
Nitric Oxide/Nitric Oxide Synthase System in the Pathogenesis of Neurodegenerative Disorders—An Overview
 ,
,
Abstract
:1. Introduction
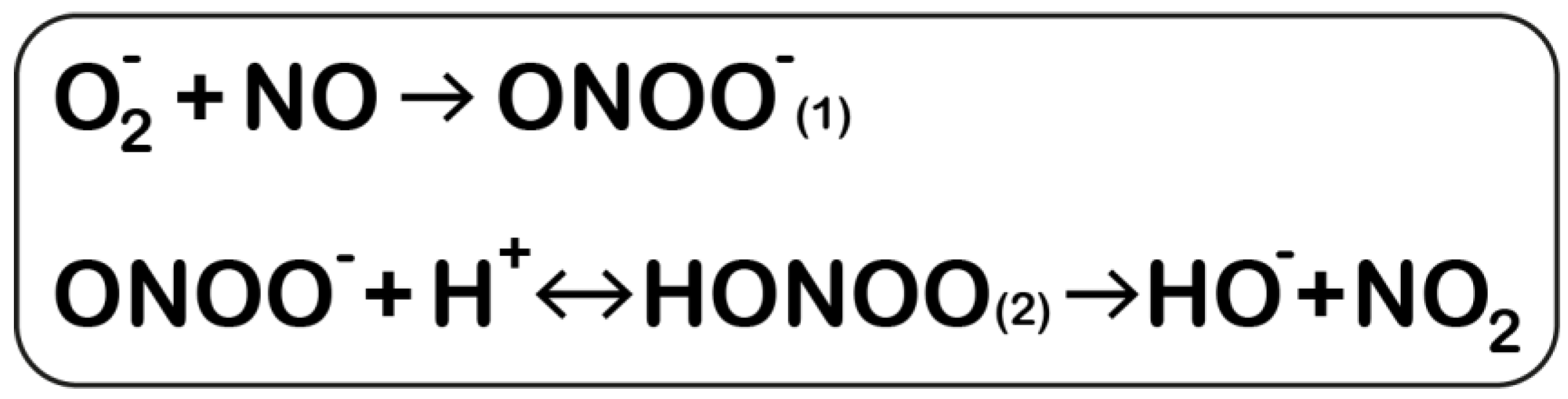
1.1. NO Formation, NOS Function, and Reactive Nitrogen Species Formation
1.2. NO/NOS in Cellular Life
1.3. NO/NOS Influence on the Blood Brain Barrier Permeability
2. Parkinson’s Disease
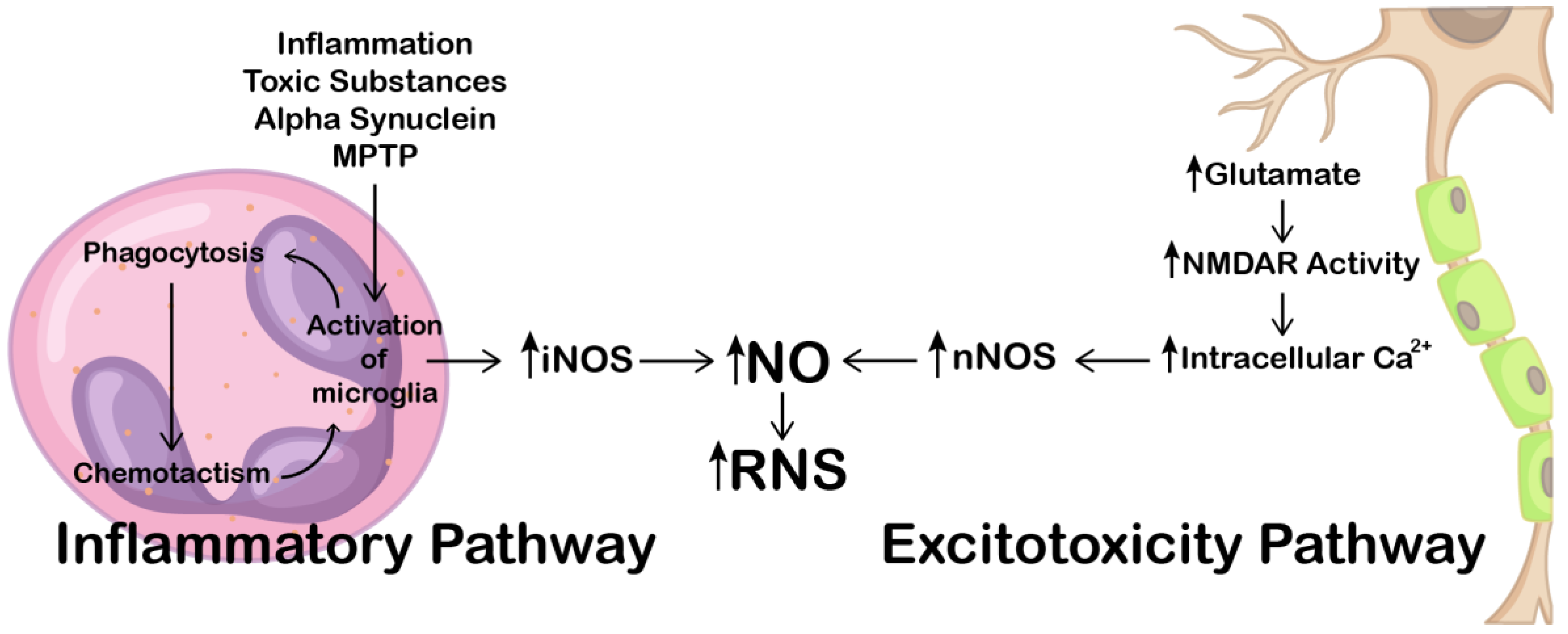
2.1. Implications of NO and NOS in Parkinson’s Disease
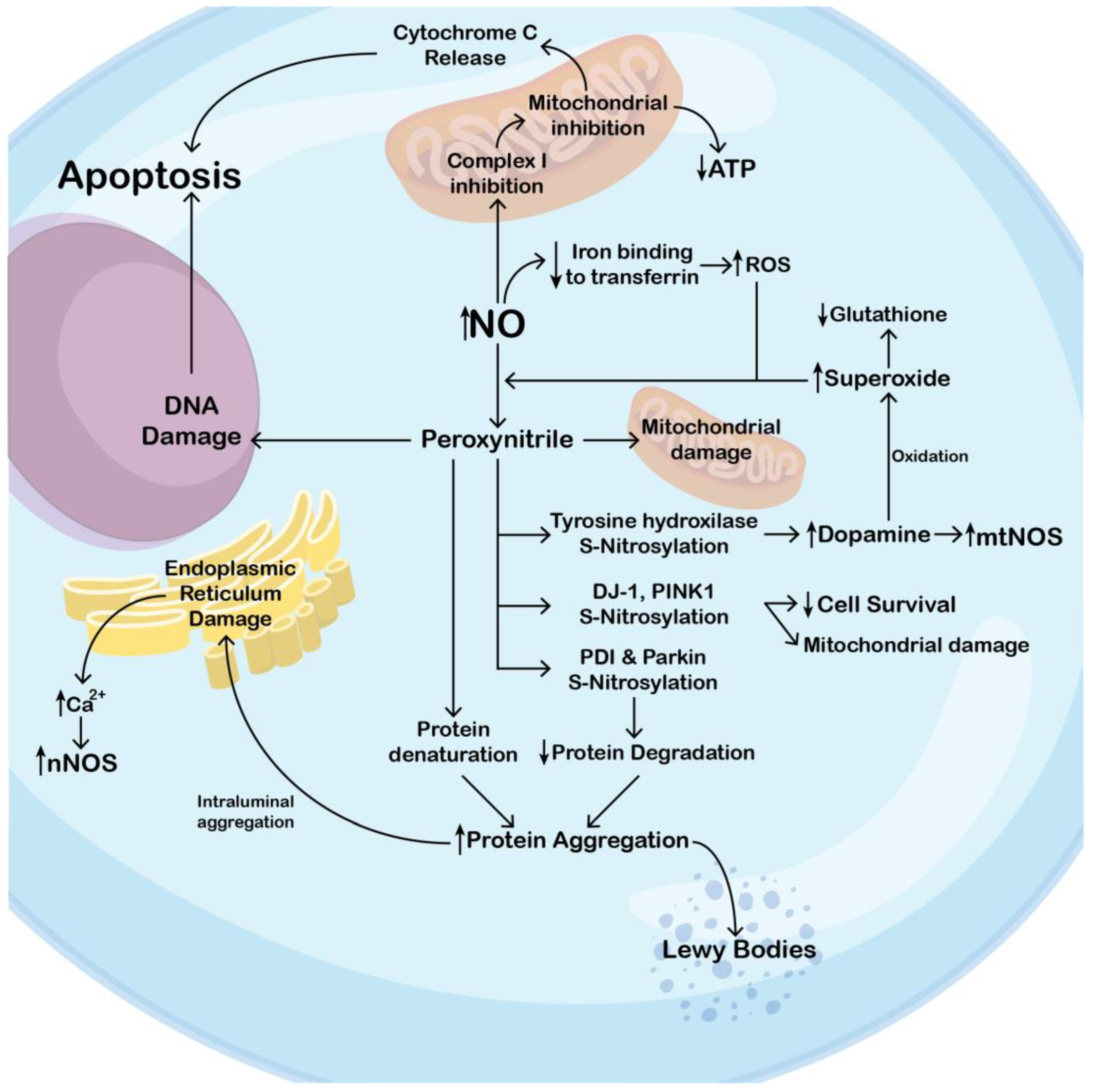
2.2. Importance of S-Nitrosylation in Parkinson’s Disease
2.3. Peroxynitrile’s Role in Parkinson’s Disease
2.4. Mitochondrial Damage in Parkinson’s Disease
2.5. Dopamine Metabolism and NO/NOS System in PD
3. Alzheimer’s Disease
3.1. S-Nitrosylation in Alzheimer’s Disease
3.2. Protein Tyrosine Nitration
3.3. Signaling via sGC and cGMP Pathway
3.4. NO in Memory and Learning
