005, 288, F988–F996. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Han, T.T.; Liu, Y.; Zheng, S.; Zhang, Y.; Liu, W.; Hu, Y.M. Insulin resistance caused by lipotoxicity is related to oxidative stress and endoplasmic reticulum stress in LPL gene knockout heterozygous mice. Atherosclerosis 2015, 239, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Mohanty, P.; Dhindsa, S.; Syed, T.; Ghanim, H.; Aljada, A.; Dandona, P. Elevation of Free Fatty Acids Induces Inflammation and Impairs Vascular Reactivity in Healthy Subjects. Diabetes 2003, 52, 2882–2887. [Google Scholar] [CrossRef] [PubMed]
- Kobayasi, R.; Akamine, E.H.; Davel, A.P.; Rodrigues, M.A.; Carvalho, C.R.; Rossoni, L.V. Oxidative stress and inflammatory mediators contribute to endothelial dysfunction in high-fat diet-induced obesity in mice. J. Hypertens. 2010, 28, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ge, X.; Li, X.; He, J.; Wei, X.; Du, J.; Sun, J.; Li, X.; Xun, Z.; Liu, W.; et al. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis. 2020, 11, 914. [Google Scholar] [CrossRef]
- Tangvarasittichai, S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J. Diabetes 2015, 6, 456–480. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.; Waked, I.; Bollipo, S.; Gomaa, A.; Ajlouni, Y.; Attia, D. What’s in a name? Renaming ‘NAFLD’ to ‘MAFLD’. Liver Int. 2020, 40, 1254–1261. [Google Scholar] [CrossRef]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022. [Google Scholar] [CrossRef]
- Teng, M.L.; Ng, C.H.; Huang, D.Q.; Chan, K.E.; Tan, D.J.; Lim, W.H.; Yang, J.D.; Tan, E.; Muthiah, M.D. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2023, 29, S32–S42. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Romero-Gomez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.C.; Itsiopoulos, C.; Thodis, T.; Ward, G.; Trost, N.; Hofferberth, S.; O’Dea, K.; Desmond, P.V.; Johnson, N.A.; Wilson, A.M. The Mediterranean diet improves hepatic steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease. J. Hepatol. 2013, 59, 138–143. [Google Scholar] [CrossRef]
- Hashida, R.; Kawaguchi, T.; Bekki, M.; Omoto, M.; Matsuse, H.; Nago, T.; Takano, Y.; Ueno, T.; Koga, H.; George, J.; et al. Aerobic vs. resistance exercise in non-alcoholic fatty liver disease: A systematic review. J. Hepatol. 2017, 66, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A.; Investigators, N.N. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Abdelmalek, M.F.; Armstrong, M.J.; Jara, M.; Kjær, M.S.; Krarup, N.; Lawitz, E.; Ratziu, V.; Sanyal, A.J.; Schattenberg, J.M.; et al. Semaglutide 2·4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: A randomised, placebo-controlled phase 2 trial. Lancet 2023, 8, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; Chaikoff, I.L.; Schusdek, A. Effect of Fructose on Lipogenesis from Lactate and Acetate in Diabetic Liver. J. Biol. Chem. 1952, 194, 435–443. [Google Scholar] [CrossRef]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef]
- Softic, S.; Gupta, M.K.; Wang, G.X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 127, 4059–4074. [Google Scholar] [CrossRef]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef]
- Morrow, M.R.; Batchuluun, B.; Wu, J.; Ahmadi, E.; Leroux, J.M.; Mohammadi-Shemirani, P.; Desjardins, E.M.; Wang, Z.; Tsakiridis, E.E.; Lavoie, D.C.T.; et al. Inhibition of ATP-citrate lyase improves NASH, liver fibrosis, and dyslipidemia. Cell Metab. 2022, 34, 919–936.e918. [Google Scholar] [CrossRef]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Bremer, A.A.; Berglund, L.; McGahan, J.P.; Havel, P.J.; et al. Consumption of fructose-sweetened beverages for 10 weeks reduces net fat oxidation and energy expenditure in overweight/obese men and women. Eur. J. Clin. Nutr. 2012, 66, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Meyer, J.G.; Wang, G.X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753 e734. [Google Scholar] [CrossRef]
- Cho, Y.E.; Kim, D.K.; Seo, W.; Gao, B.; Yoo, S.H.; Song, B.J. Fructose Promotes Leaky Gut, Endotoxemia, and Liver Fibrosis Through Ethanol-Inducible Cytochrome P450-2E1-Mediated Oxidative and Nitrative Stress. Hepatology 2021, 73, 2180–2195. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M.; Nonalcoholic Steatohepatitis Clinical Research, N. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Koek, G.H.; Liedorp, P.R.; Bast, A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Haufe, S.; Engeli, S.; Kast, P.; Bohnke, J.; Utz, W.; Haas, V.; Hermsdorf, M.; Mahler, A.; Wiesner, S.; Birkenfeld, A.L.; et al. Randomized comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology 2011, 53, 1504–1514. [Google Scholar] [CrossRef]
- Westerbacka, J.; Lammi, K.; Hakkinen, A.M.; Rissanen, A.; Salminen, I.; Aro, A.; Yki-Jarvinen, H. Dietary fat content modifies liver fat in overweight nondiabetic subjects. J. Clin. Endocrinol. Metab. 2005, 90, 2804–2809. [Google Scholar] [CrossRef]
- Bozzetto, L.; Prinster, A.; Annuzzi, G.; Costagliola, L.; Mangione, A.; Vitelli, A.; Mazzarella, R.; Longobardo, M.; Mancini, M.; Vigorito, C.; et al. Liver fat is reduced by an isoenergetic MUFA diet in a controlled randomized study in type 2 diabetic patients. Diabetes Care 2012, 35, 1429–1435. [Google Scholar] [CrossRef]
- Rezaei, S.; Akhlaghi, M.; Sasani, M.R.; Barati Boldaji, R. Olive oil lessened fatty liver severity independent of cardiometabolic correction in patients with non-alcoholic fatty liver disease: A randomized clinical trial. Nutrition 2019, 57, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Fernandez-Galilea, M.; Martinez-Fernandez, L.; Gonzalez-Muniesa, P.; Perez-Chavez, A.; Martinez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Walenta, E.; Akiyama, T.E.; Lagakos, W.S.; Lackey, D.; Pessentheiner, A.R.; Sasik, R.; Hah, N.; Chi, T.J.; Cox, J.M.; et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat. Med. 2014, 20, 942–947. [Google Scholar] [CrossRef]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef]
- Im, D.S. Omega-3 fatty acids in anti-inflammation (pro-resolution) and GPCRs. Prog. Lipid Res. 2012, 51, 232–237. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Casas, R.; Castro-Barquero, S.; Estruch, R.; Sacanella, E. Nutrition and Cardiovascular Health. Int. J. Mol. Sci. 2018, 19, 3988. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Abdelmalek, M.F.; Suzuki, A.; Cummings, O.W.; Chojkier, M.; Group, E.-A.S. No significant effects of ethyl-eicosapentanoic acid on histologic features of nonalcoholic steatohepatitis in a phase 2 trial. Gastroenterology 2014, 147, 377–384 e371. [Google Scholar] [CrossRef]
- Vega, G.L.; Chandalia, M.; Szczepaniak, L.S.; Grundy, S.M. Effects of N-3 fatty acids on hepatic triglyceride content in humans. J. Investig. Med. 2008, 56, 780–785. [Google Scholar] [CrossRef]
- Innes, J.K.; Calder, P.C. Omega-6 fatty acids and inflammation. Prostaglandins Leukot. Essent. Fatty Acids 2018, 132, 41–48. [Google Scholar] [CrossRef]
