

The NOS/NO System in Renal Programming and Reprogramming


Abstract
:1. Introduction
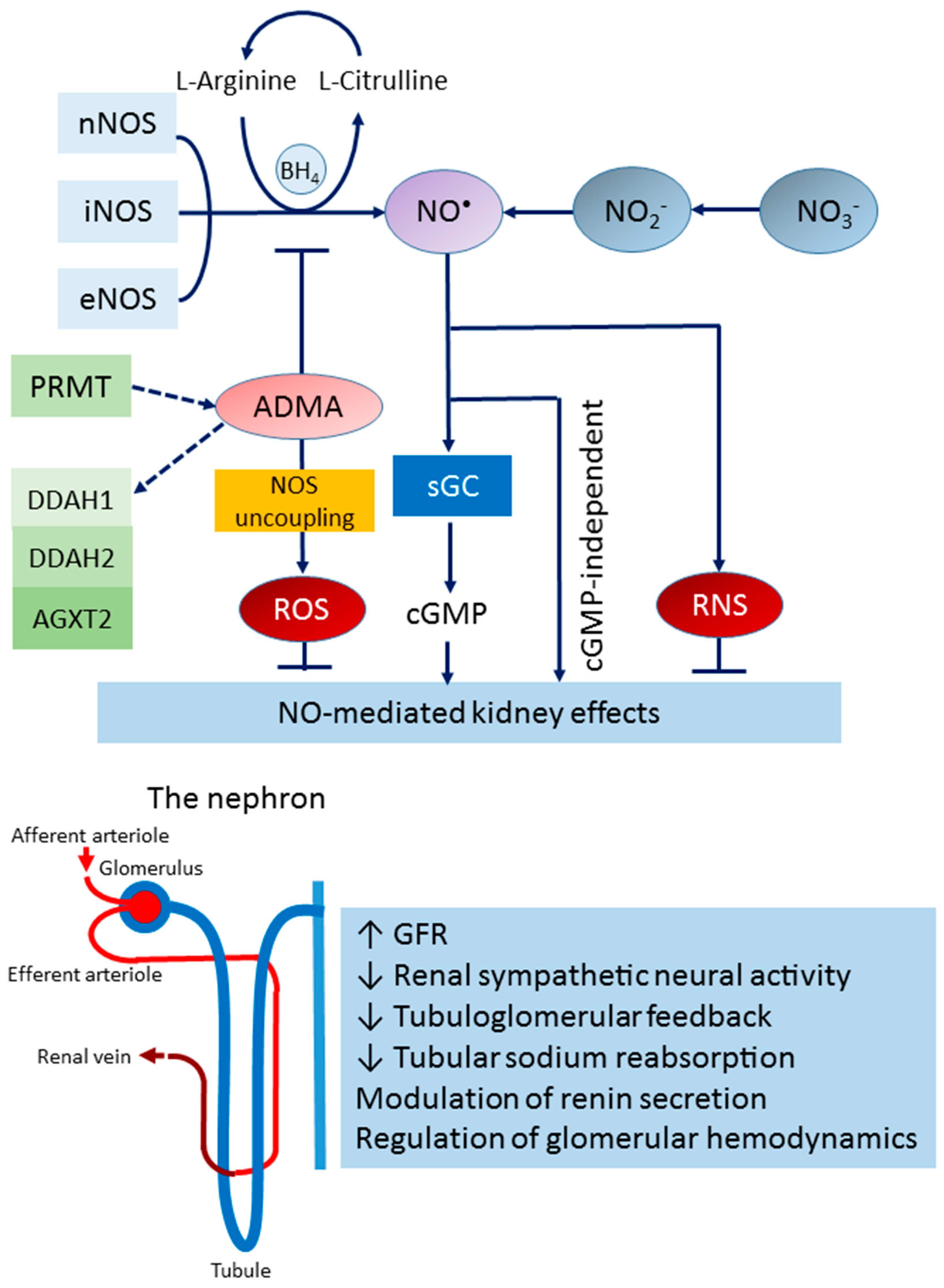
2. The NOS/NO System in the Kidney
2.1. The L-arginine–NOS–NO Pathway
2.2. The Nitrate–Nitrite–NO Pathway
2.3. Endogenous NOS Inhibitor
2.4. NO in Kidney Health and Disease
3. The NOS/NO System in Renal Programming
3.1. NO in Pregnancy
3.2. NO in Renal Programming
3.3. Animal Models of Renal Programming Linked to Impaired NOS/NO System
4. Reprogramming Strategies Targeting the NOS/NO System
4.1. NOS Substrates
4.2. ADMA-Lowering Agents
4.3. NO Donors and Nitrodilators
4.4. Others
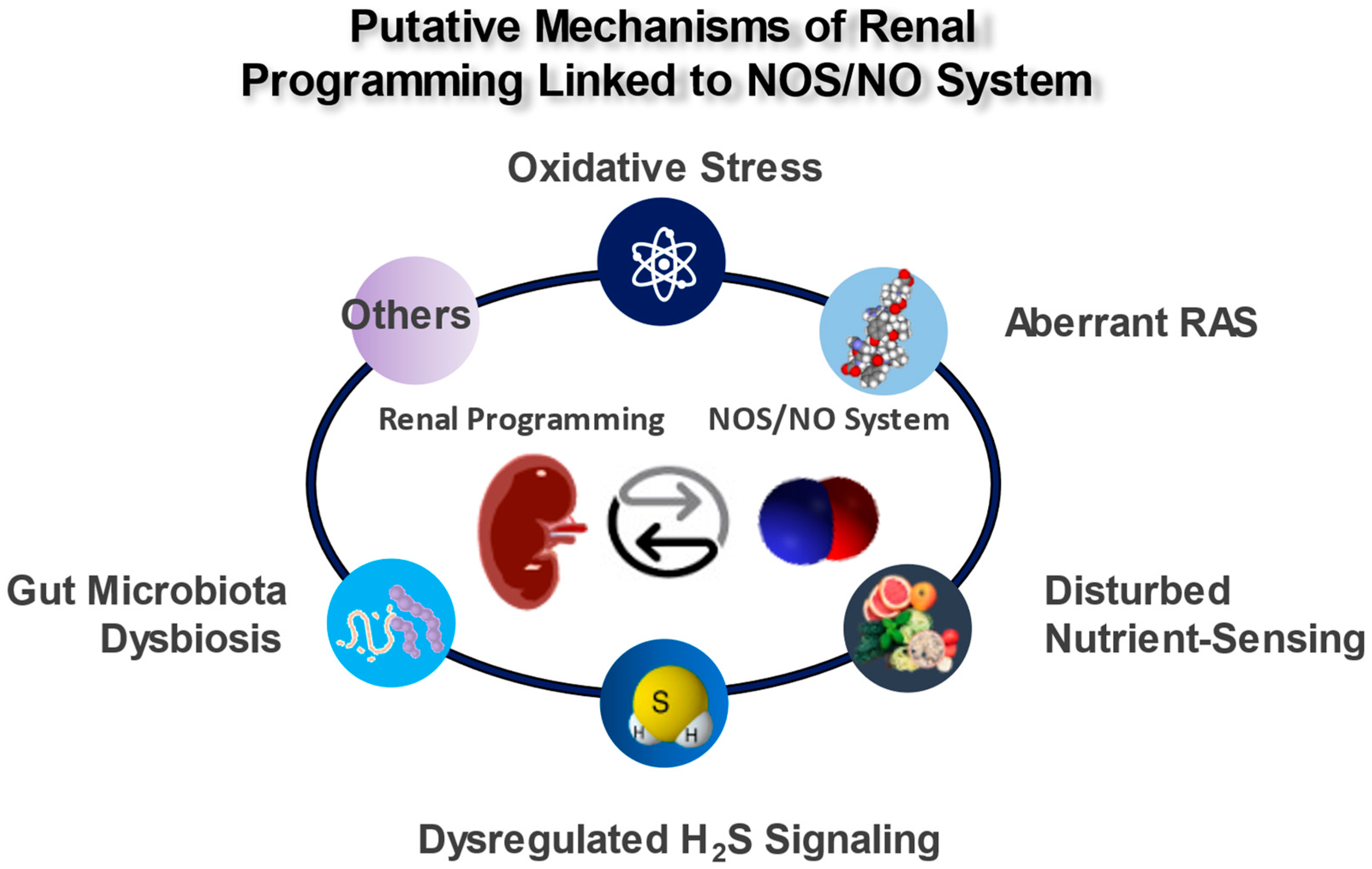
5. Mechanisms of the Renal Programming Linked to the NOS/NO System
5.1. Oxidative Stress
5.2. Aberrant RAS
5.3. Disturbed Nutrient Sensing
5.4. Dysregulated H2S Signaling
5.5. Gut Microbiota Dysbiosis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, V.A.; Bertram, J.F.; Brenner, B.M.; Fall, C.; Hoy, W.E.; Ozanne, S.E.; Vikse, B.E. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet 2013, 382, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Developmental Origins of Chronic Kidney Disease: Should We Focus on Early Life? Int. J. Mol. Sci. 2017, 18, 381. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L. Evolution, kidney development, and chronic kidney disease. Semin. Cell Dev. Biol. 2019, 91, 119–131. [Google Scholar] [CrossRef]
- Hanson, M. The birth and future health of DOHaD. J. Dev. Orig. Health Dis. 2015, 6, 434–437. [Google Scholar] [CrossRef]
- Kett, M.M.; Denton, K.M. Renal programming: Cause for concern? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R791–R803. [Google Scholar] [CrossRef]
- Nenov, V.D.; Taal, M.W.; Sakharova, O.V.; Brenner, B.M. Multi-hit nature of chronic renal disease. Curr. Opin. Nephrol. Hypertens. 2000, 9, 85–97. [Google Scholar] [CrossRef]
- Tain, Y.L.; Joles, J.A. Reprogramming: A preventive strategy in hypertension focusing on the kidney. Int. J. Mol. Sci. 2016, 17, 23. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Adverse Impact of Environmental Chemicals on Developmental Origins of Kidney Disease and Hypertension. Front. Endocrinol. 2021, 12, 745716. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Perinatal Oxidative Stress and Kidney Health: Bridging the Gap between Animal Models and Clinical Reality. Antioxidants 2022, 12, 13. [Google Scholar] [CrossRef]
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Tain, Y.L. Targeting the renin–angiotensin–aldosterone system to prevent hypertension and kidney disease of developmental origins. Int. J. Mol. Sci. 2021, 22, 2298. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Chronic Kidney Disease and Gut Microbiota: What Is Their Connection in Early Life? Int. J. Mol. Sci. 2022, 23, 3954. [Google Scholar] [CrossRef] [PubMed]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Tain, Y.L. Early-Life Programming and Reprogramming of Adult Kidney Disease and Hypertension: The Interplay between Maternal Nutrition and Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3572. [Google Scholar] [CrossRef]
- Racasan, S.; Braam, B.; Koomans, H.A.; Joles, J.A. Programming blood pressure in adult SHR by shifting perinatal balance of NO and reactive oxygen species toward NO: The inverted Barker phenomenon. Am. J. Physiol. Renal Physiol. 2005, 288, F626–F636. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Gasotransmitters for the Therapeutic Prevention of Hypertension and Kidney Disease. Int. J. Mol. Sci. 2021, 22, 7808. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Weitzberg, E. Nitric oxide signaling in health and disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef]
- Piacenza, L.; Zeida, A.; Trujillo, M.; Radi, R. The superoxide radical switch in the biology of nitric oxide and peroxynitrite. Physiol. Rev. 2022, 102, 1881–1906. [Google Scholar] [CrossRef]
- Carlström, M. Nitric oxide signalling in kidney regulation and cardiometabolic health. Nat. Rev. Nephrol. 2021, 17, 575–590. [Google Scholar] [CrossRef]
- Kone, B.C. Nitric oxide synthesis in the kidney: Isoforms, biosynthesis, and functions in health. Semin. Nephrol. 2004, 24, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Baylis, C. Nitric oxide deficiency in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2008, 294, F1–F9. [Google Scholar] [CrossRef] [PubMed]
- Solhaug, M.J.; Ballèvre, L.D.; Guignard, J.P.; Granger, J.P.; Adelman, R.D. Nitric oxide in the developing kidney. Pediatr. Nephrol. 1996, 10, 529–539. [Google Scholar] [CrossRef]