3.5. nNOS, eNOS and Cerebral Blood Flow in Alzheimer’s Disease
3.6. eNOS and Inflammation in Alzheimer’s Disease
3.7. iNOS and Inflammation in Alzheimer’s Disease
3.8. NO and Oxidative Stress-Associated Lipid Peroxidation in Alzheimer’s Disease
3.9. NO and Mitochondria in Alzheimer’s Disease
4. Amyotrophic Lateral Sclerosis
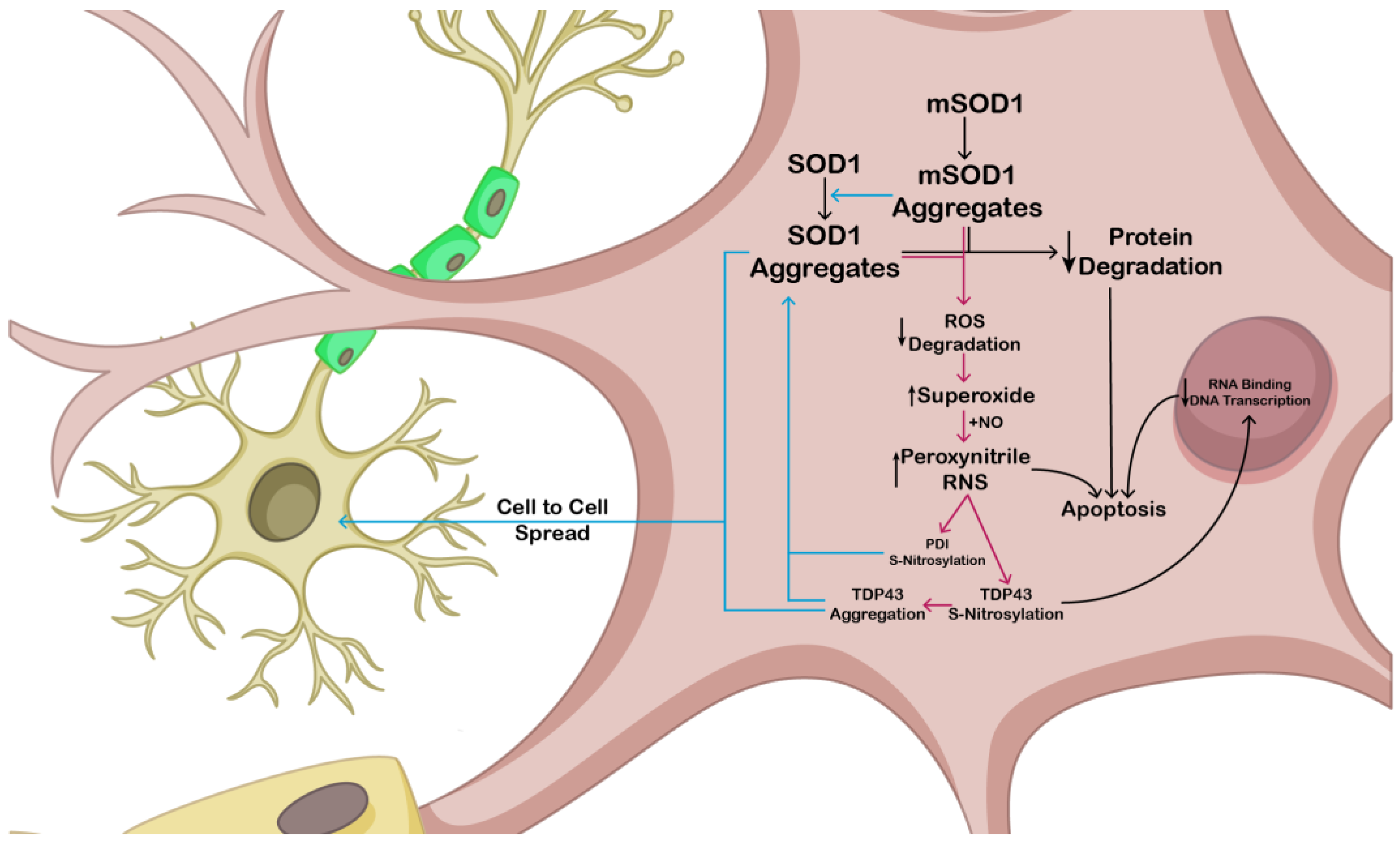
Involvment of NO in the Patogenisis of Amyotrophic Lateral Sclerosis
5. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wierońska, J.M.; Cieślik, P.; Kalinowski, L. Nitric Oxide-Dependent Pathways as Critical Factors in the Consequences and Recovery after Brain Ischemic Hypoxia. Biomolecules 2021, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Role of Nitric Oxide Synthase in Normal Brain Function and Pathophysiology of Neural Diseases | IntechOpen. Available online: https://www.intechopen.com/chapters/53963 (accessed on 31 October 2022).