- Sztolsztener, K.; Chabowski, A.; Harasim-Symbor, E.; Bielawiec, P.; Konstantynowicz-Nowicka, K. Arachidonic Acid as an Early Indicator of Inflammation during Non-Alcoholic Fatty Liver Disease Development. Biomolecules 2020, 10, 1133. [Google Scholar] [CrossRef] [PubMed]
- Bjermo, H.; Iggman, D.; Kullberg, J.; Dahlman, I.; Johansson, L.; Persson, L.; Berglund, J.; Pulkki, K.; Basu, S.; Uusitupa, M.; et al. Effects of n-6 PUFAs compared with SFAs on liver fat, lipoproteins, and inflammation in abdominal obesity: A randomized controlled trial. Am. J. Clin. Nutr. 2012, 95, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Ferolla, S.M.; Ferrari, T.C.; Lima, M.L.; Reis, T.O.; Tavares, W.C., Jr.; Couto, O.F.; Vidigal, P.V.; Fausto, M.A.; Couto, C.A. Dietary patterns in Brazilian patients with nonalcoholic fatty liver disease: A cross-sectional study. Clinics 2013, 68, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; De Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Faga, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, K.M.; Bayer-Carter, J.L.; Arbuckle, M.D.; Tidwell, J.M.; Richards, T.L.; Craft, S. Beneficial effect of a weight-stable, low-fat/low-saturated fat/low-glycaemic index diet to reduce liver fat in older subjects. Br. J. Nutr. 2013, 109, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Marina, A.; von Frankenberg, A.D.; Suvag, S.; Callahan, H.S.; Kratz, M.; Richards, T.L.; Utzschneider, K.M. Effects of dietary fat and saturated fat content on liver fat and markers of oxidative stress in overweight/obese men and women under weight-stable conditions. Nutrients 2014, 6, 4678–4690. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, M.; Kreis, R.; Debard, C.; Cariou, B.; Faeh, D.; Chetiveaux, M.; Ith, M.; Vermathen, P.; Stefanoni, N.; Le, K.A.; et al. High protein intake reduces intrahepatocellular lipid deposition in humans. Am. J. Clin. Nutr. 2009, 90, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Sadevirta, S.; Zhou, Y.; Kayser, B.; Ali, A.; Ahonen, L.; Lallukka, S.; Pelloux, V.; Gaggini, M.; Jian, C.; et al. Saturated Fat Is More Metabolically Harmful for the Human Liver Than Unsaturated Fat or Simple Sugars. Diabetes Care 2018, 41, 1732–1739. [Google Scholar] [CrossRef]
- Jensen, V.S.; Hvid, H.; Damgaard, J.; Nygaard, H.; Ingvorsen, C.; Wulff, E.M.; Lykkesfeldt, J.; Fledelius, C. Dietary fat stimulates development of NAFLD more potently than dietary fructose in Sprague-Dawley rats. Diabetol. Metab. Syndr. 2018, 10, 4. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Day, C.P. The Genetics of Nonalcoholic Fatty Liver Disease: Spotlight on PNPLA3 and TM6SF2. Semin. Liver Dis. 2015, 35, 270–290. [Google Scholar] [CrossRef] [PubMed]
- Rotman, Y.; Koh, C.; Zmuda, J.M.; Kleiner, D.E.; Liang, T.J.; Nash, C.R.N. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010, 52, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Sentinelli, F.; Cambuli, V.M.; Incani, M.; Congiu, T.; Matta, V.; Pilia, S.; Huang-Doran, I.; Cossu, E.; Loche, S.; et al. The 148M allele of the PNPLA3 gene is associated with indices of liver damage early in life. J. Hepatol. 2010, 53, 335–338. [Google Scholar] [CrossRef]
- Shen, J.H.; Li, Y.L.; Li, D.; Wang, N.N.; Jing, L.; Huang, Y.H. The rs738409 (I148M) variant of the PNPLA3 gene and cirrhosis: A meta-analysis. J. Lipid Res. 2015, 56, 167–175. [Google Scholar] [CrossRef]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef]
- Nischalke, H.D.; Berger, C.; Luda, C.; Berg, T.; Muller, T.; Grunhage, F.; Lammert, F.; Coenen, M.; Kramer, B.; Korner, C.; et al. The PNPLA3 rs738409 148M/M genotype is a risk factor for liver cancer in alcoholic cirrhosis but shows no or weak association in hepatitis C cirrhosis. PLoS ONE 2011, 6, e27087. [Google Scholar] [CrossRef]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef]
- Lake, A.C.; Sun, Y.; Li, J.L.; Kim, J.E.; Johnson, J.W.; Li, D.; Revett, T.; Shih, H.H.; Liu, W.; Paulsen, J.E.; et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J. Lipid Res. 2005, 46, 2477–2487. [Google Scholar] [CrossRef]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A.; et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef]
- Yang, A.; Mottillo, E.P.; Mladenovic-Lucas, L.; Zhou, L.; Granneman, J.G. Dynamic interactions of ABHD5 with PNPLA3 regulate triacylglycerol metabolism in brown adipocytes. Nat. Metab. 2019, 1, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chang, B.; Li, L.; Chan, L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010, 52, 1134–1142. [Google Scholar] [CrossRef]
- Basantani, M.K.; Sitnick, M.T.; Cai, L.; Brenner, D.S.; Gardner, N.P.; Li, J.Z.; Schoiswohl, G.; Yang, K.; Kumari, M.; Gross, R.W.; et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 2011, 52, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Huang, Y.; Karaman, R.; Ivanova, P.T.; Brown, H.A.; Roddy, T.; Castro-Perez, J.; Cohen, J.C.; Hobbs, H.H. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J. Clin. Investig. 2012, 122, 4130–4144. [Google Scholar] [CrossRef]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Lai, K.M.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef]
- Mitsche, M.A.; Hobbs, H.H.; Cohen, J.C. Patatin-like phospholipase domain-containing protein 3 promotes transfer of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J. Biol. Chem. 2018, 293, 6958–6968. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Yoshimura, T.; Cantley, J.L.; Majumdar, S.K.; Guebre-Egziabher, F.; Kursawe, R.; Vatner, D.F.; Fat, I.; Kahn, M.; Erion, D.M.; et al. Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatology 2013, 57, 1763–1772. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Nick, A.; Holtta-Vuori, M.; Thiele, C.; Isokuortti, E.; Lallukka-Bruck, S.; Zhou, Y.; Hakkarainen, A.; Lundbom, N.; Peltonen, M.; et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Tilson, S.G.; Morell, C.M.; Lenaerts, A.S.; Park, S.B.; Hu, Z.; Jenkins, B.; Koulman, A.; Liang, T.J.; Vallier, L. Modeling PNPLA3-Associated NAFLD Using Human-Induced Pluripotent Stem Cells. Hepatology 2021, 74, 2998–3017. [Google Scholar] [CrossRef]
- Kabbani, M.; Michailidis, E.; Steensels, S.; Fulmer, C.G.; Luna, J.M.; Le Pen, J.; Tardelli, M.; Razooky, B.; Ricardo-Lax, I.; Zou, C.; et al. Human hepatocyte PNPLA3-148M exacerbates rapid non-alcoholic fatty liver disease development in chimeric mice. Cell Rep. 2022, 40, 111321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kory, N.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology 2019, 69, 2427–2441. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Castano, G.O.; Scian, R.; Mallardi, P.; Fernandez Gianotti, T.; Burgueno, A.L.; San Martino, J.; Pirola, C.J. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 2015, 61, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; van’t Hooft, F. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918. [Google Scholar] [CrossRef] [PubMed]
- Smagris, E.; Gilyard, S.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J. Biol. Chem. 2016, 291, 10659–10676. [Google Scholar] [CrossRef]