- Zullino, S.; Buzzella, F.; Simoncini, T. Nitric oxide and the biology of pregnancy. Vascul. Pharmacol. 2018, 110, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Tain, Y.L. Regulation of Nitric Oxide Production in the Developmental Programming of Hypertension and Kidney Disease. Int. J. Mol. Sci. 2019, 20, 681. [Google Scholar] [CrossRef] [PubMed]
- Engineer, A.; Saiyin, T.; Greco, E.R.; Feng, Q. Say NO to ROS: Their Roles in Embryonic Heart Development and Pathogenesis of Congenital Heart Defects in Maternal Diabetes. Antioxidants 2019, 8, 436. [Google Scholar] [CrossRef]
- Silvagno, F.; Xia, H.; Bredt, D.S. Neuronal nitric oxide synthase-μ, an alternatively spliced isoform expressed in differentiated skeletal muscle. J. Biol. Chem. 1996, 271, 11204–11208. [Google Scholar] [CrossRef]
- Saur, D.; Paehge, H.; Schusdziarra, V.; Allescher, H.D. Distinct expression of splice variants of neuronal nitric oxide synthase in the human gastrointestinal tract. Gastroenterology 2000, 118, 849–858. [Google Scholar] [CrossRef]
- Wu, G.; Morris, S.M., Jr. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef]
- Brosnan, M.E.; Brosnan, J.T. Renal arginine metabolism. J. Nutr. 2004, 134, S2791–S2795. [Google Scholar] [CrossRef]
- Bode-Böger, S.M.; Scalera, F.; Ignarro, L.J. The L-arginine paradox: Importance of the L-arginine/asymmetrical dimethylarginine ratio. Pharmacol. Ther. 2007, 114, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008, 7, 156–167. [Google Scholar] [CrossRef]
- Baylis, C.; Vallance, P. Measurement of nitrite and nitrate (NOx) levels in plasma and urine; what does this measure tell us about the activity of the endogenous nitric oxide. Curr. Opin. Nephrol. Hypertens. 1998, 7, 59–62. [Google Scholar] [CrossRef] [PubMed]
- van Faassen, E.E.; Bahrami, S.; Feelisch, M.; Hogg, N.; Kelm, M.; Kim-Shapiro, D.B.; Kozlov, A.V.; Li, H.; Lundberg, J.O.; Mason, R.; et al. Nitrite as regulator of hypoxic signaling in mammalian physiology. Med. Res. Rev. 2009, 29, 683–741. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Toxic Dimethylarginines: Asymmetric Dimethylarginine (ADMA) and Symmetric Dimethylarginine (SDMA). Toxins 2017, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Morales, Y.; Cáceres, T.; May, K.; Hevel, J.M. Biochemistry and regulation of the protein arginine methyltransferases (PRMTs). Arch. Biochem. Biophys. 2016, 590, 138–152. [Google Scholar] [CrossRef]
- Böger, R.H.; Sydow, K.; Borlak, J.; Thum, T.; Lenzen, H.; Schubert, B.; Tsikas, D.; Bode-Böger, S.M. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: Involvement of S-adenosylmethionine-dependent methyltransferases. Circ. Res. 2000, 87, 99–105. [Google Scholar] [CrossRef]
- Raijmakers, R.; Zendman, A.J.; Egberts, W.V.; Vossenaar, E.R.; Raats, J.; Soede-Huijbregts, C.; Rutjes, F.P.; van Veelen, P.A.; Drijfhout, J.W.; Pruijn, G.J. Methylation of arginine residues interferes with citrullination by peptidylarginine deiminases in vitro. J. Mol. Biol. 2007, 367, 1118–1129. [Google Scholar] [CrossRef]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447. [Google Scholar] [CrossRef]
- Böttger, A.; Islam, M.S.; Chowdhury, R.; Schofield, C.J.; Wolf, A. The oxygenase Jmjd6—A case study in conflicting assignments. Biochem. J. 2015, 468, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol. Res. 2009, 60, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, R.N.; Martens-Lobenhoffer, J.; Brilloff, S.; Hohenstein, B.; Jarzebska, N.; Jabs, N.; Kittel, A.; Maas, R.; Weiss, N.; Bode-Böger, S.M. Role of alanine:glyoxylate aminotransferase 2 in metabolism of asymmetric dimethylarginine in the settings of asymmetric dimethylarginine overload and bilateral nephrectomy. Nephrol. Dial. Transplant. 2014, 29, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Kao, Y.H.; Hsieh, C.S.; Chen, C.C.; Sheen, J.M.; Lin, I.C.; Huang, L.T. Melatonin blocks oxidative stress-induced increased asymmetric dimethylarginine. Free Radic. Biol. Med. 2010, 49, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, V.; Mazza, F.; Campisi, A.; Vanella, L.; Li, V.G.; Di, G.C. High glucose-mediated imbalance of nitric oxide synthase and dimethylarginine dimethylaminohydrolase expression in endothelial cells. Curr. Neurovasc. Res. 2006, 3, 49–54. [Google Scholar] [CrossRef]
- Brands, M.W.; Bell, T.D.; Gibson, B. Nitric oxide may prevent hypertension early in diabetes by counteracting renal actions of superoxide. Hypertension 2004, 43, 57–63. [Google Scholar] [CrossRef]
- Palm, F.; Onozato, M.L.; Luo, Z.; Wilcox, C.S. Dimethylarginine dimethylaminohydrolase (DDAH): Expression, regulation, and function in the cardiovascular and renal systems. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3227–H3245. [Google Scholar] [CrossRef]