- Kim, T.A.; Chen, L.; Ge, S. The Interplay of Neurovasculature and Adult Hippocampal Neurogenesis. Neurosci. Lett. 2021, 760, 136071. [Google Scholar] [CrossRef] [PubMed]
- Domek-Łopacińska, K.U.; Strosznajder, J.B. Cyclic GMP and Nitric Oxide Synthase in Aging and Alzheimer’s Disease. Mol. NeuroBiol. 2010, 41, 129–137. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends Pharmacol. Sci. 2016, 37, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.-I.; Lipton, S.A. Aberrant Protein S-Nitrosylation in Neurodegenerative Diseases. Neuron 2013, 78, 596–614. [Google Scholar] [CrossRef] [Green Version]
- Picón-Pagès, P.; Garcia-Buendia, J.; Muñoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef] [PubMed]
- Ledo, A.; Lourenço, C.F.; Cadenas, E.; Barbosa, R.M.; Laranjinha, J. The bioactivity of neuronal-derived nitric oxide in aging and neurodegeneration: Switching signaling to degeneration. Free Radic. Biol. Med. 2021, 162, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Sircar, E.; Rai, S.R.; Wilson, M.A.; Schlossmacher, M.G.; Sengupta, R. Neurodegeneration: Impact of S-nitrosylated Parkin, DJ-1 and PINK1 on the pathogenesis of Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108869. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Kartawy, M.; Amal, H. The role of nitric oxide in brain disorders: Autism spectrum disorder and other psychiatric, neurological, and neurodegenerative disorders. Redox Biol. 2020, 34, 101567. [Google Scholar] [CrossRef]
- Kavyaa, R.K.; Dikshit, M. Role of Nitric Oxide/Nitric Oxide Synthase in Parkinson’s Disease. Ann. Neurosci. 2010, 12, 1–5. [Google Scholar] [CrossRef]
- Alderton, W.K.; Angell, A.D.R.; Craig, C.; Dawson, J.; Garvey, E.; Moncada, S.; Monkhouse, J.; Rees, D.; Russell, L.J.; Russell, R.J.; et al. GW274150 and GW273629 are potent and highly selective inhibitors of inducible nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 2005, 145, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Aquilano, K.; Baldelli, S.; Rotilio, G.; Ciriolo, M.R. Role of nitric oxide synthases in Parkinson’s disease: A review on the antioxidant and anti-inflammatory activity of polyphenols. Neurochem. Res. 2008, 33, 2416–2426. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; Herrero, M.T.; García-Martín, E.; Agúndez, J. An Update on the Role of Nitric Oxide in the Neurodegenerative Processes of Parkinson’s Disease. Curr. Med. Chem. 2016, 23, 2666–2679. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Chen, J.; Crow, J.P.; Zu, Y. Chapter 31 Reactions of nitric oxide, superoxide and peroxynitrite with superoxide dismutase in neurodegeneration. Prog. Brain Res. 1994, 103, 371–380. [Google Scholar] [CrossRef]
- Tohgi, H.; Abe, T.; Yamazaki, K.; Murata, T.; Ishizaki, E.; Isobe, C. Increase in oxidized NO products and reduction in oxidized glutathione in cerebrospinal fluid from patients with sporadic form of amyotrophic lateral sclerosis. Neurosci. Lett. 1999, 260, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.L.; Wu, D.; Jakes, S.; Graves, D.J. Chemical influences on the specificity of tyrosine phosphorylation. J. Biol. Chem. 1990, 265, 7108–7111. [Google Scholar] [CrossRef]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Cros, E.T. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, J.O.; Weitzberg, E. Nitric oxide signaling in health and disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. (BBA) Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemi, M.; Mayasi, Y.; Hannoun, A.; Eslami, S.M.; Carandang, R. Nitric Oxide and Mitochondrial Function in Neurological Diseases. Neuroscience 2018, 376, 48–71. [Google Scholar] [CrossRef]
- Asiimwe, N.; Yeo, S.G.; Kim, M.-S.; Jung, J.; Jeong, N.Y. Nitric Oxide: Exploring the Contextual Link with Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2016, 2016, 7205747. [Google Scholar] [CrossRef] [Green Version]
- Laranjinha, J.; Nunes, C.; Ledo, A.; Lourenço, C.; Rocha, B.; Barbosa, R.M. The Peculiar Facets of Nitric Oxide as a Cellular Messenger: From Disease-Associated Signaling to the Regulation of Brain Bioenergetics and Neurovascular Coupling. Neurochem. Res. 2021, 46, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Kourosh-Arami, M.; Hosseini, N.; Mohsenzadegan, M.; Komaki, A.; Joghataei, M.T. Neurophysiologic implications of neuronal nitric oxide synthase. Rev. Neurosci. 2020, 31, 617–636. [Google Scholar] [CrossRef]
- Lourenço, C.F.; Laranjinha, J. Nitric Oxide Pathways in Neurovascular Coupling Under Normal and Stress Conditions in the Brain: Strategies to Rescue Aberrant Coupling and Improve Cerebral Blood Flow. Front. Physiol. 2021, 12, 729201. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S193–S201. [Google Scholar] [CrossRef] [Green Version]
- Cellek, S.; Moncada, S. Nitrergic control of peripheral sympathetic responses in the human corpus cavernosum: A comparison with other species. Proc. Natl. Acad. Sci. USA 1997, 94, 8226–8231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourbagher-Shahri, A.M.; Farkhondeh, T.; Talebi, M.; Kopustinskiene, D.M.; Samarghandian, S.; Bernatoniene, J. An Overview of NO Signaling Pathways in Aging. Molecules 2021, 26, 4533. [Google Scholar] [CrossRef] [PubMed]
- Vyklicky, V.; Korinek, M.; Smejkalova, T.; Balik, A.; Krausova, B.; Kaniakova, M.; Lichnerova, K.; Černý, J.; Krusek, J.; Dittert, I.; et al. Structure, Function, and Pharmacology of NMDA Receptor Channels. Physiol. Res. 2014, 63, S191–S203. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Kelm, M. Nitric oxide metabolism and breakdown. Biochim. Biophys. Acta (BBA) Bioenerg. 1999, 1411, 273–289. [Google Scholar] [CrossRef] [Green Version]