- Luo, F.; Oldoni, F.; Das, A. TM6SF2: A Novel Genetic Player in Nonalcoholic Fatty Liver and Cardiovascular Disease. Hepatol. Commun. 2022, 6, 448–460. [Google Scholar] [CrossRef]
- Shindou, H.; Hishikawa, D.; Harayama, T.; Yuki, K.; Shimizu, T. Recent progress on acyl CoA: Lysophospholipid acyltransferase research. J. Lipid Res. 2009, 50, S46–S51. [Google Scholar] [CrossRef]
- Shindou, H.; Shimizu, T. Acyl-CoA:lysophospholipid acyltransferases. J. Biol. Chem. 2009, 284, 1–5. [Google Scholar] [CrossRef]
- Xia, M.; Chandrasekaran, P.; Rong, S.; Fu, X.; Mitsche, M.A. Hepatic deletion of Mboat7 (LPIAT1) causes activation of SREBP-1c and fatty liver. J. Lipid Res. 2021, 62, 100031. [Google Scholar] [CrossRef]
- Tanaka, Y.; Shimanaka, Y.; Caddeo, A.; Kubo, T.; Mao, Y.; Kubota, T.; Kubota, N.; Yamauchi, T.; Mancina, R.M.; Baselli, G.; et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut 2021, 70, 180–193. [Google Scholar] [CrossRef]
- Helsley, R.N.; Varadharajan, V.; Brown, A.L.; Gromovsky, A.D.; Schugar, R.C.; Ramachandiran, I.; Fung, K.; Kabbany, M.N.; Banerjee, R.; Neumann, C.K.; et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. eLife 2019, 8, e49882. [Google Scholar] [CrossRef] [PubMed]
- Thangapandi, V.R.; Knittelfelder, O.; Brosch, M.; Patsenker, E.; Vvedenskaya, O.; Buch, S.; Hinz, S.; Hendricks, A.; Nati, M.; Herrmann, A.; et al. Loss of hepatic Mboat7 leads to liver fibrosis. Gut 2021, 70, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.C.; Pyles, K.D.; Hallcox, T.; Kamm, D.R.; Piechowski, M.; Fisk, B.; Albert, C.J.; Carpenter, D.H.; Ulmasov, B.; Ford, D.A.; et al. Enhancing Hepatic MBOAT7 Expression in Mice With Nonalcoholic Steatohepatitis. Gastro Hep Adv. 2023, 2, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Dongiovanni, P.; Longo, M.; Carli, F.; Baselli, G.; Rametta, R.; Pelusi, S.; Badiali, S.; Maggioni, M.; Gaggini, M.; et al. Mboat7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. EBioMedicine 2020, 52, 102658. [Google Scholar] [CrossRef] [PubMed]
- Alharthi, J.; Bayoumi, A.; Thabet, K.; Pan, Z.; Gloss, B.S.; Latchoumanin, O.; Lundberg, M.; Twine, N.A.; McLeod, D.; Alenizi, S.; et al. A metabolic associated fatty liver disease risk variant in MBOAT7 regulates toll like receptor induced outcomes. Nat. Commun. 2022, 13, 7430. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Chen, Y.; Du, X.; Kuppa, A.; Feitosa, M.F.; Bielak, L.F.; O’Connell, J.R.; Musani, S.K.; Guo, X.; Kahali, B.; Chen, V.L.; et al. Genome-wide association meta-analysis identifies 17 loci associated with nonalcoholic fatty liver disease. Nat. Genet. 2023, 55, 1640–1650. [Google Scholar] [CrossRef]
- Stender, S.; Kozlitina, J.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Hobbs, H.H.; Cohen, J.C. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat. Genet. 2017, 49, 842–847. [Google Scholar] [CrossRef]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA 2010, 107, 7892–7897. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Arenaza, L.; Rios, C.; Plows, J.F.; Berger, P.K.; Alderete, T.L.; Fogel, J.L.; Nayak, K.; Mohamed, P.; Hwang, D.; et al. PNPLA3 Genotype, Arachidonic Acid Intake, and Unsaturated Fat Intake Influences Liver Fibrosis in Hispanic Youth with Obesity. Nutrients 2021, 13, 1621. [Google Scholar] [CrossRef] [PubMed]
- Van Name, M.A.; Savoye, M.; Chick, J.M.; Galuppo, B.T.; Feldstein, A.E.; Pierpont, B.; Johnson, C.; Shabanova, V.; Ekong, U.; Valentino, P.L.; et al. A Low omega-6 to omega-3 PUFA Ratio (n-6:n-3 PUFA) Diet to Treat Fatty Liver Disease in Obese Youth. J. Nutr. 2020, 150, 2314–2321. [Google Scholar] [CrossRef] [PubMed]
- Pirazzi, C.; Adiels, M.; Burza, M.A.; Mancina, R.M.; Levin, M.; Stahlman, M.; Taskinen, M.R.; Orho-Melander, M.; Perman, J.; Pujia, A.; et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 2012, 57, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, I.A.; Ericson, U.; Rukh, G.; Riddestrale, M.; Romeo, S.; Orho-Melander, M. The PNPLA3 Ile148Met interacts with overweight and dietary intakes on fasting triglyceride levels. Genes Nutr. 2014, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Simons, N.; Isaacs, A.; Koek, G.H.; Kuc, S.; Schaper, N.C.; Brouwers, M. PNPLA3, TM6SF2, and MBOAT7 Genotypes and Coronary Artery Disease. Gastroenterology 2017, 152, 912–913. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Boren, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230 e1216. [Google Scholar] [CrossRef]
- Krawczyk, M.; Jimenez-Aguero, R.; Alustiza, J.M.; Emparanza, J.I.; Perugorria, M.J.; Bujanda, L.; Lammert, F.; Banales, J.M. PNPLA3 p.I148M variant is associated with greater reduction of liver fat content after bariatric surgery. Surg. Obes. Relat. Dis. 2016, 12, 1838–1846. [Google Scholar] [CrossRef]
- Tabassum, R.; Ramo, J.T.; Ripatti, P.; Koskela, J.T.; Kurki, M.; Karjalainen, J.; Palta, P.; Hassan, S.; Nunez-Fontarnau, J.; Kiiskinen, T.T.J.; et al. Genetic architecture of human plasma lipidome and its link to cardiovascular disease. Nat. Commun. 2019, 10, 4329. [Google Scholar] [CrossRef]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E.K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK343489/ (accessed on 3 January 2019).
- Hu, F.; Colditz, G.; Rosner, B.; Hennekens, C.; Willett, W. Dietary Fat Intake and the Risk of Coronary Heart Disease in Women. N. Engl. J. Med. 1997, 337, 1491–1499. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Hoffmann, G. Dietary fatty acids in the secondary prevention of coronary heart disease: A systematic review, meta-analysis and meta-regression. BMJ Open 2014, 4, e004487. [Google Scholar] [CrossRef] [PubMed]
- Siri-Tarino, P.W.; Sun, Q.; Hu, F.B.; Krauss, R.M. Meta-analysis of prospective cohort studies evaluating the association of saturated fat with cardiovascular disease. Am. J. Clin. Nutr. 2010, 91, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.; Martin, N.; Jimoh, O.F.; Kirk, C.; Foster, E.; Abdelhamid, A.S. Reduction in saturated fat intake for cardiovascular disease. Cochrane Database Syst. Rev. 2020, 5, CD011737. [Google Scholar] [CrossRef]
- Maki, K.C.; Dicklin, M.R.; Kirkpatrick, C.F. Saturated fats and cardiovascular health: Current evidence and controversies. J. Clin. Lipidol. 2021, 15, 765–772. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Lin, J.; Yang, R.; Tarr, P.T.; Wu, P.H.; Handschin, C.; Li, S.; Yang, W.; Pei, L.; Uldry, M.; Tontonoz, P.; et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 2005, 120, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Rudel, L.P.J.; Sawyer, J. Compared With Dietary Monounsaturated and Saturated Fat, Polyunsaturated Fat Protects African Green Monkeys From Coronary Artery Atherosclerosis. Arterioscler. Thromb. Vase Biol. 1995, 15, 2101–2211. [Google Scholar] [CrossRef]
