
AMPK-Dependent YAP Inhibition Mediates the Protective Effect of Metformin against Obesity-Associated Endothelial Dysfunction and Inflammation
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Metformin Treatment
2.3. Cell Culture and Treatment
2.4. RNA-seq and Microarray Analyses
2.5. Functional Assay by Wire Myograph
2.6. Ex Vivo Culture of Mouse Arteries
2.7. ROS Determination by Dihydroethidium (DHE) Staining
2.8. Western Blotting
2.9. Quantitative Real-Time PCR Analysis
2.10. Data Analysis and Statistics
3. Results
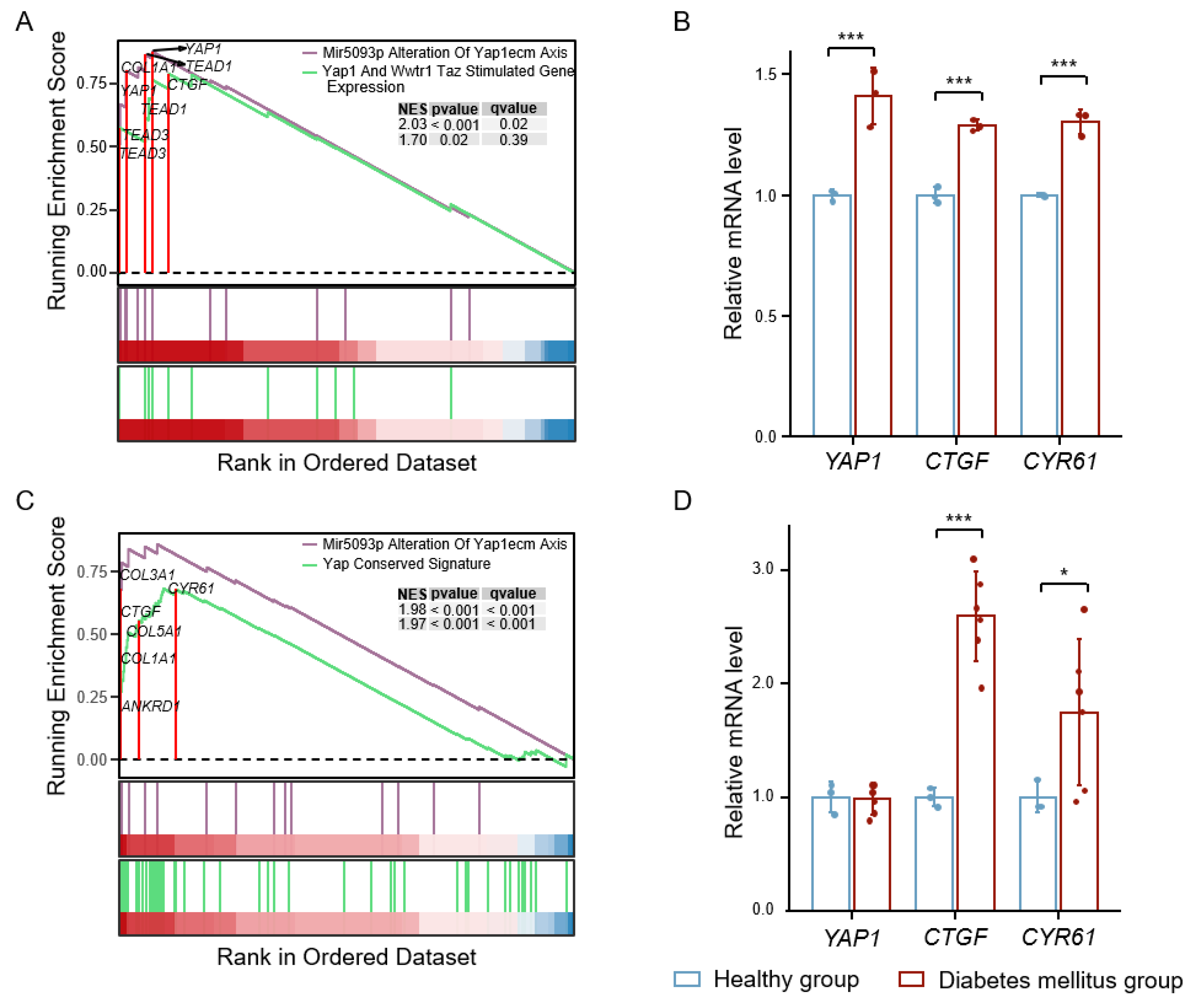
3.1. High Glucose-Induced YAP Activation Accelerates Vascular Injury
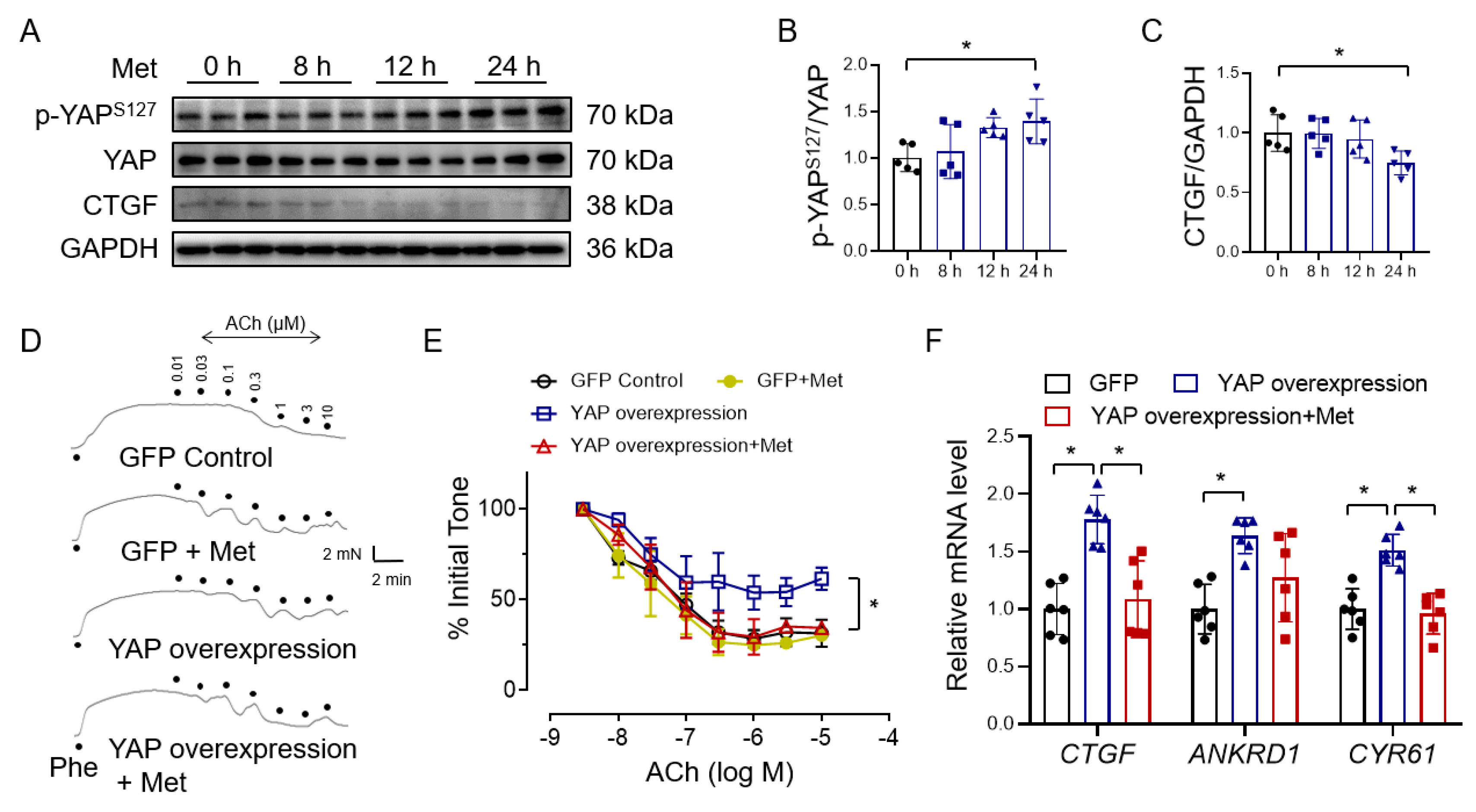
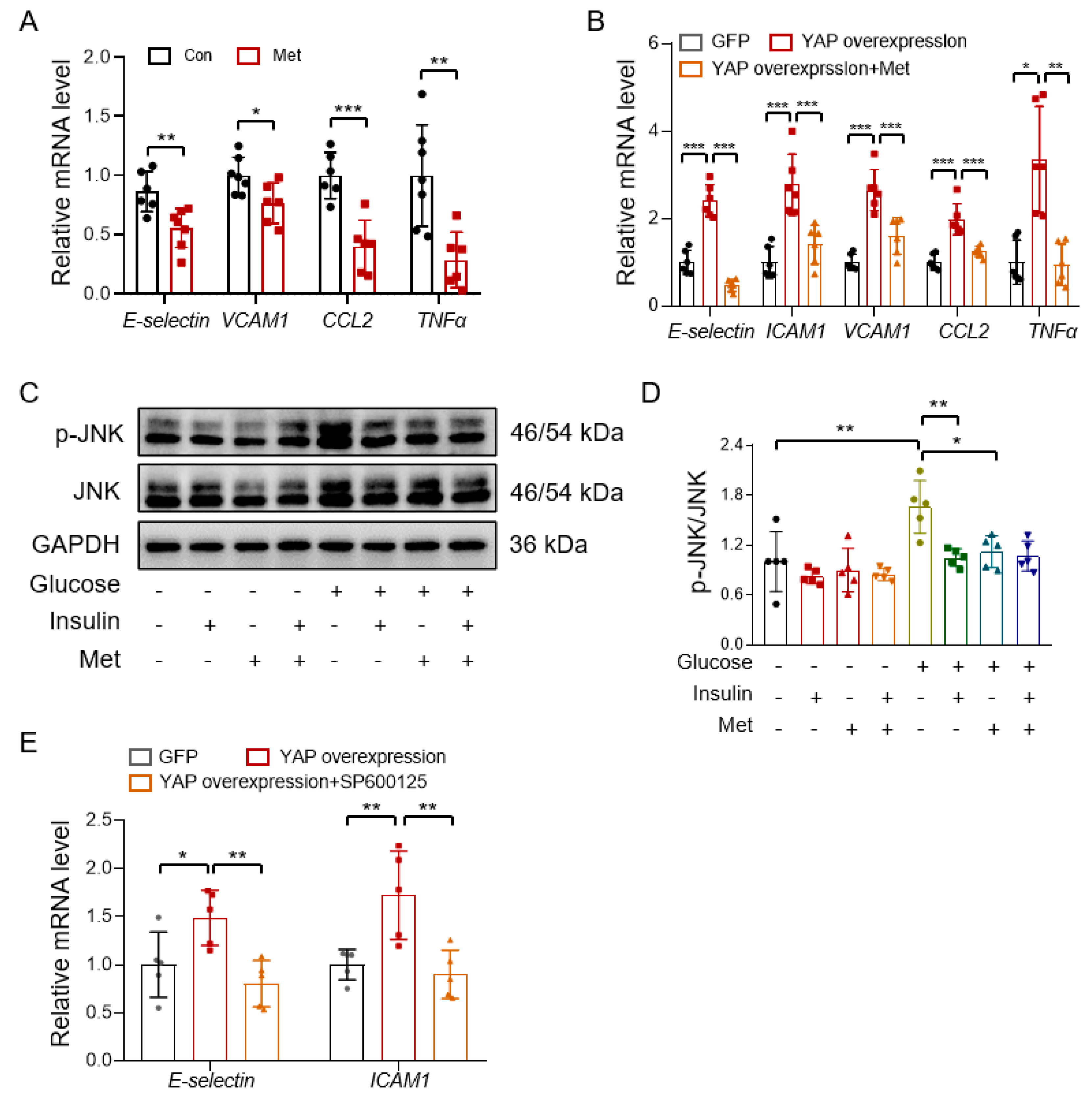
3.2. Metformin Improves Endothelial Function through Inhibiting YAP Signaling
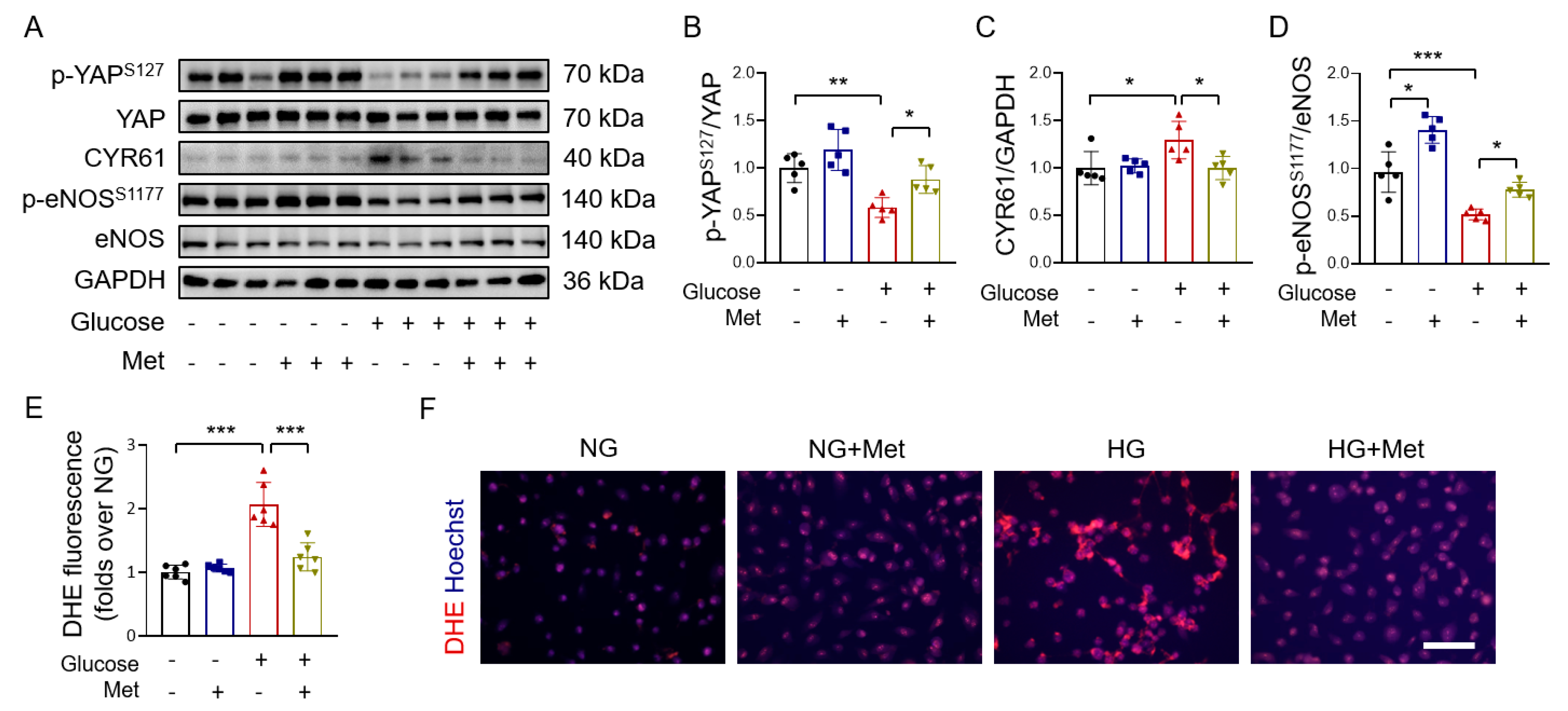
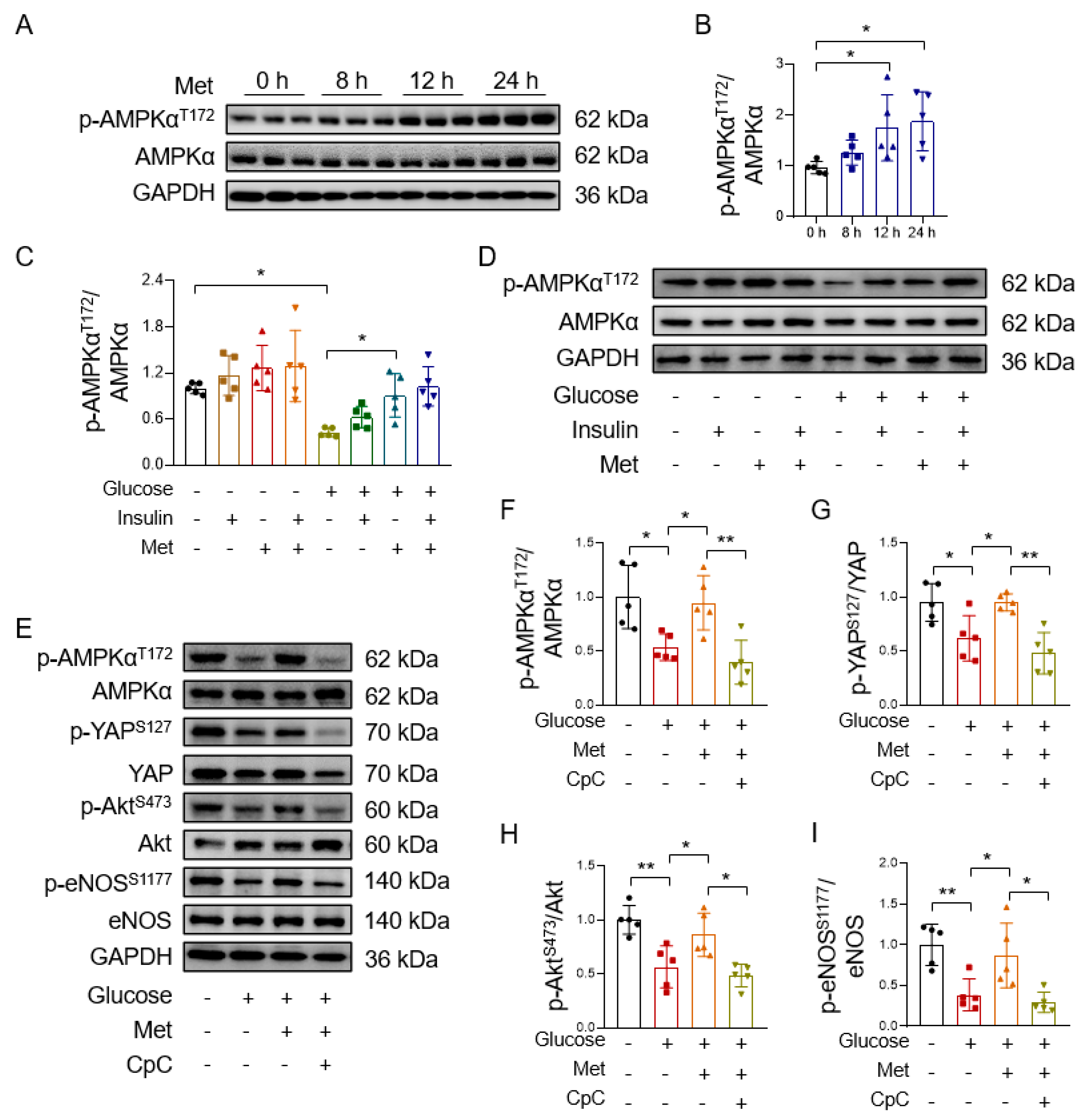
3.3. Metformin Reverses High Glucose-Induced YAP Activation
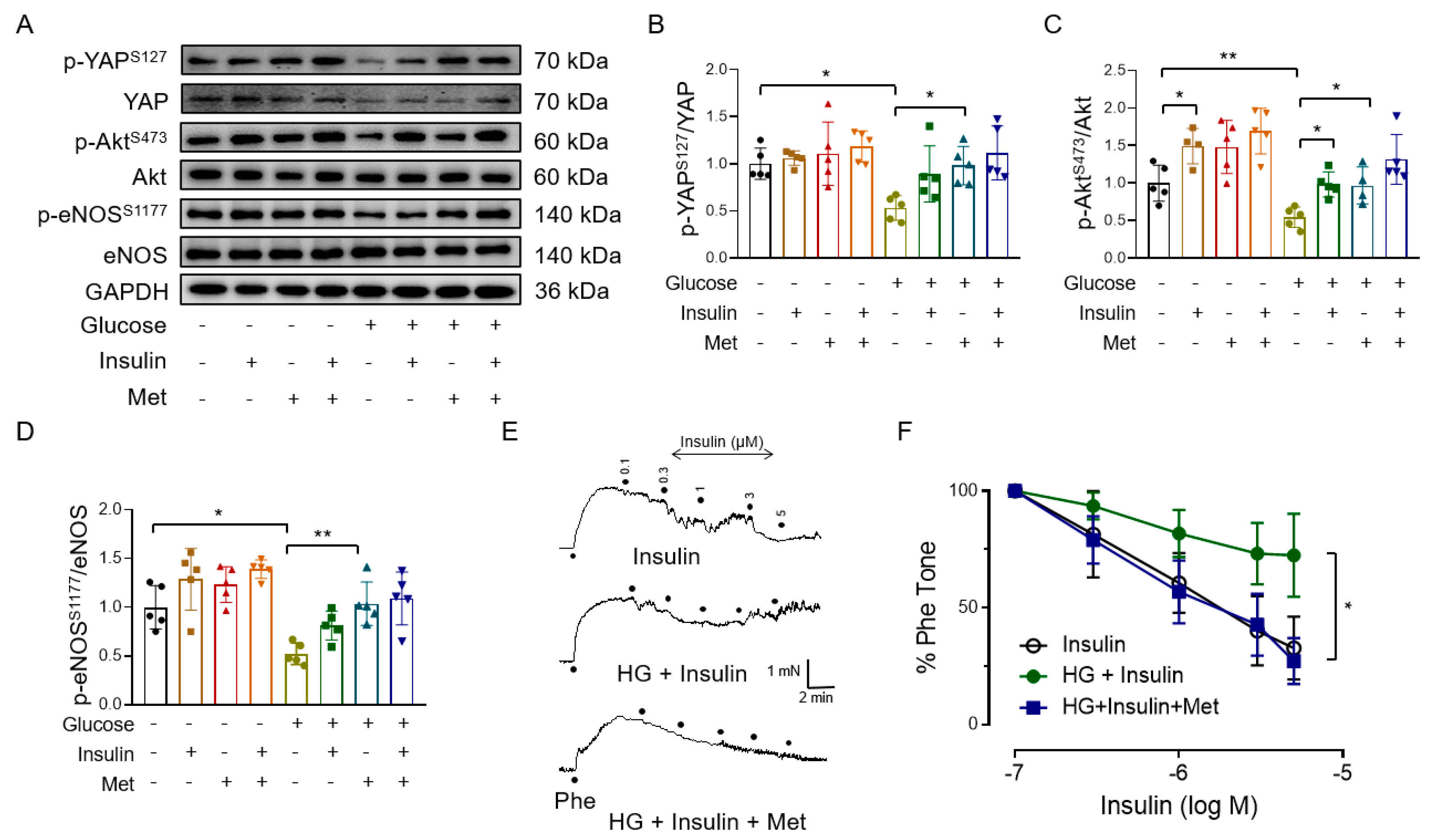
3.4. Metformin Improves Endothelial Function through AMPK-Mediated YAP Inhibition
3.5. Metformin Reduces Vascular Inflammation through Inhibiting the YAP-JNK Pathway
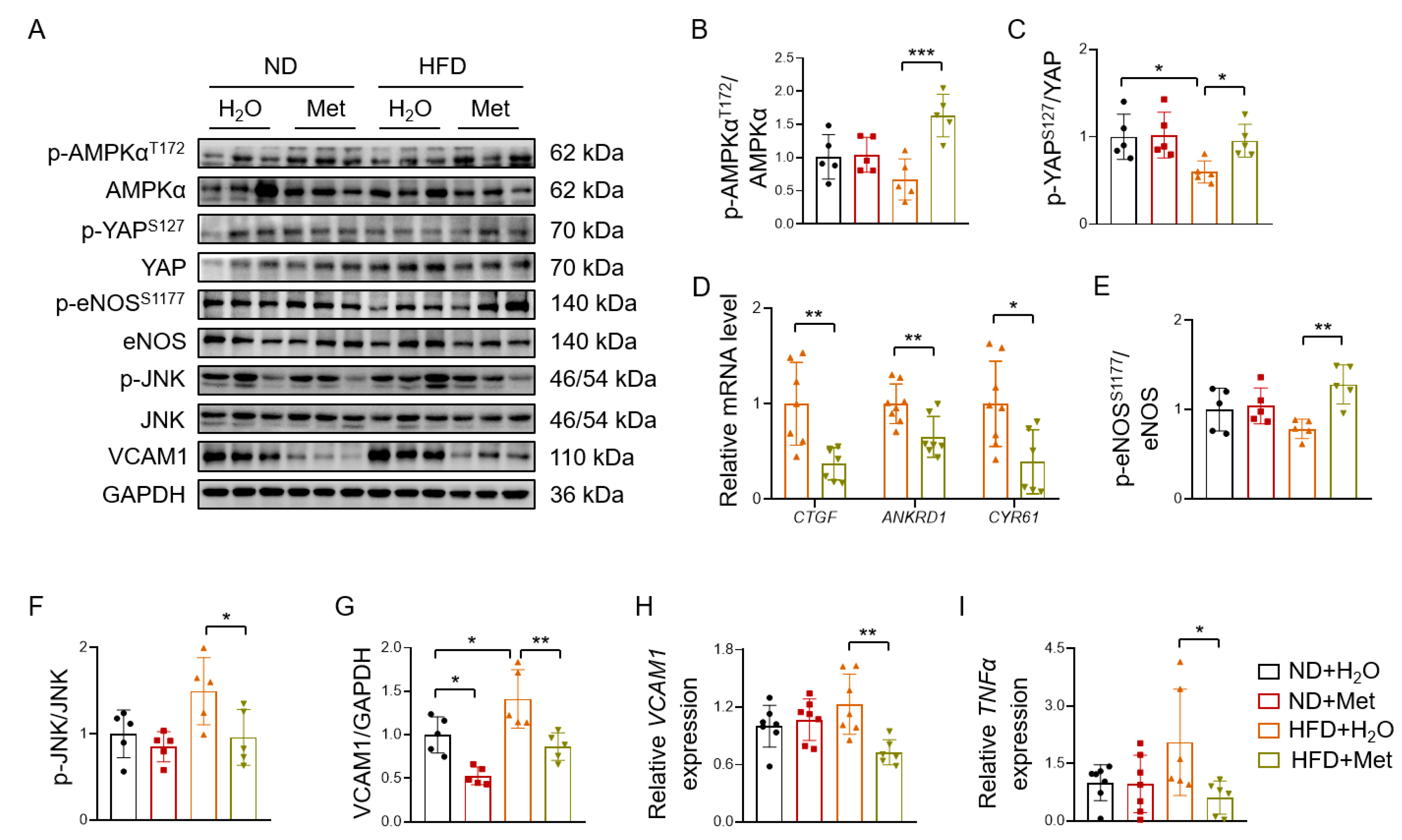
3.6. Metformin Reverses HFD-Induced YAP Activation and Vascular Inflammation
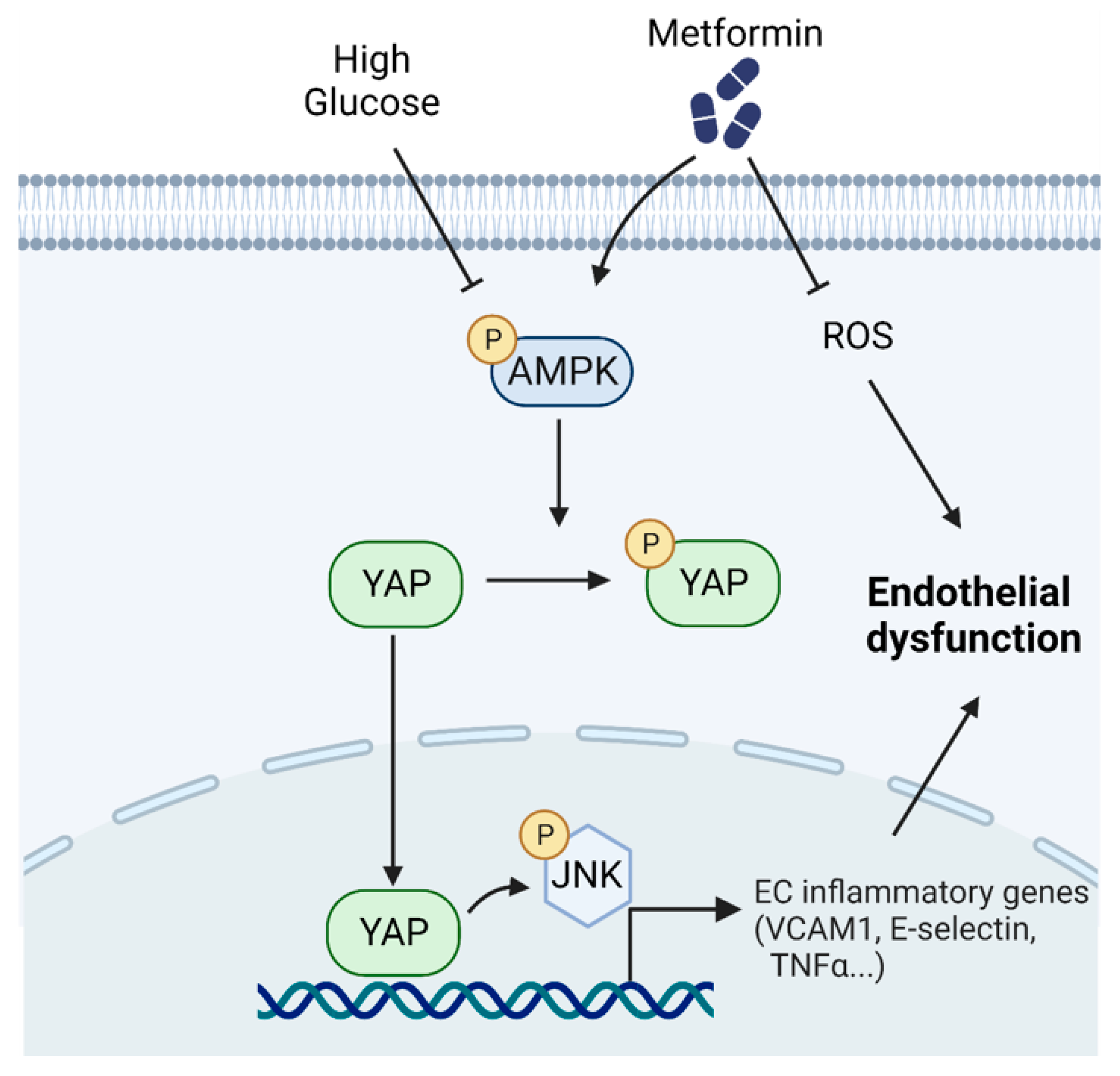
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cercato, C.; Fonseca, F.A. Cardiovascular risk and obesity. Diabetol. Metab. Syndr. 2019, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Luo, W.; Zhang, L.; Wu, W.; Yuan, L.; Xu, H.; Song, J.; Fujiwara, K.; Abe, J.-I.; Lemaire, S.A.; et al. STING–IRF3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Kwaifa, I.K.; Bahari, H.; Yong, Y.K.; Noor, S.M. Endothelial Dysfunction in Obesity-Induced Inflammation: Molecular Mechanisms and Clinical Implications. Biomolecules 2020, 10, 291. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P. Endothelium, inflammation, and diabetes. Curr. Diabetes Rep. 2002, 2, 311–315. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Y.; Lam, K.S.L.; Li, Y.; Wong, W.T.; Ye, H.; Lau, C.-W.; Vanhoutte, P.M.; Xu, A. Berberine prevents hyperglycemia-induced endothelial injury and enhances vasodilatation via adenosine monophosphate-activated protein kinase and endothelial nitric oxide synthase. Cardiovasc. Res. 2009, 82, 484–492. [Google Scholar] [CrossRef]
- Liu, F.; Fang, S.; Liu, X.; Li, J.; Wang, X.; Cui, J.; Chen, T.; Li, Z.; Yang, F.; Tian, J.; et al. Omentin-1 protects against high glucose-induced endothelial dysfunction via the AMPK/PPARδ signaling pathway. Biochem. Pharmacol. 2020, 174, 113830. [Google Scholar] [CrossRef]
- Faridvand, Y.; Kazemzadeh, H.; Vahedian, V.; Mirzajanzadeh, P.; Nejabati, H.R.; Safaie, N.; Maroufi, N.F.; Pezeshkian, M.; Nouri, M.; Jodati, A. Dapagliflozin attenuates high glucose-induced endothelial cell apoptosis and inflammation through AMPK/SIRT1 activation. Clin. Exp. Pharmacol. Physiol. 2022, 49, 643–651. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Watt, K.I.; Turner, B.J.; Hagg, A.; Zhang, X.; Davey, J.R.; Qian, H.; Beyer, C.; Winbanks, C.E.; Harvey, K.F.; Gregorevic, P. The Hippo pathway effector YAP is a critical regulator of skeletal muscle fibre size. Nat. Commun. 2015, 6, 6048. [Google Scholar] [CrossRef]
- Wang, L.; Luo, J.-Y.; Li, B.; Tian, X.Y.; Chen, L.-J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef]
- Wang, K.-C.; Yeh, Y.-T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.-L.; Li, Y.-S.