- Liy, P.M.; Puzi, N.N.A.; Jose, S.; Vidyadaran, S. Nitric oxide modulation in neuroinflammation and the role of mesenchymal stem cells. Exp. Biol. Med. 2021, 246, 2399–2406. [Google Scholar] [CrossRef]
- Verma, M.; Wills, Z.; Chu, C.T. Excitatory Dendritic Mitochondrial Calcium Toxicity: Implications for Parkinson’s and Other Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, T.N.; Arseneault, M.; Murthy, V.; Ramassamy, C. Potential role of acrolein in neurodegeneration and in Alzheimer’s disease. Curr. Mol. Pharmacol. 2010, 3, 66–78. [Google Scholar]
- Wang, Y.; Hong, F.; Yang, S. Roles of Nitric Oxide in Brain Ischemia and Reperfusion. Int. J. Mol. Sci. 2022, 23, 4243. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zhou, C.; Piao, Z.; Yuan, H.; Jiang, H.; Wei, H.; Zhou, Y.; Nan, G.; Ji, X. Cerebral edema after ischemic stroke: Pathophysiology and underlying mechanisms. Front. Neurosci. 2022, 16, 988283. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, C.; Arrick, D.M.; Yang, S.; Baluna, A.E.; Sun, H. Role of Nitric Oxide Synthases in Early Blood-Brain Barrier Disruption following Transient Focal Cerebral Ischemia. PLoS ONE 2014, 9, e93134. [Google Scholar] [CrossRef]
- Grandati, M.; Verrecchia, C.; Revaud, M.L.; Allix, M.; Boulu, R.G.; Plotkine, M. Calcium-independent NO-synthase activity and nitrites/nitrates production in transient focal cerebral ischaemia in mice. Br. J. Pharmacol. 1997, 122, 625–630. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C.; Xu, X.; Zhang, F.; el-Fakahany, E.E.; Ross, M.E. Marked induction of calcium-independent nitric oxide synthase activity after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1995, 15, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, S.; Osuka, K.; Takata, T.; Tsuchiya, Y.; Watanabe, Y. Coordination between Calcium/Calmodulin-Dependent Protein Kinase II and Neuronal Nitric Oxide Synthase in Neurons. Int. J. Mol. Sci. 2020, 21, 7997. [Google Scholar] [CrossRef]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Vemuganti, R. Mechanisms of Parkinson’s disease-related proteins in mediating secondary brain damage after cerebral ischemia. J. Cereb. Blood Flow Metab. 2017, 37, 1910–1926. [Google Scholar] [CrossRef] [Green Version]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Proteomic and Genomic Changes in Tau Protein, Which Are Associated with Alzheimer’s Disease after Ischemia-Reperfusion Brain Injury. Int. J. Mol. Sci. 2020, 21, 892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.L.; James-Kracke, M.; Sun, G.Y.; Sun, A.Y. Oxidative and Inflammatory Pathways in Parkinson’s Disease. Neurochem. Res. 2009, 34, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.; Cianciulli, A.; Trotta, T.; Lofrumento, D.D.; Calvello, R.; Panaro, M.A. Formyl-methionyl-leucyl-phenylalanine Induces Apoptosis in Murine Neurons: Evidence for NO-Dependent Caspase-9 Activation. Biology 2019, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.-J.; Kuo, W.-W.; Liu, W.-H.; Yin, M.-C. Antioxidative and anti-inflammatory protection from carnosine in the striatum of MPTP-treated mice. J. Agric. Food Chem. 2010, 58, 11510–11516. [Google Scholar] [CrossRef] [PubMed]
- Liberatore, G.T.; Jackson-Lewis, V.; Vukosavic, S.; Mandir, A.S.; Vila, M.; McAuliffe, W.G.; Dawson, V.L.; Dawson, T.M.; Przedborski, S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat. Med. 1999, 5, 1403–1409. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, H.; Liu, M.; Zhang, Z.; Wei, Z.; Sun, N.; Mao, T.; Zhang, Y. P38 MAPK signaling pathway regulates nuclear factor-κB and inducible nitric oxide synthase expressions in the substantia nigra in a mouse model of Parkinson’s disease. Nan Fang Yi Ke Da Xue Xue Bao 2014, 34, 1176–1180. [Google Scholar] [PubMed]
- Larsen, T.R.; Söderling, A.-S.; Caidahl, K.; Roepstorff, P.; Gramsbergen, J.B. Nitration of soluble proteins in organotypic culture models of Parkinson’s disease. Neurochem. Int. 2008, 52, 487–494. [Google Scholar] [CrossRef]
- Aras, S.; Tanriover, G.; Aslan, M.; Yargicoglu, P.; Agar, A. The role of nitric oxide on visual-evoked potentials in MPTP-induced Parkinsonism in mice. Neurochem. Int. 2014, 72, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V.; Yokoyama, R.; Shibata, T.; Dawson, V.L.; Dawson, T.M. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc. Natl. Acad. Sci. USA 1996, 93, 4565–4571. [Google Scholar] [CrossRef] [Green Version]
- Calvello, R.; Cianciulli, A.; Nicolardi, G.; De Nuccio, F.; Giannotti, L.; Salvatore, R.; Porro, C.; Trotta, T.; Panaro, M.A.; Lofrumento, D.D. Vitamin D Treatment Attenuates Neuroinflammation and Dopaminergic Neurodegeneration in an Animal Model of Parkinson’s Disease, Shifting M1 to M2 Microglia Responses. J. Neuroimmune Pharmacol. 2017, 12, 327–339. [Google Scholar] [CrossRef] [PubMed]
- De Nuccio, F.; Cianciulli, A.; Porro, C.; Kashyrina, M.; Ruggiero, M.; Calvello, R.; Miraglia, A.; Nicolardi, G.; Lofrumento, D.D.; Panaro, M.A. Inflammatory Response Modulation by Vitamin C in an MPTP Mouse Model of Parkinson’s Disease. Biology 2021, 10, 1155. [Google Scholar] [CrossRef]
- Singh, S.; Das, T.; Ravindran, A.; Chaturvedi, R.K.; Shukla, Y.; Agarwal, A.K.; Dikshit, M. Involvement of nitric oxide in neurodegeneration: A study on the experimental models of Parkinson’s disease. Redox Rep. 2005, 10, 103–109. [Google Scholar] [CrossRef]