- Kittel, A.; Maas, R.; König, J.; Mieth, M.; Weiss, N.; Jarzebska, N.; Hohenstein, B.; Martens-Lobenhoffer, J.; Bode-Böger, S.M.; Rodionov, R.N. In vivo evidence that Agxt2 can regulate plasma levels of dimethylarginines in mice. Biochem. Biophys. Res. Commun. 2013, 430, 84–89. [Google Scholar] [CrossRef]
- Tsikas, D. Does the inhibitory action of asymmetric dimethylarginine (ADMA) on the endothelial nitric oxide synthase activity explain its importance in the cardiovascular system? The ADMA paradox. J. Controv. Biomed. Res. 2017, 3, 16–22. [Google Scholar] [CrossRef]
- Tsikas, D.; Bollenbach, A.; Hanff, E.; Kayacelebi, A.A. Asymmetric dimethylarginine (ADMA), symmetric dimethylarginine (SDMA) and homoarginine (hArg): The ADMA, SDMA and hArg paradoxes. Cardiovasc. Diabetol. 2018, 17, 1. [Google Scholar] [CrossRef]
- Romero, J.C.; Strick, D.M. Nitric oxide and renal function. Curr. Opin. Nephrol. Hypertens. 1993, 2, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Tizianello, A.; De Ferrari, G.; Garibotto, G.; Gurreri, G.; Robaudo, C. Renal metabolism of amino acids and ammonia in subjects with normal renal function and in patients with chronic renal insufficiency. J. Clin. Investig. 1980, 65, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xiong, R.; Feng, S.; Lu, X.; Li, H.; Wang, S. Association of Circulating Levels of ADMA with Carotid Intima-Media Thickness in Patients with CKD: A Systematic Review and Meta-Analysis. Kidney Blood Press. Res. 2018, 43, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, S.; Sonntag, S.R.; Lieb, W.; Maas, R. Asymmetric and Symmetric Dimethylarginine as Risk Markers for Total Mortality and Cardiovascular Outcomes: A Systematic Review and Meta-Analysis of Prospective Studies. PLoS ONE 2016, 11, e0165811. [Google Scholar] [CrossRef]
- Wang, T.; Inglis, F.M.; Kalb, R.G. Defective fluid and HCO3− absorption in proximal tubule of neuronal nitric oxide synthase-knockout mice. Am. J. Physiol. Renal Physiol. 2000, 279, F518–F524. [Google Scholar] [CrossRef]
- Sogawa, Y.; Nagasu, H.; Itano, S.; Kidokoro, K.; Taniguchi, S.; Takahashi, M.; Kadoya, H.; Satoh, M.; Sasaki, T.; Kashihara, N. The eNOS-NO pathway attenuates kidney dysfunction via suppression of inflammasome activation in aldosterone-induced renal injury model mice. PLoS ONE 2018, 13, e0203823. [Google Scholar] [CrossRef]
- Muller, V.; Tain, Y.L.; Croker, B.; Baylis, C. Chronic nitric oxide deficiency and progression of kidney disease after renal mass reduction in the C57Bl6 mouse. Am. J. Nephrol. 2010, 32, 575–580. [Google Scholar] [CrossRef]
- Tain, Y.L.; Freshour, G.; Dikalova, A.; Griendling, K.; Baylis, C. Vitamin E reduces glomerulosclerosis, restores renal neuronal NOS, and suppresses oxidative stress in the 5/6 nephrectomized rat. Am. J. Physiol. Renal Physiol. 2007, 292, F1404–F1410. [Google Scholar] [CrossRef]
- Erdely, A.; Freshour, G.; Tain, Y.L.; Engels, K.; Baylis, C. DOCA/NaCl-induced chronic kidney disease: A comparison of renal nitric oxide production in resistant and susceptible rat strains. Am. J. Physiol. Renal Physiol. 2007, 292, F192–F196. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Wilcox, C.S. Oxidative stress and nitric oxide deficiency in the kidney: A critical link to hypertension? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R913–R935. [Google Scholar] [CrossRef] [PubMed]
- Krause, B.J.; Hanson, M.A.; Casanello, P. Role of nitric oxide in placental vascular development and function. Placenta 2011, 32, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Purcell, T.L.; Given, R.; Chwalisz, K.; Garfield, R.E. Nitric oxide synthase distribution during implantation in the mouse. Mol. Hum. Reprod. 1999, 5, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Seligman, S.P.; Buyon, J.P.; Clancy, R.M.; Young, B.K.; Abramson, S.B. The role of nitric oxide in the pathogenesis of preeclampsia. Am. J. Obstet. Gynecol. 1994, 171, 944–948. [Google Scholar] [CrossRef]
- Huang, L.T.; Hsieh, C.S.; Chang, K.A.; Tain, Y.L. Roles of nitric oxide and asymmetric dimethylarginine in pregnancy and fetal programming. Int. J. Mol. Sci. 2012, 13, 14606–14622. [Google Scholar] [CrossRef]
- Bavoux, F.; Georges, P.; Bouy, M.; Leroy, B. Growth retardation and amino acids. Analysis of maternal plasma and amniotic fluid. J. Gynecol. Obstet. Biol. Reprod. 1977, 6, 931–940. [Google Scholar]
- Kim, Y.J.; Park, H.S.; Lee, H.Y.; Ha, E.H.; Suh, S.H.; Oh, S.K.; Yoo, H.S. Reduced L-arginine level and decreased placental eNOS activity in preeclampsia. Placenta 2006, 27, 438–444. [Google Scholar] [CrossRef]
- Pettersson, A.; Hedner, T.; Milsom, I. Increased circulating concentrations of asymmetric dimethyl arginine (ADMA), an endogenous inhibitor of nitric oxide synthesis, in preeclampsia. Acta Obstet. Gynecol. Scand. 1998, 77, 808–813. [Google Scholar]