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, S.; Chen, L.; Wang, Z.; Chen, J.; Ni, Q.; Guo, X.; Zhang, L.; Xue, G. Neutrophil Extracellular Traps Delay Diabetic Wound Healing by Inducing Endothelial-to-Mesenchymal Transition via the Hippo pathway. Int. J. Biol. Sci. 2023, 19, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Heng, C.; Zhou, Y.; Hu, Y.; Chen, S.; Wang, H.; Yang, H.; Jiang, Z.; Qian, S.; Wang, Y.; et al. Targeting mammalian serine/threonine-protein kinase 4 through Yes-associated protein/TEA domain transcription factor-mediated epithelial-mesenchymal transition ameliorates diabetic nephropathy orchestrated renal fibrosis. Metabolism 2020, 108, 154258. [Google Scholar] [CrossRef]
- Ortillon, J.; Le Bail, J.-C.; Villard, E.; Léger, B.; Poirier, B.; Girardot, C.; Beeske, S.; Ledein, L.; Blanchard, V.; Brieu, P.; et al. High Glucose Activates YAP Signaling to Promote Vascular Inflammation. Front. Physiol. 2021, 12, 994. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Shi, Y.; Li, R.; Günther, S.; Ong, Y.T.; Potente, M.; Yuan, Z.; Liu, E.; Offermanns, S. YAP and TAZ protect against white adipocyte cell death during obesity. Nat. Commun. 2020, 11, 5455. [Google Scholar] [CrossRef] [PubMed]
- Han, D.J.; Aslam, R.; Misra, P.S.; Chiu, F.; Ojha, T.; Chowdhury, A.; Chan, C.K.; Sung, H.-K.; Yuen, D.A.; Luk, C.T. Disruption of adipocyte YAP improves glucose homeostasis in mice and decreases adipose tissue fibrosis. Mol. Metab. 2022, 66, 101594. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.-S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.-S.; Guan, K.-L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef]
- DeRan, M.; Yang, J.; Shen, C.-H.; Peters, E.C.; Fitamant, J.; Chan, P.; Hsieh, M.; Zhu, S.; Asara, J.M.; Zheng, B.; et al. Energy Stress Regulates Hippo-YAP Signaling Involving AMPK-Mediated Regulation of Angiomotin-like 1 Protein. Cell Rep. 2014, 9, 495–503. [Google Scholar] [CrossRef]