- Barthwal, M.K.; Srivastava, N.; Dikshit, M. Role of nitric oxide in a progressive neurodegeneration model of Parkinson’s disease in the rat. Redox Rep. 2001, 6, 297–302. [Google Scholar] [CrossRef]
- Eve, D.J.; Nisbet, A.P.; Kingsbury, A.E.; Hewson, E.L.; Daniel, S.E.; Lees, A.J.; Marsden, C.D.; Foster, O.J.F. Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Mol. Brain Res. 1998, 63, 62–71. [Google Scholar] [CrossRef]
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 2000, 16, 724–739. [Google Scholar] [CrossRef]
- Li, L.; Li, L. Recent advances in multinuclear metal nitrosyl complexes. Coord. Chem. Rev. 2016, 306, 678–700. [Google Scholar] [CrossRef] [Green Version]
- Shergill, J.K.; Cammack, R.; Cooper, C.E.; Cooper, J.M.; Mann, V.M.; Schapira, A.H. Detection of nitrosyl complexes in human substantia nigra, in relation to Parkinson’s disease. Biochem. Biophys. Res. Commun. 1996, 228, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.M.; Riobó, N.A.; Carreras, M.C.; Cherñavsky, A.; Rubio, A.; Satz, M.L.; Poderoso, J.J. Overexpression of Neutrophil Neuronal Nitric Oxide Synthase in Parkinson’s Disease. Nitric Oxide 2000, 4, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Pahan, K. Regulation of Inducible Nitric Oxide Synthase Gene in Glial Cells. Antioxid. Redox Signal. 2006, 8, 929–947. [Google Scholar] [CrossRef] [PubMed]
- Hancock, D.B.; Martin, E.R.; Vance, J.M.; Scott, W.K. Nitric oxide synthase genes and their interactions with environmental factors in Parkinson’s disease. Neurogenetics 2008, 9, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Nunes, C.; Laranjinha, J. Nitric oxide and dopamine metabolism converge via mitochondrial dysfunction in the mechanisms of neurodegeneration in Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108877. [Google Scholar] [CrossRef] [PubMed]
- Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/12446870/ (accessed on 12 November 2022).
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.; Singh, S. Reciprocal Upshot of Nitric Oxide, Endoplasmic Reticulum Stress, and Ubiquitin Proteasome System in Parkinson’s Disease Pathology. Neuroscientist 2021, 27, 340–354. [Google Scholar] [CrossRef]
- Chung, K.K.K.; Thomas, B.; Li, X.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, V.L.; Dawson, T.M. S-Nitrosylation of Parkin Regulates Ubiquitination and Compromises Parkin’s Protective Function. Science 2004, 304, 1328–1331. [Google Scholar] [CrossRef]
- Sunico, C.R.; Nakamura, T.; Rockenstein, E.; Mante, M.; Adame, A.; Chan, S.F.; Newmeyer, T.F.; Masliah, E.; Nakanishi, N.; Lipton, S.A. S-Nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson’s disease. Mol. Neurodegener. 2013, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.-Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deas, E.; Plun-Favreau, H.; Wood, N.W. PINK1 function in health and disease. EMBO Mol. Med. 2009, 1, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta (BBA) Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Y.; Tang, H.; Chen, W.-X.; Ji, G.-J.; Ye, J.; Wang, N.; Wu, J.-T.; Guan, B. Mapping the functional connectivity of the substantia nigra, red nucleus and dentate nucleus: A network analysis hypothesis associated with the extrapyramidal system. Neurosci. Lett. 2015, 606, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Takagi, S.; Watanabe, Y.; Kato, H.; Araki, T. Role of reactive nitrogen and reactive oxygen species against MPTP neurotoxicity in mice. J. Neural Transm. 2008, 115, 831–842. [Google Scholar] [CrossRef]
- McConkey, D.J.; Orrenius, S. The Role of Calcium in the Regulation of Apoptosis. Biochem. Biophys. Res. Commun. 1997, 239, 357–366. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Nitric Oxide-Dependent Protein Post-Translational Modifications Impair Mitochondrial Function and Metabolism to Contribute to Neurodegenerative Diseases. Antioxidants Redox Signal. 2020, 32, 817–833. [Google Scholar] [CrossRef]
- Iljina, M.; Garcia, G.A.; Horrocks, M.H.; Tosatto, L.; Choi, M.L.; Ganzinger, K.A.; Abramov, A.Y.; Gandhi, S.; Wood, N.W.; Cremades, N.; et al. Kinetic model of the aggregation of alpha-synuclein provides insights into prion-like spreading. Proc. Natl. Acad. Sci. USA 2016, 113, E1206–E1215. [Google Scholar] [CrossRef] [Green Version]
- Crichton, R.; Ward, R. (Eds.) Oxidative Stress in Neurodegenerative Diseases. In Metal-Based Neurodegeneration; John Wiley and Sons Ltd.: Chichester, UK, 2013; pp. 75–109. ISBN 978-1-118-55348-0. [Google Scholar]
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine Oxidation and Autophagy. Park. Dis. 2012, 2012, e920953. [Google Scholar] [CrossRef] [Green Version]
- Kavya, R.; Saluja, R.; Singh, S.; Dikshit, M. Nitric oxide synthase regulation and diversity: Implications in Parkinson’s disease. Nitric Oxide 2006, 15, 280–294. [Google Scholar] [CrossRef]
- Dubey, H.; Gulati, K.; Ray, A. Alzheimer’s Disease: A Contextual Link with Nitric Oxide Synthase. Curr. Mol. Med. 2020, 20, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Aliev, G.; Palacios, H.H.; Lipsitt, A.E.; Fischbach, K.; Lamb, B.T.; Obrenovich, M.E.; Morales, L.; Gasimov, E.; Bragin, V. Nitric Oxide as an Initiator of Brain Lesions During the Development of Alzheimer Disease. Neurotox. Res. 2009, 16, 293–305. [Google Scholar] [CrossRef]