- Braekke, K.; Ueland, P.M.; Harsem, N.K.; Staff, A.C. Asymmetric dimethylarginine in the maternal and fetal circulation in preeclampsia. Pediatr. Res. 2009, 66, 411–415. [Google Scholar] [CrossRef]
- Noris, M.; Todeschini, M.; Cassis, P.; Pasta, F.; Cappellini, A.; Bonazzola, S.; Macconi, D.; Maucci, R.; Porrati, F.; Benigni, A.; et al. L-arginine depletion in preeclampsia orients nitric oxide synthase toward oxidant species. Hypertension 2004, 43, 614–622. [Google Scholar] [CrossRef]
- Little, M.H.; McMahon, A.P. Mammalian kidney development: Principles, progress, and projections. Cold Spring Harb. Perspect. Biol. 2012, 4, a008300. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, V.A.; Brenner, B.M. The clinical importance of nephron mass. J. Am. Soc. Nephrol. 2010, 21, 898–910. [Google Scholar] [CrossRef]
- Hartman, H.A.; Lai, H.L.; Patterson, L.T. Cessation of renal morphogenesis in mice. Dev. Biol. 2007, 310, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.M.; Sampogna, R.V.; Sakurai, H.; Bush, K.T.; Nigam, S.K. Branching morphogenesis and kidney disease. Development 2004, 131, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Murugapoopathy, V.; Gupta, I.R. A primer on congenital anomalies of the kidneys and urinary tracts (CAKUT). Clin. J. Am. Soc. Nephrol. 2020, 15, 723–731. [Google Scholar] [CrossRef]
- Bertram, J.F.; Douglas-Denton, R.N.; Diouf, B.; Hughson, M.; Hoy, W. Human nephron number: Implications for health and disease. Pediatr. Nephrol. 2011, 26, 1529–1533. [Google Scholar] [CrossRef]
- Luyckx, V.A.; Brenner, B.M. Birth weight, malnutrition and kidney-associated outcomes—A global concern. Nat. Rev. Nephrol. 2015, 11, 135–149. [Google Scholar] [CrossRef]
- White, S.L.; Perkovic, V.; Cass, A.; Chang, C.L.; Poulter, N.R.; Spector, T.; Haysom, L.; Craig, J.C.; Salmi, I.A.; Chadban, S.J.; et al. Is low birth weight an antecedent of CKD in later life? A systematic review of observational studies. Am. J. Kidney Dis. 2009, 54, 248–261. [Google Scholar] [CrossRef]
- Hsu, C.W.; Yamamoto, K.T.; Henry, R.K.; de Roos, A.J.; Flynn, J.T. Prenatal risk factors for childhood CKD. J. Am. Soc. Nephrol. 2014, 25, 2105–2111. [Google Scholar] [CrossRef]
- Tain, Y.L.; Luh, H.; Lin, C.Y.; Hsu, C.N. Incidence and risks of congenital anomalies of kidney and urinary tract in newborns: A population-based case-control study in Taiwan. Medicine 2016, 95, e2659. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal N(G)-Nitro-L-argininemethyl ester (L-NAME)-induced fetal programming of hypertension in adult male offspring. Am. J. Obstet. Gynecol. 2016, 215, 636. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.F.; Tofteng, S.S.; Madsen, K.; Jensen, B.L. Role of the renin-angiotensin system in kidney development and programming of adult blood pressure. Clin. Sci. (Lond.) 2020, 134, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, W.C.; Wu, K.L.; Leu, S.; Chan, J.Y. Targeting arachidonic acid pathway to prevent programmed hypertension in maternal fructose-fed male adult rat offspring. J. Nutr. Biochem. 2016, 38, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Paixão, A.D.; Alexander, B.T. How the kidney is impacted by the perinatal maternal environment to develop hypertension. Biol. Reprod. 2013, 89, 144. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y.H. PPARs Link Early Life Nutritional Insults to Later Programmed Hypertension and Metabolic Syndrome. Int. J. Mol. Sci. 2016, 17, 20. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lee, W.C.; Hsu, C.N.; Lee, W.C.; Huang, L.T.; Lee, C.T.; Lin, C.Y. Asymmetric dimethylarginine is associated with developmental programming of adult kidney disease and hypertension in offspring of streptozotocin-treated mothers. PLoS ONE 2013, 8, e55420. [Google Scholar] [CrossRef]
- Tain, Y.L.; Huang, L.T.; Chan, J.Y.; Lee, C.T. Transcriptome analysis in rat kidneys: Importance of genes involved in programmed hypertension. Int. J. Mol. Sci. 2015, 16, 4744–4758. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y.; Huang, L.T. Renal Transcriptome Analysis of Programmed Hypertension Induced by Maternal Nutritional Insults. Int. J. Mol. Sci. 2015, 16, 17826–17837. [Google Scholar] [CrossRef]
- Tomat, A.L.; Veiras, L.C.; Aguirre, S.; Fasoli, H.; Elesgaray, R.; Caniffi, C.; Costa, M.Á.; Arranz, C.T. Mild zinc deficiency in male and female rats: Early postnatal alterations in renal nitric oxide system and morphology. Nutrition 2013, 29, 568–573. [Google Scholar] [CrossRef]