- Xiong, J.; Dong, X.; Li, S.; Jiang, F.; Chen, J.; Yu, S.; Dong, B.; Su, Q. Effects of (Pro)renin Receptor on Diabetic Cardiomyopathy Pathological Processes in Rats via the PRR-AMPK-YAP Pathway. Front. Physiol. 2021, 12, 657378. [Google Scholar] [CrossRef]
- Katakam, P.V.G.; Ujhelyi, M.R.; Hoenig, M.; Miller, A.W. Metformin Improves Vascular Function in Insulin-Resistant Rats. Hypertension 2000, 35, 108–112. [Google Scholar] [CrossRef]
- Mather, K.J.; Verma, S.; Anderson, T.J. Improved endothelial function with metformin in type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2001, 37, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.; Letiexhe, M.; Lefèbvre, P. Short Administration of Metformin Improves Insulin Sensitivity in Android Obese Subjects with Impaired Glucose Tolerance. Diabet. Med. 1995, 12, 985–989. [Google Scholar] [CrossRef]
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin Inhibits Cytokine-Induced Nuclear Factor κB Activation Via AMP-Activated Protein Kinase Activation in Vascular Endothelial Cells. Hypertension 2006, 47, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef]
- Ding, Y.; Zhou, Y.; Ling, P.; Feng, X.; Luo, S.; Zheng, X.; Little, P.J.; Xu, S.; Weng, J. Metformin in cardiovascular diabetology: A focused review of its impact on endothelial function. Theranostics 2021, 11, 9376–9396. [Google Scholar] [CrossRef] [PubMed]
- UPDSU Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Ziqubu, K.; Mazibuko-Mbeje, S.E.; Mthembu, S.X.H.; Mabhida, S.E.; Jack, B.U.; Nyambuya, T.M.; Nkambule, B.B.; Basson, A.K.; Tiano, L.; Dludla, P.V. Anti-Obesity Effects of Metformin: A Scoping Review Evaluating the Feasibility of Brown Adipose Tissue as a Therapeutic Target. Int. J. Mol. Sci. 2023, 24, 2227. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.; De Sanctis, V.; Alaaraj, N.; Hamed, N. The clinical application of metformin in children and adolescents: A short update. Acta Biomed. 2020, 91, e2020086. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, J.; Yang, O.; Kong, J.; Lin, G. Effect of metformin on insulin-resistant endothelial cell function. Oncol. Lett. 2015, 9, 1149–1153. [Google Scholar] [CrossRef]