- Hannibal, L. Nitric Oxide Homeostasis in Neurodegenerative Diseases. Curr. Alzheimer Res. 2016, 13, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Xiao, B.; Wang, L.-P.; Li, Y.-S.; Jin, H.; Jin, Q.-H. Nitric oxide impairs spatial learning and memory in a rat model of Alzheimer’s disease via disturbance of glutamate response in the hippocampal dentate gyrus during spatial learning. Behav. Brain Res. 2022, 422, 113750. [Google Scholar] [CrossRef] [PubMed]
- Balez, R.; Ooi, L. Getting to NO Alzheimer’s Disease: Neuroprotection versus Neurotoxicity Mediated by Nitric Oxide. Oxid Med. Cell. Longev. 2016, 2016, 3806157. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, S. Role of Neuron and Glia in Alzheimer’s Disease and Associated Vascular Dysfunction. Front. Aging Neurosci. 2021, 13, 653334. [Google Scholar] [CrossRef]
- Zhu, X.; Smith, M.A.; Honda, K.; Aliev, G.; Moreira, P.I.; Nunomura, A.; Casadesus, G.; Harris, P.L.R.; Siedlak, S.L.; Perry, G. Vascular oxidative stress in Alzheimer disease. J. Neurol. Sci. 2007, 257, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Lane, R.M.; Farlow, M.R. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. J. Lipid Res. 2005, 46, 949–968. [Google Scholar] [CrossRef] [Green Version]
- Spiers, J.G.; Chen, H.-J.C.; Bourgognon, J.-M.; Steinert, J.R. Dysregulation of stress systems and nitric oxide signaling underlies neuronal dysfunction in Alzheimer’s disease. Free Radic. Biol. Med. 2019, 134, 468–483. [Google Scholar] [CrossRef]
- Abrams, A.J.; Farooq, A.; Wang, G. S-Nitrosylation of ApoE in Alzheimer’s Disease. Biochemistry 2011, 50, 3405–3407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Vizarra, P.; Fernández, A.P.; Castro-Blanco, S.; Encinas, J.M.; Serrano, J.; Bentura, M.L.; Muñoz, P.; Martínez-Murillo, R.; Rodrigo, J. Expression of nitric oxide system in clinically evaluated cases of Alzheimer’s disease. Neurobiol. Dis. 2004, 15, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Vallebuona, F.; Raiteri, M. Age-related Changes in the NMDA Receptor/Nitric Oxide/cGMP Pathway in the Hippocampus and Cerebellum of Freely Moving Rats Subjected to Transcerebral Microdialysis. Eur. J. Neurosci. 1995, 7, 694–701. [Google Scholar] [CrossRef]
- Chalimoniuk, M.; Strosznajder, J.B. Aging modulates nitric oxide synthesis and cGMP levels in hippocampus and cerebellum. Mol. Chem. Neuropathol. 1998, 35, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.-S.; Kim, K.-M.; Kwon, Y.-G.; Bai, S.-K.; Nam, W.-D.; Yoo, Y.-M.; Kim, P.K.M.; Chung, H.-T.; Billiar, T.R.; Kim, Y.-M. Nitric oxide prevents 6-hydroxydopamine-induced apoptosis in PC12 cells through cGMP-dependent PI3 kinase/Akt activation. FASEB J. 2003, 17, 1036–1047. [Google Scholar] [CrossRef]
- Bobba, A.; Atlante, A.; Moro, L.; Calissano, P.; Marra, E. Nitric oxide has dual opposite roles during early and late phases of apoptosis in cerebellar granule neurons. Apoptosis 2007, 12, 1597–1610. [Google Scholar] [CrossRef]
- Dias, C.; Lourenço, C.F.; Ferreiro, E.; Barbosa, R.M.; Laranjinha, J.; Ledo, A. Age-dependent changes in the glutamate-nitric oxide pathway in the hippocampus of the triple transgenic model of Alzheimer’s disease: Implications for neurometabolic regulation. NeuroBiol. Aging 2016, 46, 84–95. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, D.-Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009, 20, 223–230. [Google Scholar] [CrossRef]
- Huang, E.P. Synaptic plasticity: A role for nitric oxide in LTP. Curr. Biol. 1997, 7, R141–R143. [Google Scholar] [CrossRef] [Green Version]
- Collingridge, G. The role of NMDA receptors in learning and memory. Nature 1987, 330, 604–605. [Google Scholar] [CrossRef]
- Böhme, G.A.; Bon, C.; Lemaire, M.; Reibaud, M.; Piot, O.; Stutzmann, J.M.; Doble, A.; Blanchard, J.C. Altered synaptic plasticity and memory formation in nitric oxide synthase inhibitor-treated rats. Proc. Natl. Acad. Sci. USA 1993, 90, 9191–9194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickard, N.S.; Gibbs, M.E. Effects of nitric oxide inhibition on avoidance learning in the chick are lateralized and localized. Neurobiol. Learn. Mem. 2003, 79, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, D.T.; Dewar, D.; Graham, D.I.; Brooks, D.N.; McCulloch, J. Differential alterations of cortical glutamatergic binding sites in senile dementia of the Alzheimer type. Proc. Natl. Acad. Sci. USA 1990, 87, 1352–1356. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.R.P.; Wyper, D.J.; Owens, J.; Patterson, J.; Kelly, R.C.; Hunter, R.; McCulloch, J. 123Iodo-MK-801: A spect agent for imaging the pattern and extent of glutamate (NMDA) receptor activation in Alzheimer’s disease. J. Psychiatr. Res. 1997, 31, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Nitric oxide and neuronal death. Nitric Oxide 2010, 23, 153–165. [Google Scholar] [CrossRef]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.J.; MacVicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298 Pt 2, 249–258. [Google Scholar] [CrossRef]
- Fanet, H.; Capuron, L.; Castanon, N.; Calon, F.; Vancassel, S. Tetrahydrobioterin (BH4) Pathway: From Metabolism to Neuropsychiatry. Curr. Neuropharmacol. 2021, 19, 591–609. [Google Scholar] [CrossRef]
- Iadecola, C. Regulation of the cerebral microcirculation during neural activity: Is nitric oxide the missing link? Trends Neurosci. 1993, 16, 206–214. [Google Scholar] [CrossRef]
- Price, J.; Sutton, T.; Hellermann, A.; Thomas, T. β-Amyloid induces cerebrovascular endothelial dysfunction in the rat brain. Neurol. Res. 1997, 19, 534–538. [Google Scholar] [CrossRef]
- Lourenço, C.F.; Ledo, A.; Barbosa, R.M.; Laranjinha, J. Neurovascular uncoupling in the triple transgenic model of Alzheimer’s disease: Impaired cerebral blood flow response to neuronal-derived nitric oxide signaling. Exp. Neurol. 2017, 291, 36–43. [Google Scholar] [CrossRef]