- Alves, G.M.; Barão, M.A.; Odo, L.N.; Nascimento Gomes, G.; Franco Md Mdo, C.; Nigro, D.; Lucas, S.R.; Laurindo, F.R.; Brandizzi, L.I.; Zaladek Gil, F. L-Arginine effects on blood pressure and renal function of intrauterine restricted rats. Pediatr. Nephrol. 2002, 17, 856–862. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsieh, C.S.; Lin, I.C.; Chen, C.C.; Sheen, J.M.; Huang, L.T. Effects of maternal L-citrulline supplementation on renal function and blood pressure in offspring exposed to maternal caloric restriction: The impact of nitric oxide pathway. Nitric Oxide 2010, 23, 34–41. [Google Scholar] [CrossRef]
- Tain, Y.L.; Huang, L.T.; Hsu, C.N.; Lee, C.T. Melatonin therapy prevents programmed hypertension and nitric oxide deficiency in offspring exposed to maternal caloric restriction. Oxid. Med. Cell Longev. 2014, 2014, 283180. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N.; Lee, C.T.; Lin, Y.J.; Tsai, C.C. N-Acetylcysteine prevents programmed hypertension in male rat offspring born to suramin-treated mothers. Biol. Reprod. 2016, 95, 8. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal citrulline supplementation prevents prenatal N(G)-nitro-L-arginine-methyl ester (L-NAME)-induced programmed hypertension in rats. Biol. Reprod. 2015, 92, 7. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tzeng, H.T.; Lee, W.C.; Wu, K.L.H.; Yu, H.R.; Chan, J.Y.H.; Hsu, C.N. Reprogramming Effects of Postbiotic Butyrate and Propionate on Maternal High-Fructose Diet-Induced Offspring Hypertension. Nutrients 2023, 15, 1682. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Chan, J.Y.H.; Lee, C.T.; Tain, Y.L. Maternal resveratrol therapy protected adult rat offspring against hypertension programmed by combined exposures to asymmetric dimethylarginine and trimethylamine-N-oxide. J. Nutr. Biochem. 2021, 93, 108630. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Yang, H.W.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal Adenine-Induced Chronic Kidney Disease Programs Hypertension in Adult Male Rat Offspring: Implications of Nitric Oxide and Gut Microbiome Derived Metabolites. Int. J. Mol. Sci. 2020, 21, 7237. [Google Scholar] [CrossRef]
- Sato, S.; Mukai, Y.; Norikura, T. Maternal low-protein diet suppresses vascular and renal endothelial nitric oxide synthase phosphorylation in rat offspring independent of a postnatal fructose diet. J. Dev. Orig. Health Dis. 2011, 2, 168–175. [Google Scholar] [CrossRef]
- Tain, Y.L.; Sheen, J.M.; Chen, C.C.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Maternal citrulline supplementation prevents prenatal dexamethasone-induced programmed hypertension. Free Radic. Res. 2014, 48, 580–586. [Google Scholar] [CrossRef]
- Tsai, W.L.; Hsu, C.N.; Tain, Y.L. Whether AICAR in Pregnancy or Lactation Prevents Hypertension Programmed by High Saturated Fat Diet: A Pilot Study. Nutrients 2020, 12, 448. [Google Scholar] [CrossRef]
- Uson-Lopez, R.A.; Kataoka, S.; Mukai, Y.; Sato, S.; Kurasaki, M. Melinjo (Gnetum gnemon) Seed Extract Consumption during Lactation Improved Vasodilation and Attenuated the Development of Hypertension in Female Offspring of Fructose-Fed Pregnant Rats. Birth Defects Res. 2018, 110, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lin, Y.J.; Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Tsai, C.C.; Huang, L.T.; Hsu, C.N. High fat diets sex-specifically affect the renal transcriptome and program obesity, kidney injury, and hypertension in the offspring. Nutrients 2017, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Woodman, A.G.; Mah, R.; Keddie, D.L.; Noble, R.M.N.; Holody, C.D.; Panahi, S.; Gragasin, F.S.; Lemieux, H.; Bourque, S.L. Perinatal iron deficiency and a high salt diet cause long-term kidney mitochondrial dysfunction and oxidative stress. Cardiovasc. Res. 2020, 116, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Easton, L.K.; Booth, L.C.; Schlaich, M.P.; Head, G.A.; Moritz, K.M.; Denton, K.M. Renal Nitric Oxide Deficiency and Chronic Kidney Disease in Young Sheep Born with a Solitary Functioning Kidney. Sci. Rep. 2016, 6, 26777. [Google Scholar] [CrossRef]
- Gwathmey, T.M.; Shaltout, H.A.; Rose, J.C.; Diz, D.I.; Chappell, M.C. Glucocorticoid-induced fetal programming alters the functional complement of angiotensin receptor subtypes within the kidney. Hypertension 2011, 57, 620–626. [Google Scholar] [CrossRef]
- Sengupta, P. The laboratory rat: Relating its age with human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar]
- Koeners, M.P.; Braam, B.; van der Giezen, D.M.; Goldschmeding, R.; Joles, J.A. Perinatal micronutrient supplements ameliorate hypertension and proteinuria in adult fawn-hooded hypertensive rats. Am. J. Hypertens. 2010, 23, 802–808. [Google Scholar] [CrossRef]