- Ramsden, C.E.; Zamora, D.; Majchrzak-Hong, S.; Faurot, K.R.; Broste, S.K.; Frantz, R.P.; Davis, J.M.; Ringel, A.; Suchindran, C.M.; Hibbeln, J.R. Re-evaluation of the traditional diet-heart hypothesis: Analysis of recovered data from Minnesota Coronary Experiment (1968-73). BMJ Open 2016, 353, 1756–1833. [Google Scholar] [CrossRef]
- Ramsden, C.E.; Zamora, D.; Leelarthaepin, B.; Majchrzak-Hong, S.F.; Faurot, K.R.; Suchindran, C.M.; Ringel, A.; Hibbeln, J.R. Use of dietary linoleic acid for secondary prevention of coronary heart disease and death: Evaluation of recovered data from the Sydney Diet Heart Study and updated meta-analysis. FASEB 2013, 27, 1756–1833. [Google Scholar] [CrossRef]
- Leren, P. The effect of plasma-cholesterol-lowering diet in male survivors of myocardial infarction. A controlled clinical trial. Bull. N. Y. Acad. Med. 1968, 44, 1012–1020. [Google Scholar]
- Wang, Y.; Fang, Y.; Witting, P.K.; Charchar, F.J.; Sobey, C.G.; Drummond, G.R.; Golledge, J. Dietary fatty acids and mortality risk from heart disease in US adults: An analysis based on NHANES. Sci. Rep. 2023, 13, 1614. [Google Scholar] [CrossRef] [PubMed]
- Dayton, S.; Pearce, M.L.; Goldman, H.; Harnish, A.; Plotkin, D.; Shickman, M.; Winfield, M.; Zager, A.; Dixon, W. Controlled trial of a diet high in unsaturated fat for prevention of atherosclerotic complications. Lancet 1968, 2, 1060–1062. [Google Scholar] [CrossRef] [PubMed]
- Karvonen, M.J.; Pekkarinen, M.; Miettinen, M.; Elosuo, R.; Paavilainen, E.; Paavilainen, E. Dietary prevention of coronary heart disease: The Finnish Mental Hospital Study. Int. J. Epidemiol. 1979, 8, 99–118. [Google Scholar] [CrossRef]
- Sacks, F.M.; Lichtenstein, A.H.; Wu, J.H.Y.; Appel, L.J.; Creager, M.A.; Kris-Etherton, P.M.; Miller, M.; Rimm, E.B.; Rudel, L.L.; Robinson, J.G.; et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation 2017, 136, e1–e23. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, M.U.; O’Reilly, E.J.; Heitmann, B.L.; Pereira, M.A.; Balter, K.; Fraser, G.E.; Goldbourt, U.; Hallmans, G.; Knekt, P.; Liu, S.; et al. Major types of dietary fat and risk of coronary heart disease: A pooled analysis of 11 cohort studies. Am. J. Clin. Nutr. 2009, 89, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liang, X.; Wang, L.; Lu, X.; Huang, J.; Cao, J.; Li, H.; Gu, D. Effect of omega-3 fatty acids supplementation on endothelial function: A meta-analysis of randomized controlled trials. Atherosclerosis 2012, 221, 536–543. [Google Scholar] [CrossRef]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef]
- Heydari, B.; Abdullah, S.; Pottala, J.V.; Shah, R.; Abbasi, S.; Mandry, D.; Francis, S.A.; Lumish, H.; Ghoshhajra, B.B.; Hoffmann, U.; et al. Effect of Omega-3 Acid Ethyl Esters on Left Ventricular Remodeling After Acute Myocardial Infarction: The OMEGA-REMODEL Randomized Clinical Trial. Circulation 2016, 134, 378–391. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. 2008, 233, 674–688. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Khoo, J.C.; Miller, E.; Barnett, J.; Witztum, J.L.; Steinberg, D. Low density lipoprotein rich in oleic acid is protected against oxidative modification: Implications for dietary prevention of atherosclerosis. Proc. Natl. Acad. Sci. USA 1990, 87, 3894–3898. [Google Scholar] [CrossRef]
- Reaven, P.D.; Grasse, B.J.; Tribble, D.L. Effects of linoleate-enriched and oleate-enriched diets in combination with alpha-tocopherol on the susceptibility of LDL and LDL subfractions to oxidative modification in humans. Arterioscler. Thromb. 1994, 14, 557–566. [Google Scholar] [CrossRef]
- Louheranta, A.M.; Porkkala-Sarataho, E.K.; Nyyssonen, M.K.; Salonen, R.M.; Salonen, J.T. Linoleic acid intake and susceptibility of very-low-density and low density lipoproteins to oxidation in men. Am. J. Clin. Nutr. 1996, 63, 698–703. [Google Scholar] [CrossRef]
- Regnstrom, J.; Nilsson, J.; Tornvall, P.; Hamsten, A.; Landou, C. Susceptibility to low-density lipoprotein oxidation and coronary atherosclerosis in man. Lancet 1992, 339, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Mariamenatu, A.H.; Abdu, E.M.; Kostner, G.M. Overconsumption of Omega-6 Polyunsaturated Fatty Acids (PUFAs) versus Deficiency of Omega-3 PUFAs in Modern-Day Diets: The Disturbing Factor for Their “Balanced Antagonistic Metabolic Functions” in the Human Body. J. Lipids 2021, 2021, 8848161. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J.; et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 2016, 7, 12859. [Google Scholar] [CrossRef] [PubMed]
- Viola, J.R.; Lemnitzer, P.; Jansen, Y.; Csaba, G.; Winter, C.; Neideck, C.; Silvestre-Roig, C.; Dittmar, G.; Doring, Y.; Drechsler, M.; et al. Resolving Lipid Mediators Maresin 1 and Resolvin D2 Prevent Atheroprogression in Mice. Circ. Res. 2016, 119, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Edwards-Glenn, J.M.; Fontes, M.T.; Waigi, E.W.; Costa, T.J.; Maiseyeu, A.; Webb, R.C.; McCarthy, C.G.; Wenceslau, C.F. Specialized Pro-resolving Mediator Improves Vascular Relaxation via Formyl Peptide Receptor-2. Am. J. Hypertens. 2023, 36, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Massaro, M.; Habib, A.; Lubrano, L.; Del Turco, S.; Lazzerini, G.; Bourcier, T.; Weksler, B.B.; De Caterina, R. The omega-3 fatty acid docosahexaenoate attenuates endothelial cyclooxygenase-2 induction through both NADP(H) oxidase and PKC epsilon inhibition. Proc. Natl. Acad. Sci. USA 2006, 103, 15184–15189. [Google Scholar] [CrossRef]
- Emken, E.A.; Rohwedder, W.K.; Adlof, R.O.; Rakoff, H.; Gulley, R.M. Metabolism in humans ofcis-12,rans-15-octadecadienoic acid relative to palmitic, stearic, oleic and linoleic acids. Lipids 1987, 22, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Indu, M. Ghafoorunissa. n-3 fatty acids in Indian diets—Comparison of the effects of precursor (alpha-linolenic acid) Vs product (long chain n-3 poly unsaturated fatty acids). Nutr. Res. 1992, 12, 569–582. [Google Scholar] [CrossRef]
- Yam, D.; Berry, E.M.; Berry, E.M. Diet and disease--the Israeli paradox: Possible dangers of a high omega-6 polyunsaturated fatty acid diet. Isr. J. Med. Sci. 1996, 32, 1134–1143. [Google Scholar] [PubMed]
- Zhao, Z.W.; Zhang, M.; Zou, J.; Wan, X.J.; Zhou, L.; Wu, Y.; Liu, S.M.; Liao, L.X.; Li, H.; Qin, Y.S.; et al. TIGAR mitigates atherosclerosis by promoting cholesterol efflux from macrophages. Atherosclerosis 2021, 327, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.S.; Mozaffarian, D.; Rimm, E.; Kris-Etherton, P.; Rudel, L.L.; Appel, L.J.; Engler, M.M.; Engler, M.B.; Sacks, F. Omega-6 fatty acids and risk for cardiovascular disease: A science advisory from the American Heart Association Nutrition Subcommittee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Cardiovascular Nursing; and Council on Epidemiology and Prevention. Circulation 2009, 119, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Ahn, H.; Park, Y.K. High Dietary Fructose Intake on Cardiovascular Disease Related Parameters in Growing Rats. Nutrients 2016, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Despres, J.P.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: A meta-analysis. Diabetes Care 2010, 33, 2477–2483. [Google Scholar] [CrossRef] [PubMed]