- Chrissobolis, S.; Faraci, F.M. The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends Mol. Med. 2008, 14, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, N.; Okamura, T. Cerebral blood flow regulation by nitric oxide in Alzheimer’s disease. J. Alzheimers Dis. 2012, 32, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Lüth, H.-J.; Münch, G.; Arendt, T. Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res. 2002, 953, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Lüth, H.-J.; Holzer, M.; Gärtner, U.; Staufenbiel, M.; Arendt, T. Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer’s disease, in APP23 transgenic mice and after experimental brain lesion in rat: Evidence for an induction by amyloid pathology. Brain Res. 2001, 913, 57–67. [Google Scholar] [CrossRef]
- Katusic, Z.S.; Austin, S.A. Endothelial nitric oxide: Protector of a healthy mind. Eur. Heart J. 2014, 35, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Minhas, R.; Bansal, Y.; Bansal, G. Inducible nitric oxide synthase inhibitors: A comprehensive update. Med. Res. Rev. 2020, 40, 823–855. [Google Scholar] [CrossRef]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Grad, L.I.; Rouleau, G.A.; Ravits, J.; Cashman, N.R. Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harb. Perspect. Med. 2017, 7, a024117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amyotrophic Lateral Sclerosis (ALS). Fact Sheet|National Institute of Neurological Disorders and Stroke. Available online: https://www.ninds.nih.gov/amyotrophic-lateral-sclerosis-als-fact-sheet (accessed on 5 November 2022).
- Motor Neuron Diseases. Fact Sheet|National Institute of Neurological Disorders and Stroke. Available online: https://www.ninds.nih.gov/motor-neuron-diseases-fact-sheet (accessed on 5 November 2022).
- Ghatak, N.R.; Campbell, W.W.; Lippman, R.H.; Hadfield, M.G. Anterior horn changes of motor neuron disease associated with demyelinating radiculopathy. J. Neuropathol. Exp. Neurol. 1986, 45, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.T. Pathology of amyotrophic lateral sclerosis. Adv. Neurol. 1982, 36, 61–74. [Google Scholar] [PubMed]
- Hammer, R.P.; Tomiyasu, U.; Scheibel, A.B. Degeneration of the human Betz cell due to amyotrophic lateral sclerosis. Exp. Neurol. 1979, 63, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Udaka, F.; Kameyama, M.; Tomonaga, M. Degeneration of Betz cells in motor neuron disease. A Golgi study. Acta Neuropathol. 1986, 70, 289–295. [Google Scholar] [CrossRef]
- Maekawa, S.; Al-Sarraj, S.; Kibble, M.; Landau, S.; Parnavelas, J.; Cotter, D.; Everall, I.; Leigh, P.N. Cortical selective vulnerability in motor neuron disease: A morphometric study. Brain 2004, 127, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- A Unique Pattern of Astrocytosis in the Primary Motor Area in Amyotrophic Lateral Sclerosis—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/1785258/ (accessed on 5 November 2022).
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992, 140, 691–707. [Google Scholar]
- Schiffer, D.; Cordera, S.; Cavalla, P.; Migheli, A. Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J. Neurol. Sci. 1996, 139, 27–33. [Google Scholar] [CrossRef]
- Lowe, J.; Lennox, G.; Jefferson, D.; Morrell, K.; McQuire, D.; Gray, T.; Landon, M.; Doherty, F.J.; Mayer, R.J. A filamentous inclusion body within anterior horn neurones in motor neurone disease defined by immunocytochemical localisation of ubiquitin. Neurosci. Lett. 1988, 94, 203–210. [Google Scholar] [CrossRef]
- Leigh, P.N.; Anderton, B.H.; Dodson, A.; Gallo, J.-M.; Swash, M.; Power, D.M. Ubiquitin deposits in anterior horn cells in motor neurone disease. Neurosci. Lett. 1988, 93, 197–203. [Google Scholar] [CrossRef]
- Kato, S.; Takikawa, M.; Nakashima, K.; Hirano, A.; Cleveland, D.W.; Kusaka, H.; Shibata, N.; Kato, M.; Nakano, I.; Ohama, E. New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: Inclusions containing SOD1 in neurons and astrocytes. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 163–184. [Google Scholar] [CrossRef] [PubMed]
- Pokrishevsky, E.; Grad, L.I.; Yousefi, M.; Wang, J.; Mackenzie, I.R.; Cashman, N.R. Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e35050. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, K.; Jonsson, P.A.; Andersen, P.M.; Bergemalm, D.; Graffmo, K.S.; Hultdin, M.; Jacobsson, J.; Rosquist, R.; Marklund, S.L.; Brännström, T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE 2010, 5, e11552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.M.; Wang, H.S.; Taniguchi, A. Role of SOD-1 and nitric oxide/cyclic GMP cascade on neurofilament aggregation in ALS/MND. J. Neurol. Sci. 1996, 139, 16–26. [Google Scholar] [CrossRef]
- Rotilio, G.; Aquilano, K.; Ciriolo, M.R. Interplay of Cu, Zn superoxide dismutase and nitric oxide synthase in neurodegenerative processes. IUBMB Life 2003, 55, 629–634. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Li, C.; Guan, T.; Shang, H.; Cui, L.; Li, X.-M.; Kong, J. S-nitrosylated protein disulfide isomerase contributes to mutant SOD1 aggregates in amyotrophic lateral sclerosis. J. Neurochem. 2013, 124, 45–58. [Google Scholar] [CrossRef]
- Pirie, E.; Oh, C.-K.; Zhang, X.; Han, X.; Cieplak, P.; Scott, H.R.; Deal, A.K.; Ghatak, S.; Martinez, F.J.; Yeo, G.W.; et al. S-nitrosylated TDP-43 triggers aggregation, cell-to-cell spread, and neurotoxicity in hiPSCs and in vivo models of ALS/FTD. Proc. Natl. Acad. Sci. USA 2021, 118, e2021368118. [Google Scholar] [CrossRef]