- Cheang, W.S.; Tian, X.Y.; Wong, W.T.; Lau, C.W.; Lee, S.S.; Chen, Z.Y.; Yao, X.; Wang, N.; Huang, Y. Metformin Protects Endothelial Function in Diet-Induced Obese Mice by Inhibition of Endoplasmic Reticulum Stress Through 5′ Adenosine Monophosphate–Activated Protein Kinase–Peroxisome Proliferator–Activated Receptor δ Pathway. Arter. Thromb. Vasc. Biol. 2014, 34, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, J.; Chen, H.; Wang, R.; Li, P.; Miao, Y.; Liu, P. Metformin suppresses proliferation and invasion of drug-resistant breast cancer cells by activation of the Hippo pathway. J. Cell. Mol. Med. 2020, 24, 5786–5796. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Li, H.; Wang, J. Mechanisms of metformin inhibiting cancer invasion and migration. Am. J. Transl. Res. 2020, 12, 4885–4901. [Google Scholar] [PubMed]
- Zhang, Q.; Li, W.; Zhu, Y.; Wang, Q.; Zhai, C.; Shi, W.; Feng, W.; Wang, J.; Yan, X.; Chai, L.; et al. Activation of AMPK inhibits Galectin-3-induced pulmonary artery smooth muscle cells proliferation by upregulating hippo signaling effector YAP. Mol. Cell. Biochem. 2021, 476, 3037–3049. [Google Scholar] [CrossRef] [PubMed]
- Lós, D.B.; de Oliveira, W.H.; Duarte-Silva, E.; Sougey, W.W.D.; Freitas, E.d.S.R.d.; de Oliveira, A.G.V.; Braga, C.F.; de França, M.E.R.; Araújo, S.M.d.R.; Rodrigues, G.B.; et al. Preventive role of metformin on peripheral neuropathy induced by diabetes. Int. Immunopharmacol. 2019, 74, 105672. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Cheng, C.-K.; Shang, W.; Liu, J.; Cheang, W.-S.; Wang, Y.; Xiang, L.; Lau, C.-W.; Luo, J.-Y.; Ng, C.-F.; Huang, Y.; et al. Activation of AMPK/miR-181b Axis Alleviates Endothelial Dysfunction and Vascular Inflammation in Diabetic Mice. Antioxidants 2022, 11, 1137. [Google Scholar] [CrossRef]