- Kritchevsky, D. The effects of feeding various carbohydrates on the development of hypercholesterolemia and atherosclerosis. Adv. Exp. Med. Biol. 1975, 60, 231–249. [Google Scholar] [CrossRef]
- Gugliucci, A.; Lustig, R.H.; Caccavello, R.; Erkin-Cakmak, A.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Mulligan, K.; Schwarz, J.M. Short-term isocaloric fructose restriction lowers apoC-III levels and yields less atherogenic lipoprotein profiles in children with obesity and metabolic syndrome. Atherosclerosis 2016, 253, 171–177. [Google Scholar] [CrossRef]
- Tokita, Y.; Hirayama, Y.; Sekikawa, A.; Kotake, H.; Toyota, T.; Miyazawa, T.; Sawai, T.; Oikawa, S. Fructose Ingestion Enhances Atherosclerosis and Deposition of Advanced Glycated End-products in Cholesterol-fed Rabbits. J. Atheroscler. Thromb. 2005, 12, 260–267. [Google Scholar] [CrossRef]
- Chaurasia, B.; Tippetts, T.S.; Mayoral Monibas, R.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 2019, 365, 386–392. [Google Scholar] [CrossRef]
- Olson, E.; Suh, J.H.; Schwarz, J.M.; Noworolski, S.M.; Jones, G.M.; Barber, J.R.; Erkin-Cakmak, A.; Mulligan, K.; Lustig, R.H.; Mietus-Snyder, M. Effects of Isocaloric Fructose Restriction on Ceramide Levels in Children with Obesity and Cardiometabolic Risk: Relation to Hepatic De Novo Lipogenesis and Insulin Sensitivity. Nutrients 2022, 14, 1432. [Google Scholar] [CrossRef]
- Lian, Y.G.; Zhao, H.Y.; Wang, S.J.; Xu, Q.L.; Xia, X.J. NLRP4 is an essential negative regulator of fructose-induced cardiac injury in vitro and in vivo. Biomed. Pharmacother. 2017, 91, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, Z.; Chang, R.; Zeng, C.; Zhao, Y. High-Fructose Diet Induces Cardiac Dysfunction via Macrophage Recruitment in Adult Mice. J. Cardiovasc. Pharmacol. Ther. 2023, 28, 10742484231162249. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, K.; Senador, D.D.; Mostarda, C.; Irigoyen, M.C.; Morris, M. Sympathetic overactivity precedes metabolic dysfunction in a fructose model of glucose intolerance in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R950–R957. [Google Scholar] [CrossRef] [PubMed]
- Cannizzo, B.; Lujan, A.; Estrella, N.; Lembo, C.; Cruzado, M.; Castro, C. Insulin resistance promotes early atherosclerosis via increased proinflammatory proteins and oxidative stress in fructose-fed ApoE-KO mice. Exp. Diabetes Res. 2012, 2012, 941304. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef]
- Utermann, G.; Hees, M.; Steinmetz, A. Polymorphism of apolipoprotein E and occurrence of dysbetalipoproteinaemia in man. Nature 1977, 269, 604–607. [Google Scholar] [CrossRef]
- Carrasquilla, G.D.; Christiansen, M.R.; Kilpelainen, T.O. The Genetic Basis of Hypertriglyceridemia. Curr. Atheroscler. Rep. 2021, 23, 39. [Google Scholar] [CrossRef]
- Boren, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Zhang, D.W.; Lagace, T.A.; Garuti, R.; Zhao, Z.; McDonald, M.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 2007, 282, 18602–18612. [Google Scholar] [CrossRef]
- Meng, F.H.; Liu, S.; Xiao, J.; Zhou, Y.X.; Dong, L.W.; Li, Y.F.; Zhang, Y.Q.; Li, W.H.; Wang, J.Q.; Wang, Y.; et al. New Loss-of-Function Mutations in PCSK9 Reduce Plasma LDL Cholesterol. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Orringer, C.E.; Jacobson, T.A.; Saseen, J.J.; Brown, A.S.; Gotto, A.M.; Ross, J.L.; Underberg, J.A. Update on the use of PCSK9 inhibitors in adults: Recommendations from an Expert Panel of the National Lipid Association. J. Clin. Lipidol. 2017, 11, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Wiebe, N.; Culleton, B.; House, A.; Rabbat, C.; Fok, M.; McAlister, F.; Garg, A.X. Chronic kidney disease and mortality risk: A systematic review. J. Am. Soc. Nephrol. 2006, 17, 2034–2047. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, M.; Li, Y.; Li, M.; Sieber, K.B.; Feitosa, M.F.; Gorski, M.; Tin, A.; Wang, L.; Chu, A.Y.; Hoppmann, A.; et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 2019, 51, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, J.C.; Ng, K.F.; Aung, H.H.; Wilson, D.W. Role of triglyceride-rich lipoproteins in diabetic nephropathy. Nat. Rev. Nephrol. 2010, 6, 361–370. [Google Scholar] [CrossRef]
- van Herpen, N.A.; Schrauwen-Hinderling, V.B. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 2008, 94, 231–241. [Google Scholar] [CrossRef]
- Rada, P.; Gonzalez-Rodriguez, A.; Garcia-Monzon, C.; Valverde, A.M. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef]
- Moorhead, J.F.; Chan, M.K.; El-Nahas, M.; Varghese, Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet 1982, 2, 1309–1311. [Google Scholar] [CrossRef]
- Sharma, M.; Singh, V.; Sharma, R.; Koul, A.; McCarthy, E.T.; Savin, V.J.; Joshi, T.; Srivastava, T. Glomerular Biomechanical Stress and Lipid Mediators during Cellular Changes Leading to Chronic Kidney Disease. Biomedicines 2022, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Badr, K.F.; Lakkis, F.G. Lipoxygenase products in normal and diseased glomeruli. Ann. N. Y. Acad. Sci. 1994, 744, 216–228. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.T.; Sharma, R.; Sharma, M. Protective effect of 20-hydroxyeicosatetraenoic acid (20-HETE) on glomerular protein permeability barrier. Kidney Int. 2005, 67, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; McCarthy, E.T.; Reddy, D.S.; Patel, P.K.; Savin, V.J.; Medhora, M.; Falck, J.R. 8,9-Epoxyeicosatrienoic acid protects the glomerular filtration barrier. Prostaglandins Other Lipid Mediat. 2009, 89, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Bobulescu, I.A. Renal lipid metabolism and lipotoxicity. Curr. Opin. Nephrol. Hypertens. 2010, 19, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.E.; Chen, Y.; Huang, A.; Varghese, Z.; Moorhead, J.F.; Yan, F.; Powis, S.H.; Li, Q.; Ruan, X.Z. Inflammatory stress exacerbates lipid-mediated renal injury in ApoE/CD36/SRA triple knockout mice. Am. J. Physiol. Renal Physiol. 2011, 301, F713–F722. [Google Scholar] [CrossRef]
- Rinaldi, A.; Lazareth, H.; Poindessous, V.; Nemazanyy, I.; Sampaio, J.L.; Malpetti, D.; Bignon, Y.; Naesens, M.; Rabant, M.; Anglicheau, D.; et al. Impaired fatty acid metabolism perpetuates lipotoxicity along the transition to chronic kidney injury. JCI Insight 2022, 7, e161783. [Google Scholar] [CrossRef]
- Hansell, P.; Welch, W.J.; Blantz, R.C.; Palm, F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 2013, 40, 123–137. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Hoeks, J.; Hesselink, M.K.; Russell, A.P.; Mensink, M.; Saris, W.H.; Mensink, R.P.; Schrauwen, P. Peroxisome proliferator-activated receptor-gamma coactivator-1 and insulin resistance: Acute effect of fatty acids. Diabetologia 2006, 49, 2419–2426. [Google Scholar] [CrossRef]