- Koeners, M.P.; van Faassen, E.E.; Wesseling, S.; Sain-vander Velden, M.; Koomans, H.A.; Braam, B.; Joles, J.A. Maternal supplementation with citrulline increases renal nitric oxide in young spontaneously hypertensive rats and has long-term antihypertensive effects. Hypertension 2007, 50, 1077–1084. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lin, Y.J.; Lu, P.C.; Tain, Y.L. Maternal Resveratrol Therapy Protects Male Rat Offspring against Programmed Hypertension Induced by TCDD and Dexamethasone Exposures: Is It Relevant to Aryl Hydrocarbon Receptor? Int. J. Mol. Sci. 2018, 19, 2459. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Hsu, C.N. Perinatal Garlic Oil Supplementation Averts Rat Offspring Hypertension Programmed by Maternal Chronic Kidney Disease. Nutrients 2022, 14, 4624. [Google Scholar] [CrossRef]
- Tai, I.H.; Sheen, J.M.; Lin, Y.J.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Tain, Y.L. Maternal N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and prevents programmed hypertension in male offspring exposed to prenatal dexamethasone and postnatal high-fat diet. Nitric Oxide 2016, 53, 6–12. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lin, Y.J.; Yu, H.R.; Lin, I.C.; Sheen, J.M.; Huang, L.T.; Tain, Y.L. Protection of Male Rat Offspring against Hypertension Programmed by Prenatal Dexamethasone Administration and Postnatal High-Fat Diet with the Nrf2 Activator Dimethyl Fumarate during Pregnancy. Int. J. Mol. Sci. 2019, 20, 3957. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Siuda, D.; Xia, N.; Reifenberg, G.; Daiber, A.; Münzel, T.; Förstermann, U.; Li, H. Maternal treatment of spontaneously hypertensive rats with pentaerythritoltetranitrate reduces blood pressure in female offspring. Hypertension 2015, 65, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, S.; Essers, P.B.; Koeners, M.P.; Pereboom, T.C.; Braam, B.; van Faassen, E.E.; Macinnes, A.W.; Joles, J.A. Perinatal exogenous nitric oxide in fawn-hooded hypertensive rats reduces renal ribosomal biogenesis in early life. Front. Genet. 2011, 2, 52. [Google Scholar] [CrossRef] [PubMed]
- Gokce, N. L-Arginine and hypertension. J. Nutr. 2004, 134, 2807S–2811S. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.J.; Platt, D.H.; Caldwell, R.B.; Caldwell, R.W. Therapeutic use of citrulline in cardiovascular disease. Cardiovasc. Drug Rev. 2006, 24, 275–290. [Google Scholar] [CrossRef]
- Beltowski, J.; Kedra, A. Asymmetric dimethylarginine (ADMA) as a target for pharmacotherapy. Pharmacol. Rep. 2006, 58, 159–178. [Google Scholar]
- Wang, K.; Wang, Y.; Zhang, H.; Li, X.; Han, W. A Review of the Synthesis of Nitric Oxide Donor and Donor Derivatives with Pharmacological Activities. Mini. Rev. Med. Chem. 2022, 22, 873–883. [Google Scholar] [CrossRef]
- Tain, Y.L.; Yang, H.W.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Hsu, C.N. Anti-Hypertensive Property of an NO Nanoparticle in an Adenine-Induced Chronic Kidney Disease Young Rat Model. Antioxidants 2023, 12, 513. [Google Scholar] [CrossRef]
- Bonini, M.G.; Stadler, K.; Silva, S.O.; Corbett, J.; Dore, M.; Petranka, J.; Fernandes, D.C.; Tanaka, L.Y.; Duma, D.; Laurindo, F.R.; et al. Constitutive nitric oxide synthase activation is a significant route for nitroglycerin-mediated vasodilation. Proc. Natl. Acad. Sci. USA 2008, 105, 8569–8574. [Google Scholar] [CrossRef]
- Liu, T.; Schroeder, H.; Power, G.G.; Blood, A.B. A physiologically relevant role for NO stored in vascular smooth muscle cells: A novel theory of vascular NO signaling. Redox Biol. 2022, 53, 102327. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.J.; Lin, K.M.; Kuo, H.C.; Huang, C.F.; Lin, Y.J.; Huang, L.T.; Tain, Y.L. Two different approaches to restore renal nitric oxide and prevent hypertension in young spontaneously hypertensive rats: L-Citrulline and nitrate. Transl. Res. 2014, 163, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef] [PubMed]
- Gubler, M.C.; Antignac, C. Renin–angiotensin system in kidney development: Renal tubular dysgenesis. Kidney Int. 2010, 77, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Yosypiv, I.V. Renin-angiotensin system in mammalian kidney development. Pediatr. Nephrol. 2020, 36, 479–489. [Google Scholar] [CrossRef]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBOMol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef]
- Schulman, I.H.; Zhou, M.S.; Raij, L. Interaction between nitric oxide and angiotensin II in the endothelium: Role in atherosclerosis and hypertension. J. Hypertens. 2006, 24, S45–S50. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N.; Lin, C.Y.; Huang, L.T.; Lau, Y.T. Aliskiren prevents hypertension and reduces asymmetric dimethylarginine in young spontaneously hypertensive rats. Eur. J. Pharmacol. 2011, 670, 561–565. [Google Scholar] [CrossRef]