- Zhang, J.; Velmeshev, D.; Hashimoto, K.; Huang, Y.-H.; Hofmann, J.W.; Shi, X.; Chen, J.; Leidal, A.M.; Dishart, J.G.; Cahill, M.K.; et al. Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature 2020, 588, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.A.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Zhao, W.; Beers, D.R.; Yen, A.A.; Xie, W.; Henkel, J.S.; Appel, S.H. Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J. Neurochem. 2007, 102, 2008–2019. [Google Scholar] [CrossRef]
- Almer, G.; Vukosavic, S.; Romero, N.; Przedborski, S. Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 1999, 72, 2415–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassina, P.; Peluffo, H.; Pehar, M.; Martinez-Palma, L.; Ressia, A.; Beckman, J.S.; Estévez, A.G.; Barbeito, L. Peroxynitrite triggers a phenotypic transformation in spinal cord astrocytes that induces motor neuron apoptosis. J. Neurosci. Res. 2002, 67, 21–29. [Google Scholar] [CrossRef]
- Zhao, W.; Xie, W.; Le, W.; Beers, D.R.; He, Y.; Henkel, J.S.; Simpson, E.P.; Yen, A.A.; Xiao, Q.; Appel, S.H. Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J. Neuropathol. Exp. Neurol. 2004, 63, 964–977. [Google Scholar] [CrossRef] [Green Version]
- Pehar, M.; Cassina, P.; Vargas, M.R.; Castellanos, R.; Viera, L.; Beckman, J.S.; Estévez, A.G.; Barbeito, L. Astrocytic production of nerve growth factor in motor neuron apoptosis: Implications for amyotrophic lateral sclerosis. J. Neurochem. 2004, 89, 464–473. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible Nitric Oxide Synthase: Regulation, Structure, and Inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Southan, G.J.; Szabó, C.; Thiemermann, C. Isothioureas: Potent inhibitors of nitric oxide synthases with variable isoform selectivity. Br. J. Pharmacol. 1995, 114, 510–516. [Google Scholar] [CrossRef]
- Tewari, D.; Sah, A.N.; Bawari, S.; Nabavi, S.F.; Dehpour, A.R.; Shirooie, S.; Braidy, N.; Fiebich, B.L.; Vacca, R.A. Role of Nitric Oxide in Neurodegeneration: Function, Regulation, and Inhibition. Curr. NeuroPharmacol. 2021, 19, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Tinker, A.C.; Wallace, A.V. Selective inhibitors of inducible nitric oxide synthase: Potential agents for the treatment of inflammatory diseases? Curr. Top. Med. Chem. 2006, 6, 77–92. [Google Scholar] [CrossRef]
- Witte, A.V.; Kerti, L.; Margulies, D.S.; Flöel, A. Effects of Resveratrol on Memory Performance, Hippocampal Functional Connectivity, and Glucose Metabolism in Healthy Older Adults. J. Neurosci. 2014, 34, 7862–7870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behl, T.; Rana, T.; Sehgal, A.; Makeen, H.A.; Albratty, M.; Alhazmi, H.A.; Meraya, A.M.; Bhatia, S.; Sachdeva, M. Phytochemicals targeting nitric oxide signaling in neurodegenerative diseases. Nitric Oxide 2023, 130, 1–11. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Iacomino, A.; Steardo, L.; Scuderi, C. Targeting neuroinflammation in Alzheimer’s disease. J. Inflamm. Res. 2016, 9, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Wolff, D.J.; Lubeskie, A. Aminoguanidine is an isoform-selective, mechanism-based inactivator of nitric oxide synthase. Arch. Biochem. Biophys. 1995, 316, 290–301. [Google Scholar] [CrossRef]
- Hagmann, W.K.; Caldwell, C.G.; Chen, P.; Durette, P.L.; Esser, C.K.; Lanza, T.J.; Kopka, I.E.; Guthikonda, R.; Shah, S.K.; MacCoss, M.; et al. Substituted 2-aminopyridines as inhibitors of nitric oxide synthases. Bioorg. Med. Chem. Lett. 2000, 10, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Causal Trigger | Effect |
|---|---|
| S-Nitrosylation of tyrosine hydroxylase | ↑ Dopamine synthesis Indirect increase in dopamine metabolites |
| Dopamine activation of mtNOS | ↑ NO synthesis |
| ↑ Peroxynitrile | ↓ Dopamine re-uptake |
| ↑ Glutamate | ↑ NO synthesis ↑ Dopamine oxidation |
| ↑ NO ↑ Peroxynitrile | ↑ Dopamine oxidation |
| Trigger Factor | Effect | Result | Ref. |
|---|---|---|---|
| LPS | Activation of mSOD1 bearing astrocytes | ↑ iNOS ↑ NO | [150] |
| Mutant SOD1 | Activation of astrocytes and microglial cells | ↑ iNOS ↑ NO | [151] |
| Peroxynitrile or LPS | Activation of astrocytes | ↑ iNOS ↑ NO Cytotoxic phenotype transformation | [152] |
| Peroxynitrile | Activation of astrocytes and microglial cells | ↓ Astrocytic glutamate uptake ↑ Neuronal excitotoxicity | [153] |
| Peroxynitrile or LPS | Activation of astrocytes | ↑ iNOS ↑ NO ↑ nerve growth factor ↑ nitrotyrosine ↑ death of p75(NTR)+ neurons | [154] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iova, O.-M.; Marin, G.-E.; Lazar, I.; Stanescu, I.; Dogaru, G.; Nicula, C.A.; Bulboacă, A.E. Nitric Oxide/Nitric Oxide Synthase System in the Pathogenesis of Neurodegenerative Disorders—An Overview. Antioxidants 2023, 12, 753. https://doi.org/10.3390/antiox12030753
Iova O-M, Marin G-E, Lazar I, Stanescu I, Dogaru G, Nicula CA, Bulboacă AE. Nitric Oxide/Nitric Oxide Synthase System in the Pathogenesis of Neurodegenerative Disorders—An Overview. Antioxidants. 2023; 12(3):753. https://doi.org/10.3390/antiox12030753
Chicago/Turabian StyleIova, Olga-Maria, Gheorghe-Eduard Marin, Izabella Lazar, Ioana Stanescu, Gabriela Dogaru, Cristina Ariadna Nicula, and Adriana Elena Bulboacă. 2023. "Nitric Oxide/Nitric Oxide Synthase System in the Pathogenesis of Neurodegenerative Disorders—An Overview" Antioxidants 12, no. 3: 753. https://doi.org/10.3390/antiox12030753