- Koyama, T.; Kume, S.; Koya, D.; Araki, S.; Isshiki, K.; Chin-Kanasaki, M.; Sugimoto, T.; Haneda, M.; Sugaya, T.; Kashiwagi, A.; et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic. Biol. Med. 2011, 51, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T.; Wang, M.Y.; Rutkowski, J.M.; Deja, S.; Chen, S.; Balzer, M.S.; Kim, D.S.; Sun, X.; An, Y.A.; Field, B.C.; et al. Endogenous renal adiponectin drives gluconeogenesis through enhancing pyruvate and fatty acid utilization. Nat. Commun. 2023, 14, 6531. [Google Scholar] [CrossRef] [PubMed]
- Brinkkoetter, P.T.; Bork, T.; Salou, S.; Liang, W.; Mizi, A.; Ozel, C.; Koehler, S.; Hagmann, H.H.; Ising, C.; Kuczkowski, A.; et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell Rep. 2019, 27, 1551–1566 e1555. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, J.; Lee, H.S. Palmitate induces mitochondrial superoxide generation and activates AMPK in podocytes. J. Cell Physiol. 2017, 232, 3209–3217. [Google Scholar] [CrossRef]
- Xu, S.; Nam, S.M.; Kim, J.H.; Das, R.; Choi, S.K.; Nguyen, T.T.; Quan, X.; Choi, S.J.; Chung, C.H.; Lee, E.Y.; et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015, 6, e1976. [Google Scholar] [CrossRef] [PubMed]
- Lennon, R.; Pons, D.; Sabin, M.A.; Wei, C.; Shield, J.P.; Coward, R.J.; Tavare, J.M.; Mathieson, P.W.; Saleem, M.A.; Welsh, G.I. Saturated fatty acids induce insulin resistance in human podocytes: Implications for diabetic nephropathy. Nephrol. Dial. Transplant. 2009, 24, 3288–3296. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P. Nutrients and Oxidative Stress: Friend or Foe? Oxid. Med. Cell Longev. 2018, 2018, 9719584. [Google Scholar] [CrossRef]
- Neurohr, J.M.; Paulson, E.T.; Kinsey, S.T. A higher mitochondrial content is associated with greater oxidative damage, oxidative defenses, protein synthesis and ATP turnover in resting skeletal muscle. J. Exp. Biol. 2021, 224, jeb242462. [Google Scholar] [CrossRef]
- Opazo-Rios, L.; Mas, S.; Marin-Royo, G.; Mezzano, S.; Gomez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 3632. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Ishimoto, T.; Cicerchi, C.; Tamura, Y.; Roncal-Jimenez, C.A.; Chen, W.; Tanabe, K.; Andres-Hernando, A.; Orlicky, D.J.; Finol, E.; et al. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 2526–2538. [Google Scholar] [CrossRef]
- Hu, Z.; Ren, L.; Wang, C.; Liu, B.; Song, G. Effect of chenodeoxycholic acid on fibrosis, inflammation and oxidative stress in kidney in high-fructose-fed Wistar rats. Kidney Blood Press. Res. 2012, 36, 85–97. [Google Scholar] [CrossRef]
- Li, Q.; Xu, Q.; Tan, J.; Hu, L.; Ge, C.; Xu, M. Carminic acid supplementation protects against fructose-induced kidney injury mainly through suppressing inflammation and oxidative stress via improving Nrf-2 signaling. Aging 2021, 13, 10326–10353. [Google Scholar] [CrossRef] [PubMed]
- Gherghina, M.E.; Peride, I.; Tiglis, M.; Neagu, T.P.; Niculae, A.; Checherita, I.A. Uric Acid and Oxidative Stress-Relationship with Cardiovascular, Metabolic, and Renal Impairment. Int. J. Mol. Sci. 2022, 23, 3188. [Google Scholar] [CrossRef] [PubMed]
- Roncal, C.A.; Mu, W.; Croker, B.; Reungjui, S.; Ouyang, X.; Tabah-Fisch, I.; Johnson, R.J.; Ejaz, A.A. Effect of elevated serum uric acid on cisplatin-induced acute renal failure. Am. J. Physiol. Renal Physiol. 2007, 292, F116–F122. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Perkins, B.A.; Cherney, D.Z. Uric acid as a biomarker and a therapeutic target in diabetes. Can. J. Diabetes 2015, 39, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Abuja, P.M. Ascorbate prevents prooxidant effects of urate in oxidation of human low density lipoprotein. FEBS Lett. 1999, 446, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Zharikov, S.; Krotova, K.; Hu, H.; Baylis, C.; Johnson, R.J.; Block, E.R.; Patel, J. Uric acid decreases NO production and increases arginase activity in cultured pulmonary artery endothelial cells. Am. J. Physiol. Cell Physiol. 2008, 295, C1183–C1190. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef]
- Nakagawa, T.; Johnson, R.J.; Andres-Hernando, A.; Roncal-Jimenez, C.; Sanchez-Lozada, L.G.; Tolan, D.R.; Lanaspa, M.A. Fructose Production and Metabolism in the Kidney. J. Am. Soc. Nephrol. 2020, 31, 898–906. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J.; et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Gansevoort, R.T.; Matsushita, K.; van der Velde, M.; Astor, B.C.; Woodward, M.; Levey, A.S.; de Jong, P.E.; Coresh, J.; Chronic Kidney Disease Prognosis, C. Lower estimated GFR and higher albuminuria are associated with adverse kidney outcomes. A collaborative meta-analysis of general and high-risk population cohorts. Kidney Int. 2011, 80, 93–104. [Google Scholar] [CrossRef]
- Astor, B.C.; Matsushita, K.; Gansevoort, R.T.; van der Velde, M.; Woodward, M.; Levey, A.S.; Jong, P.E.; Coresh, J.; Chronic Kidney Disease Prognosis, C.; Astor, B.C.; et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney Int. 2011, 79, 1331–1340. [Google Scholar] [CrossRef]
- Liu, W.J.; Xu, B.H.; Ye, L.; Liang, D.; Wu, H.L.; Zheng, Y.Y.; Deng, J.K.; Li, B.; Liu, H.F. Urinary proteins induce lysosomal membrane permeabilization and lysosomal dysfunction in renal tubular epithelial cells. Am. J. Physiol. Renal Physiol. 2015, 308, F639–F649. [Google Scholar] [CrossRef]
- Nolin, A.C.; Mulhern, R.M.; Panchenko, M.V.; Pisarek-Horowitz, A.; Wang, Z.; Shirihai, O.; Borkan, S.C.; Havasi, A. Proteinuria causes dysfunctional autophagy in the proximal tubule. Am. J. Physiol. Renal Physiol. 2016, 311, F1271–F1279. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Abbate, M.; Zoja, C.; Remuzzi, G. How does proteinuria cause progressive renal damage? J. Am. Soc. Nephrol. 2006, 17, 2974–2984. [Google Scholar] [CrossRef] [PubMed]
- Westenfelder, C.; Gooch, A. Heme Protein-Induced Acute Kidney Injury Is Caused by Disruption of Mitochondrial Homeostasis in Proximal Tubular Cells. Kidney360 2022, 3, 2140–2142. [Google Scholar] [CrossRef]
- Zager, R.A. Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney Int. 1996, 49, 314–326. [Google Scholar] [CrossRef]
- Nath, K.A.; Singh, R.D.; Croatt, A.J.; Adams, C.M. Heme Proteins and Kidney Injury: Beyond Rhabdomyolysis. Kidney360 2022, 3, 1969–1979. [Google Scholar] [CrossRef]
- Deng, J.K.; Zhang, X.; Wu, H.L.; Gan, Y.; Ye, L.; Zheng, H.; Zhu, Z.; Liu, W.J.; Liu, H.F. ROS-ERK Pathway as Dual Mediators of Cellular Injury and Autophagy-Associated Adaptive Response in Urinary Protein-Irritated Renal Tubular Epithelial Cells. J. Diabetes Res. 2021, 2021, 6614848. [Google Scholar] [CrossRef]
- Kao, W.H.; Klag, M.J.; Meoni, L.A.; Reich, D.; Berthier-Schaad, Y.; Li, M.; Coresh, J.; Patterson, N.; Tandon, A.; Powe, N.R.; et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat. Genet. 2008, 40, 1185–1192. [Google Scholar] [CrossRef]
- Kottgen, A.; Glazer, N.L.; Dehghan, A.; Hwang, S.J.; Katz, R.; Li, M.; Yang, Q.; Gudnason, V.; Launer, L.J.; Harris, T.B.; et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet. 2009, 41, 712–717. [Google Scholar] [CrossRef]