- Jansson, T.; Powell, T.L. Role of placental nutrient sensing in developmental programming. Clin. Obstet. Gynecol. 2013, 56, 591–601. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Interplay between oxidative stress and nutrient sensing signaling in the developmental origins of cardiovascular disease. Int. J. Mol. Sci. 2017, 18, 841. [Google Scholar] [CrossRef]
- Grahame Hardie, D. AMP-activated protein kinase: A key regulator of energy balance with many roles in human disease. J. Intern. Med. 2014, 276, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. AMP-Activated Protein Kinase as a Reprogramming Strategy for Hypertension and Kidney Disease of Developmental Origin. Int. J. Mol. Sci. 2018, 19, 1744. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. Molecular mechanisms underlying the activation of eNOS. Pflugers. Arch. 2010, 459, 793–806. [Google Scholar] [CrossRef]
- Parsamanesh, N.; Asghari, A.; Sardari, S.; Tasbandi, A.; Jamialahmadi, T.; Xu, S.; Sahebkar, A. Resveratrol and endothelial function: A literature review. Pharmacol. Res. 2021, 170, 105725. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lin, Y.J.; Sheen, J.M.; Lin, I.C.; Yu, H.R.; Huang, L.T.; Hsu, C.N. Resveratrol prevents the combined maternal plus postweaning high-fat-diets-induced hypertension in male offspring. J. Nutr. Biochem. 2017, 48, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef]
- Feliers, D.; Lee, H.J.; Kasinath, B.S. Hydrogen sulfide in renal physiology and disease. Antioxid. Redox Signal. 2016, 25, 720–731. [Google Scholar] [CrossRef]
- Cirino, G.; Vellecco, V.; Bucci, M. Nitric oxide and hydrogen sulfide: The gasotransmitter paradigm of the vascular system. Br. J. Pharmacol. 2017, 174, 4021–4031. [Google Scholar] [CrossRef]
- Zhong, G.; Chen, F.; Cheng, Y.; Tang, C.; Du, J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J. Hypertens. 2003, 21, 1879–1885. [Google Scholar] [CrossRef]
- Yuan, S.; Patel, R.P.; Kevil, C.G. Working with nitric oxide and hydrogen sulfide in biological systems. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L403–L415. [Google Scholar] [CrossRef]
- Szabo, C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: Mechanisms and implications. Am. J. Physiol. Cell Physiol. 2017, 312, C3–C15. [Google Scholar] [CrossRef]
- Hsu, C.N.; Tain, Y.L. Preventing Developmental Origins of Cardiovascular Disease: Hydrogen Sulfide as a Potential Target? Antioxidants 2021, 10, 247. [Google Scholar] [CrossRef]
- Hobby, G.P.; Karaduta, O.; Dusio, G.F.; Singh, M.; Zybailov, B.L.; Arthur, J.M. Chronic kidney disease and the gut microbiome. Am. J. Physiol. Renal Physiol. 2019, 316, F1211–F1217. [Google Scholar] [CrossRef]
- Arrieta, M.C.; Stiemsma, L.T.; Amenyogbe, N.; Brown, E.M.; Finlay, B. The intestinal microbiome in early life: Health and disease. Front. Immunol. 2014, 5, 427. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Yoo, J.Y.; Valeria Ozorio Dutra, S.; Morgan, K.H.; Groer, M. The Association between Early-Life Gut Microbiota and Long-Term Health and Diseases. J. Clin. Med. 2021, 10, 459. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal Garlic Oil Supplementation Prevents High-Fat Diet-Induced Hypertension in Adult Rat Offspring: Implications of H2S-Generating Pathway in the Gut and Kidneys. Mol. Nutr. Food Res. 2021, 65, e2001116. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L. Microbial short-chain fatty acids and blood pressure regulation. Curr. Hypertens. Rep. 2017, 19, 25. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hou, C.Y.; Hsu, W.H.; Tain, Y.L. Cardiovascular Diseases of Developmental Origins: Preventive Aspects of Gut Microbiota-Targeted Therapy. Nutrients 2021, 13, 2290. [Google Scholar] [CrossRef]
- Morikawa, A.; Sugiyama, T.; Koide, N.; Mori, I.; Mu, M.M.; Yoshida, T.; Hassan, F.; Islam, S.; Yokochi, T. Butyrate enhances the production of nitric oxide in mouse vascular endothelial cells in response to gamma interferon. J. Endotoxin Res. 2004, 10, 32–38. [Google Scholar] [CrossRef]
- Gao, B.; Jose, A.; Alonzo-Palma, N.; Malik, T.; Shankaranarayanan, D.; Regunathan-Shenk, R.; Raj, D.S. Butyrate producing microbiota are reduced in chronic kidney diseases. Sci. Rep. 2021, 11, 23530. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Animal Models | Species/ Gender | Age at Measure (Weeks) | NOS/NO System | Renal Programming | Ref. |
|---|---|---|---|---|---|