- Qu, D.; Liu, J.; Lau, C.W.; Huang, Y. Differential mechanisms for insulin-induced relaxations in mouse posterior tibial arteries and main mesenteric arteries. Vasc. Pharmacol. 2014, 63, 173–177. [Google Scholar] [CrossRef]
- Albrecht, E.W.; Stegeman, C.A.; Heeringa, P.; Henning, R.H.; van Goor, H. Protective role of endothelial nitric oxide synthase. J. Pathol. 2003, 199, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, M.J.; Ceradini, D.J.; Gurtner, G.C. Hyperglycemia-Induced Reactive Oxygen Species and Impaired Endothelial Progenitor Cell Function. Antioxidants Redox Signal. 2005, 7, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Fan, X.; Sun, J.; Zhang, Z. Metformin prevented high glucose-induced endothelial reactive oxygen species via OGG1 in an AMPKα-Lin-28 dependent pathway. Life Sci. 2021, 268, 119015. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathna, P.; Su, Y. The critical role of Akt in cardiovascular function. Vasc. Pharmacol. 2015, 74, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Chao, M.-L.; Luo, S.; Zhang, C.; Zhou, X.; Zhou, M.; Wang, J.; Kong, C.; Chen, J.; Lin, Z.; Tang, X.; et al. S-nitrosylation-mediated coupling of G-protein alpha-2 with CXCR5 induces Hippo/YAP-dependent diabetes-accelerated atherosclerosis. Nat. Commun. 2021, 12, 4452. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; He, Q.; Bulus, N.; Fogo, A.B.; Zhang, M.-Z.; Harris, R.C. YAP Activation in Renal Proximal Tubule Cells Drives Diabetic Renal Interstitial Fibrogenesis. Diabetes 2020, 69, 2446–2457. [Google Scholar] [CrossRef]