- McKnight, A.J.; Currie, D.; Maxwell, A.P. Unravelling the genetic basis of renal diseases; from single gene to multifactorial disorders. J. Pathol. 2010, 220, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Marzuillo, P.; Di Sessa, A.; Guarino, S.; Capalbo, D.; Umano, G.R.; Pedulla, M.; La Manna, A.; Cirillo, G.; Miraglia Del Giudice, E. Nonalcoholic fatty liver disease and eGFR levels could be linked by the PNPLA3 I148M polymorphism in children with obesity. Pediatr. Obes. 2019, 14, e12539. [Google Scholar] [CrossRef] [PubMed]
- Di Sessa, A.; Russo, M.C.; Arienzo, M.R.; Umano, G.R.; Cozzolino, D.; Cirillo, G.; Guarino, S.; Miraglia Del Giudice, E.; Marzuillo, P. PNPLA3 I148M Polymorphism Influences Renal Function in Children With Obesity and Prediabetes. J. Ren. Nutr. 2022, 32, 670–676. [Google Scholar] [CrossRef]
- Verma, S.B.; Mittal, A.; Wollina, U.; Eckstein, G.H.; Gohel, K.; Giehl, K. Chanarin-Dorfman syndrome with rare renal involvement. Br. J. Dermatol. 2017, 176, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Zusi, C.; Sani, E.; Colecchia, A.; Lippi, G.; Zaza, G.L.; Valenti, L.; Byrne, C.D.; Maffeis, C.; Bonora, E.; et al. Association between PNPLA3rs738409 polymorphism decreased kidney function in postmenopausal type 2 diabetic women with or without non-alcoholic fatty liver disease. Diabetes Metab. 2019, 45, 480–487. [Google Scholar] [CrossRef]
- Zhao, J.; Rui, H.L.; Yang, M.; Sun, L.J.; Dong, H.R.; Cheng, H. CD36-Mediated Lipid Accumulation and Activation of NLRP3 Inflammasome Lead to Podocyte Injury in Obesity-Related Glomerulopathy. Mediators Inflamm. 2019, 2019, 3172647. [Google Scholar] [CrossRef]
- Koo, B.K.; An, J.N.; Joo, S.K.; Kim, D.; Lee, S.; Bae, J.M.; Park, J.H.; Kim, J.H.; Chang, M.S.; Kim, W. Association Between a Polymorphism in MBOAT7 and Chronic Kidney Disease in Patients With Biopsy-Confirmed Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2020, 18, 2837–2839.e2832. [Google Scholar] [CrossRef]
- Varadharajan, V.; Massey, W.J.; Brown, J.M. Membrane-bound O-acyltransferase 7 (MBOAT7)-driven phosphatidylinositol remodeling in advanced liver disease. J. Lipid Res. 2022, 63, 100234. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Gambino, R. PNPLA3 rs738409 and TM6SF2 rs58542926 gene variants affect renal disease and function in nonalcoholic fatty liver disease. Hepatology 2015, 62, 658–659. [Google Scholar] [CrossRef] [PubMed]
- Di Sessa, A.; Guarino, S.; Umano, G.R.; Arenella, M.; Alfiero, S.; Quaranta, G.; Miraglia Del Giudice, E.; Marzuillo, P. MAFLD in Obese Children: A Challenging Definition. Children 2021, 8, 247. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, S.; Zhao, H.; Yu, M.; Deng, X.; Jiang, Y.; Cao, Y.; Li, P.; Niu, W. Susceptibility of ApoB and PCSK9 Genetic Polymorphisms to Diabetic Kidney Disease Among Chinese Diabetic Patients. Front. Med. 2021, 8, 659188. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, B.; Lin, L.; Lei, F.; Sun, T.; Zhang, X.; Song, X.; Huang, X.; Zeng, Q.; Cai, J.; et al. The association of apolipoprotein B with chronic kidney disease in the Chinese population. Front. Endocrinol. 2023, 14, 1083614. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, F.; Nordestgaard, B.G.; Benn, M. Familial Hypercholesterolemia and Risk of Peripheral Arterial Disease and Chronic Kidney Disease. J. Clin. Endocrinol. Metab. 2018, 103, 4491–4500. [Google Scholar] [CrossRef]
- Vaseghi, G.; Javanmard, S.H.; Heshmat-Ghahdarijani, K.; Sarrafzadegan, N.; Amerizadeh, A. Comorbidities with Familial Hypercholesterolemia (FH): A Systematic Review. Curr. Probl. Cardiol. 2023, 48, 101109. [Google Scholar] [CrossRef]
- Barbagallo, C.M.; Cefalu, A.B.; Giammanco, A.; Noto, D.; Caldarella, R.; Ciaccio, M.; Averna, M.R.; Nardi, E. Lipoprotein Abnormalities in Chronic Kidney Disease and Renal Transplantation. Life 2021, 11, 315. [Google Scholar] [CrossRef]
- Ge, S.; Hertel, B.; Koltsova, E.K.; Sorensen-Zender, I.; Kielstein, J.T.; Ley, K.; Haller, H.; von Vietinghoff, S. Increased atherosclerotic lesion formation and vascular leukocyte accumulation in renal impairment are mediated by interleukin-17A. Circ. Res. 2013, 113, 965–974. [Google Scholar] [CrossRef]
- Targher, G.; Chonchol, M.B.; Byrne, C.D. CKD and nonalcoholic fatty liver disease. Am. J. Kidney Dis. 2014, 64, 638–652. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Tabibian, J.H.; Ekstedt, M.; Kechagias, S.; Hamaguchi, M.; Hultcrantz, R.; Hagström, H.; Yoon, S.K.; Charatcharoenwitthaya, P.; et al. Association of Non-alcoholic Fatty Liver Disease with Chronic Kidney Disease: A Systematic Review and Meta-analysis. PLoS Med. 2014, 11, e12539. [Google Scholar] [CrossRef]
- Targher, G.; Mantovani, A.; Alisi, A.; Mosca, A.; Panera, N.; Byrne, C.D.; Nobili, V. Relationship Between PNPLA3 rs738409 Polymorphism and Decreased Kidney Function in Children with NAFLD. Hepatology 2019, 70, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.H.; Kim, J.Y.; Choi, E.; Kim, J.S.; Chang, Y.; Sung, K.C. The fatty liver index as a predictor of incident chronic kidney disease in a 10-year prospective cohort study. PLoS ONE 2017, 12, e0180951. [Google Scholar] [CrossRef]
- Jang, H.R.; Kang, D.; Sinn, D.H.; Gu, S.; Cho, S.J.; Lee, J.E.; Huh, W.; Paik, S.W.; Ryu, S.; Chang, Y.; et al. Nonalcoholic fatty liver disease accelerates kidney function decline in patients with chronic kidney disease: A cohort study. Sci. Rep. 2018, 8, 4718. [Google Scholar] [CrossRef] [PubMed]
- Onnerhag, K.; Dreja, K.; Nilsson, P.M.; Lindgren, S. Increased mortality in non-alcoholic fatty liver disease with chronic kidney disease is explained by metabolic comorbidities. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Dawwas, G.K.; Liu, X.; Nguyen, M.H. Nonalcoholic fatty liver disease increases risk of incident advanced chronic kidney disease: A propensity-matched cohort study. J. Intern. Med. 2019, 286, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Zaza, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Bonora, E.; Targher, G. Nonalcoholic fatty liver disease increases risk of incident chronic kidney disease: A systematic review and meta-analysis. Metabolism 2018, 79, 64–76. [Google Scholar] [CrossRef]
- Marcuccilli, M.; Chonchol, M. NAFLD and Chronic Kidney Disease. Int. J. Mol. Sci. 2016, 17, 562. [Google Scholar] [CrossRef]
- Massiera, F.; Bloch-Faure, M.; Ceiler, D.; Murakami, K.; Fukamizu, A.; Gasc, J.M.; Quignard-Boulange, A.; Negrel, R.; Ailhaud, G.; Seydoux, J.; et al. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J. 2001, 15, 2727–2729. [Google Scholar] [CrossRef]
- Frederich, R.; Kahn, B.; Peach, M.; Flier, J. Tissue-specific nutritional regulation of angiotensinogen in adipose tissue. Hypertension 1992, 19, 339–344. [Google Scholar] [CrossRef]