| Maternal zinc deficiency during gestation and lactation | Wistar rat/M and F | 3 | ↓ Renal NOS activity | ↓ Nephron number and glomerular hypertrophy | [89] |
| Maternal caloric restriction during gestation and lactation | SD rat/M and F | 8 | ↓ Urinary NOx level | ↑ BP and ↓ GFR | [90] |
| Maternal caloric restriction during gestation and lactation | SD rat/M | 12 | ↑ ADMA, ↓ AAR | ↓ Nephron number, ↑ tubulointerstitial injury, ↑ BP and glomerular hypertrophy, | [91,92] |
| Streptozotocin-induced maternal diabetes | SD rat/M | 12 | ↑ ADMA, ↓ AAR | ↓ nephron number, ↑ tubulointerstitial injury and ↑ BP | [86] |
| Maternal suramin exposure during gestation and lactation | SD rat/M | 12 | ↑ ADMA, ↓ AAR | ↑ BP | [93] |
| Maternal L-NAME administration during gestation and lactation | SD rat/M | 12 | ↓ Renal NO, ↓ urinary cGMP level | ↑BP, ↑ renal NHE3 protein level, and altered renal transcriptome | [81,94] |
| Maternal high-fructose diet during gestation and lactation | SD rat/M | 12 | ↑ ADMA, ↓ L-arginine, and ↓ AAR | ↑ BP and altered renal transcriptome | [83,95] |
| Maternal ADMA administration in gestation | SD rat/M | 12 | ↓ L-arginine and ↓ AAR | ↑ BP | [96] |
| Maternal TMAO administration in gestation | SD rat/M | 12 | ↓ L-arginine | ↑ BP and ↑ plasma Cr concentration | [96] |
| Maternal CKD | SD rat/M | 12 | ↑ ADMA, ↓ AAR | ↑ BP and renal hypertrophy | [97] |
| Maternal low-protein diet during gestation and lactation | Wistar rat/M | 14 | ↓ Renal eNOS phosphorylated protein level and ↓ urinary NOx level | ↑ BP | [98] |
| Prenatal dexamethasone administration at gestational days 15 and 16 | SD rat/M | 16 | ↑ ADMA, ↓ renal NO | ↑ BP and renal hypertrophy | [99] |
| Maternal high-fat diet during gestation and lactation | SD rat/M | 16 | ↓ L-arginine and ↓ AAR | ↑ BP | [100] |
| Maternal high-fructose diet in gestation | Wistar rat/F | 17 | ↓ Renal eNOS protein level | ↑ BP | [101] |
| Maternal high-fat diet during gestation and lactation | SD rat/M | 24 | ↑ ADMA and ↑ SDMA | ↑ Plasma Cr concentration | [102] |
| Maternal iron deficiency diet in pregnancy | SD rat/M | 24 | ↓ Renal NO | ↑ BP, ↑ renal collagen deposition, and glomerular hypertrophy | [103] |
| Fetal unilateral nephrectomy model | Sheep/M and F | 24 | ↓ Urinary NOx level | ↑ BP and ↓ GFR | [104] |
| Prenatal betamethasone exposure at gestational days 80 and 81 | Sheep/M and F | 72 | ↓ NO | ↑ BP | [105] |
| Interventions | Animal Models | Species/ Gender | Age at Measure (Weeks) | Protective Effect | Ref. |
|---|---|---|---|---|---|
| L-arginine + antioxidants | Genetic hypertension | FHH/M and F | 36 | Prevented high BP, proteinuria, and glomerulosclerosis | [107] |
| L-citrulline | Streptozotocin-induced maternal diabetes | SD rat/M | 12 | Prevented kidney damage and high BP and protected against reduced nephron number | [86] |
| L-citrulline | Maternal caloric restriction | SD rat/M | 12 | Prevented kidney damage and protected against reduced nephron number | [91] |
| L-citrulline | Maternal L-NAME administration | SD rat/M | 12 | Prevented high BP | [94] |
| L-citrulline | Prenatal dexamethasone exposure | SD rat/M | 12 | Prevented high BP and protected against reduced nephron number | [99] |
| L-citrulline | Genetic hypertension | SHR/M and F | 50 | Prevented high BP | [108] |
| Melatonin | Maternal caloric restriction | SD rat/M | 12 | Prevented high BP | [92] |
| Resveratrol | Prenatal dexamethasone plus TCDD exposure | SD rat/M | 12 | Prevented high BP | [109] |
| Garlic oil | Maternal CKD | SD rat/M | 12 | Prevented high BP | [110] |
| Butyrate | Maternal high-fructose diet | SD rat/M | 12 | Prevented high BP | [95] |
| N-acetylcysteine | Prenatal dexamethasone plus postnatal high-fat diet | SD rat/M | 16 | Prevented high BP | [111] |
| Dimethyl fumarate | Prenatal dexamethasone plus postnatal high-fat diet | SD rat/M | 16 | Prevented high BP | [112] |
| Pentaerythritol tetranitrate | Genetic hypertension | SHR/M and F | 24 | Prevented high BP | [113] |
| Molsidomine | Genetic hypertension | FHH/M and F | 42 | Prevented high BP | [114] |
| Melinjo (Gnetum gnemon) seed extract | Maternal high-fructose diet | Wistar rat/F | 17 | Prevented high BP | [101] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tain, Y.-L.; Hsu, C.-N. The NOS/NO System in Renal Programming and Reprogramming. Antioxidants 2023, 12, 1629. https://doi.org/10.3390/antiox12081629
Tain Y-L, Hsu C-N. The NOS/NO System in Renal Programming and Reprogramming. Antioxidants. 2023; 12(8):1629. https://doi.org/10.3390/antiox12081629
Chicago/Turabian StyleTain, You-Lin, and Chien-Ning Hsu. 2023. "The NOS/NO System in Renal Programming and Reprogramming" Antioxidants 12, no. 8: 1629. https://doi.org/10.3390/antiox12081629