- Rinschen, M.M.; Grahammer, F.; Hoppe, A.-K.; Kohli, P.; Hagmann, H.; Kretz, O.; Bertsch, S.; Höhne, M.; Göbel, H.; Bartram, M.P.; et al. YAP-mediated mechanotransduction determines the podocyte’s response to damage. Sci. Signal. 2017, 10, eaaf8165. [Google Scholar] [CrossRef]
- Song, X.; Xu, H.; Wang, P.; Wang, J.; Affo, S.; Wang, H.; Xu, M.; Liang, B.; Che, L.; Qiu, W.; et al. Focal adhesion kinase (FAK) promotes cholangiocarcinoma development and progression via YAP activation. J. Hepatol. 2021, 75, 888–899. [Google Scholar] [CrossRef]
- Xu, D.; Chen, P.-P.; Zheng, P.-Q.; Yin, F.; Cheng, Q.; Zhou, Z.-L.; Xie, H.-Y.; Li, J.-Y.; Ni, J.-Y.; Wang, Y.-Z.; et al. KLF4 initiates sustained YAP activation to promote renal fibrosis in mice after ischemia-reperfusion kidney injury. Acta Pharmacol. Sin. 2021, 42, 436–450. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, T.; Lin, Q. Non-hippo kinases: Indispensable roles in YAP/TAZ signaling and implications in cancer therapy. Mol. Biol. Rep. 2023, 50, 4565–4578. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xiao, Z.-D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′–3′) |
|---|---|
| Human CTGF forward | ACCGACTGGAAGACACGTTTG |
| Human CTGF reverse | CCAGGTCAGCTTCGCAAGG |
| Human CYR61 forward | TGAAGCGGCTCCCTGTTTT |
| Human CYR61 reverse | CGGGTTTCTTTCACAAGGCG |
| Human ANKRD1 forward | AGAACTGTGCTGGGAAGACG |
| Human ANKRD1 reverse | GCCATGCCTTCAAAATGCCA |
| Human E-selectin forward | TGTGGGTCTGGGTAGGAACC |
| Human E-selectin reverse | AGCTGTGTAGCATAGGGCAAG |
| Human ICAM1 forward | TTGGGCATAGAGACCCCGTT |
| Human ICAM1 reverse | GCACATTGCTCAGTTCATACACC |
| Human VCAM1 forward | CAGTAAGGCAGGCTGTAAAAGA |
| Human VCAM1 reverse | TGGAGCTGGTAGACCCTCG |
| Human CCL2 forward | TCAAACTGAAGCTCGCACTC |
| Human CCL2 reverse | TGGGGCATTGATTGCATCTGG |
| Human TNFα forward | CCTCTCTCTAATCAGCCCTCTG |