- Pahlavani, M.; Kalupahana, N.S.; Ramalingam, L.; Moustaid-Moussa, N. Regulation and Functions of the Renin-Angiotensin System in White and Brown Adipose Tissue. Compr. Physiol. 2017, 7, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.B.; Pagadala, M.R.; Dasarathy, J.; Unalp-Arida, A.; Sargent, R.; Hawkins, C.; Sourianarayanane, A.; Khiyami, A.; Yerian, L.; Pai, R.; et al. Renin-angiotensin system and fibrosis in non-alcoholic fatty liver disease. Liver Int. 2015, 35, 979–985. [Google Scholar] [CrossRef] [PubMed]
- de Vries, A.P.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Pongrac Barlovic, D.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.E.; Ford, J.L.; Boston, R.C.; Savinova, O.V.; Harris, W.S.; Green, M.H.; Shearer, G.C. Trafficking of nonesterified fatty acids in insulin resistance and relationship to dysglycemia. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E392–E404. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D. Non-alcoholic fatty liver disease: An emerging driving force in chronic kidney disease. Nat. Rev. Nephrol. 2017, 13, 297–310. [Google Scholar] [CrossRef]
- Verweij, N.; Haas, M.E.; Nielsen, J.B.; Sosina, O.A.; Kim, M.; Akbari, P.; De, T.; Hindy, G.; Bovijn, J.; Persaud, T.; et al. Germline Mutations in CIDEB and Protection against Liver Disease. N. Engl. J. Med. 2022, 387, 332–344. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Model | Subject (Gene/Nutrient) | Major Findings | Citation |
|---|---|---|---|
| In vitro | Fructose | Activates chREBP and DNL | [60] |
| PNPLA3 I148M | promotes MAFLD through reducing hepatic TAG hydrolysis; sequestration of ABHD5 | [93,100]; [104,114] | |
| TM6SF2 | Involved in secretion of hepatic TAGs | [117] | |
| MBOAT7 | depletion increases hepatic TAGs | [123] | |
| In vivo | Fructose | Provides lactate and acetate for DNL | [59] |
| Metabolites activate chREBP | [33] | ||
| Increases lipogenesis independent of ACLY | [62] | ||
| ATP-citrate Lyase (ACLY) | Inhibition reduces liver fat and ballooning; reduces blood glucose, TAGs and cholesterol | [63] | |
| High fat vs. high fructose diet | Dietary fat and cholesterol are primary drivers of MAFLD | [91] | |
| PNPLA3 | PNPLA3 deficiency does not promote hepatic steatosis; nor does overexpression, I148M is gain of function | [105,106]; [107] | |
| TM6SF2 | TM6SF2 is required for VLDL assembly | [118] | |
| MBOAT7 | Loss of MBOAT7 promotes MAFLD while overexpression improves | [122,124,125,127]; [126] | |
| Clinical | Fructose | High consumption of fructose associates with greater fibrosis | [67] |
| Mitochondrial activity | MAFLD reduces mitochondrial activity | [68] | |
| Fatty Acids | Hypocaloric diet low in fat harbors same benefits as hypocaloric diet low in carbs | [70] | |
| MUFA enriched diet reduces hepatic steatosis | [72] | ||
| n-3 PUFA supplementation improves MAFLD n-3 PUFA supplementation improves MAFLD | [73,80,81,128] [73,80,81,128] | ||
| HFD increases AA in phospholipid fraction of liver | [83] | ||
| N-6 supplementation reduced liver fat relative to high saturated fat diet | [84] | ||
| Amount of dietary fat influences liver fat content | [71] | ||
| Low fat diet reduced liver TAGs. No effect of HFD | [88] | ||
| Saturated fat is more metabolically harmful for liver | [90] | ||
| Dietary patterns in MAFLD patients | MAFLD patients consume diets rich in saturated fat | [86] | |
| MAFLD patients consume a diet rich in sat. fat and majority are deficient in PUFAs and MUFAs | [85] | ||
| TM6SF2 | rs58542926 promotes MAFLD progression | [115] | |
| PNPLA3 | Association with MAFLD, MASH, cirrhosis | [92,93,94,95,96,97,98,99] | |
| PNPLA3 I148M affects VLDL secretion | [136] |
| Model | Subject (Gene/Nutrient) | Major Findings | Citation |
|---|---|---|---|
| In vivo | PUFAs and CVD | PUFAs protect against CAD (non-human primates) | [150] |
| Fructose and CVD | Fructose consumption exerts negative effects on CV health | [177,179,180,181,184,185,186,187] | |
| Human studies | Dietary Fat and CAD in women | Saturated and trans fats increase risk of CAD | [143] |
| Clinical | PUFAs and CVD | Replacement of sat. fat with veg oil reduces risk of CHD | [153,155,156] |
| n-3 PUFAs negate adverse LV remodeling after MI | [161] | ||
| Meta-analysis | Dietary fat modulation and risk of CVD | No effect observed | [144,145,151] |
| Replacing Sat. fat with PUFAs lowers risk of CVD | [146,147,157,158] | ||
| Replace sat. fat with n-6 PUFA increases CVD death | [152] | ||
| n-3 PUFAs and endothelial function | n-3 supplementation improves endothelial function | [159] | |
| n-3/n-6 ratio and CVD risk | n-3/n-6 ratio important for CVD risk | [162,167,173,174,176] | |
| APOE; LDLr; PCSK9 | LDL promotes atherosclerosis and CVD | [189]; [192]; [194,195] | |
| GWAS | Lipid metabolism genes | Genetic variants influence risk of CVD | [141,189] |
| Model | Subject (Gene/Nutrient) | Major Findings | Citation |
|---|---|---|---|
| In vitro | Lipid-derived mediators | Exaggerates or protects against CKD depending on mediator and context | [204]; [205]; [207] |
| Palmitic acid | Increased mitochondrial ROS and decreased oxidative capacity in RPTEC; decreased cytosolic and mitochondrial ROS, ER stress, apoptosis, and insulin resistance in podocytes | [214]; [217]; [218]; [219] | |
| Albumin | Tubule apoptosis; decreased autophagosome number | [237]; [238]; [244] | |
| Urinary protein | Increases ROS-mediated activation of ERK, leading to tubule damage and apoptosis | ||
| In vivo | Lipid | Increased renal fibrosis | [209] |
| Lipid-derived mediators | Exaggerates or protects against CKD depending on mediator and context | [204]; [205]; [206]; | |
| Fructose | Increased renal damage | [223] | |
| Uric acid | Exacerbates tubule injury | [227] | |
| Meta-analyses | Albumin | Higher risk of CKD when increased in urine | [235]; [236] |
| Gene Mutation | MAFLD | CVD | CKD | Result of Mutation on Function |
|---|---|---|---|---|
| PNPLA3 rs738409 | Increase | Decrease | Increase | Gain of function /Neomorph |
| TM6SF2 | Increase | Decrease | Decrease | Loss of function |
| MBOAT7 | Increase | No effect | Increase | Loss of function |
| APOB | Increase | Increase | Loss of function | |
| LDLr | Increase | Increase | Unknown | Loss of function |
| PCSK9 | GOF = increase LOF = no effect | GOF = increase LOF = decrease | Unknown | Both GOF and LOF identified |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butcko, A.J.; Putman, A.K.; Mottillo, E.P. The Intersection of Genetic Factors, Aberrant Nutrient Metabolism and Oxidative Stress in the Progression of Cardiometabolic Disease. Antioxidants 2024, 13, 87. https://doi.org/10.3390/antiox13010087
Butcko AJ, Putman AK, Mottillo EP. The Intersection of Genetic Factors, Aberrant Nutrient Metabolism and Oxidative Stress in the Progression of Cardiometabolic Disease. Antioxidants. 2024; 13(1):87. https://doi.org/10.3390/antiox13010087
Chicago/Turabian StyleButcko, Andrew J., Ashley K. Putman, and Emilio P. Mottillo. 2024. "The Intersection of Genetic Factors, Aberrant Nutrient Metabolism and Oxidative Stress in the Progression of Cardiometabolic Disease" Antioxidants 13, no. 1: 87. https://doi.org/10.3390/antiox13010087