| Human TNFα reverse | GAGGACCTGGGAGTAGATGAG |
| Human CXCL1 forward | CTGGCTTAGAACAAAGGGGCT |
| Human CXCL1 reverse | TAAAGGTAGCCCTTGTTTCCCC |
| Human GAPDH forward | TTCGTCATGGGTGTGAACCA |
| Human GAPDH reverse | TGATGGCATGGACTGTGGTC |
| Mouse CTGF forward | TCAACCTCAGACACTGGTTTCG |
| Mouse CTGF reverse | TAGAGCAGGTCTGTCTGCAAGC |
| Mouse CYR61 forward | GCCGTGGGCTGCATTCCTCT |
| Mouse CYR61 reverse | GCGGTTCGGTGCCAAAGACAGG |
| Mouse ANKRD1 forward | GCTGGTAACAGGCAAAAAGAAC |
| Mouse ANKRD1 reverse | CCTCTCGCAGTTTCTCGCT |
| Mouse TNFα forward | GCGGTGCCTATGTCTCAGC |
| Mouse TNFα reverse | CACTTGGTGGTTTGTGAGTGT |
| Mouse VCAM1 forward | GTTCCAGCGAGGGTCTACC |
| Mouse VCAM1 reverse | AACTCTTGGCAAACATTAGGTGT |
| Mouse GAPDH forward | AGGTCGGTGTGAACGGATTTG |
| Mouse GAPDH reverse | TGTAGACCATGTAGTTGAGGTCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, L.; Yi, J.; Lau, C.-W.; He, L.; Chen, Q.; Xu, S.; Li, J.; Xia, Y.; Zhang, Y.; Huang, Y.; et al. AMPK-Dependent YAP Inhibition Mediates the Protective Effect of Metformin against Obesity-Associated Endothelial Dysfunction and Inflammation. Antioxidants 2023, 12, 1681. https://doi.org/10.3390/antiox12091681
Kang L, Yi J, Lau C-W, He L, Chen Q, Xu S, Li J, Xia Y, Zhang Y, Huang Y, et al. AMPK-Dependent YAP Inhibition Mediates the Protective Effect of Metformin against Obesity-Associated Endothelial Dysfunction and Inflammation. Antioxidants. 2023; 12(9):1681. https://doi.org/10.3390/antiox12091681
Chicago/Turabian StyleKang, Lijing, Juanjuan Yi, Chi-Wai Lau, Lei He, Qinghua Chen, Suowen Xu, Jun Li, Yin Xia, Yuanting Zhang, Yu Huang, and et al. 2023. "AMPK-Dependent YAP Inhibition Mediates the Protective Effect of Metformin against Obesity-Associated Endothelial Dysfunction and Inflammation" Antioxidants 12, no. 9: 1681. https://doi.org/10.3390/antiox12091681