
Biosynthesis, Deficiency, and Supplementation of Coenzyme Q
 , , and
, , and 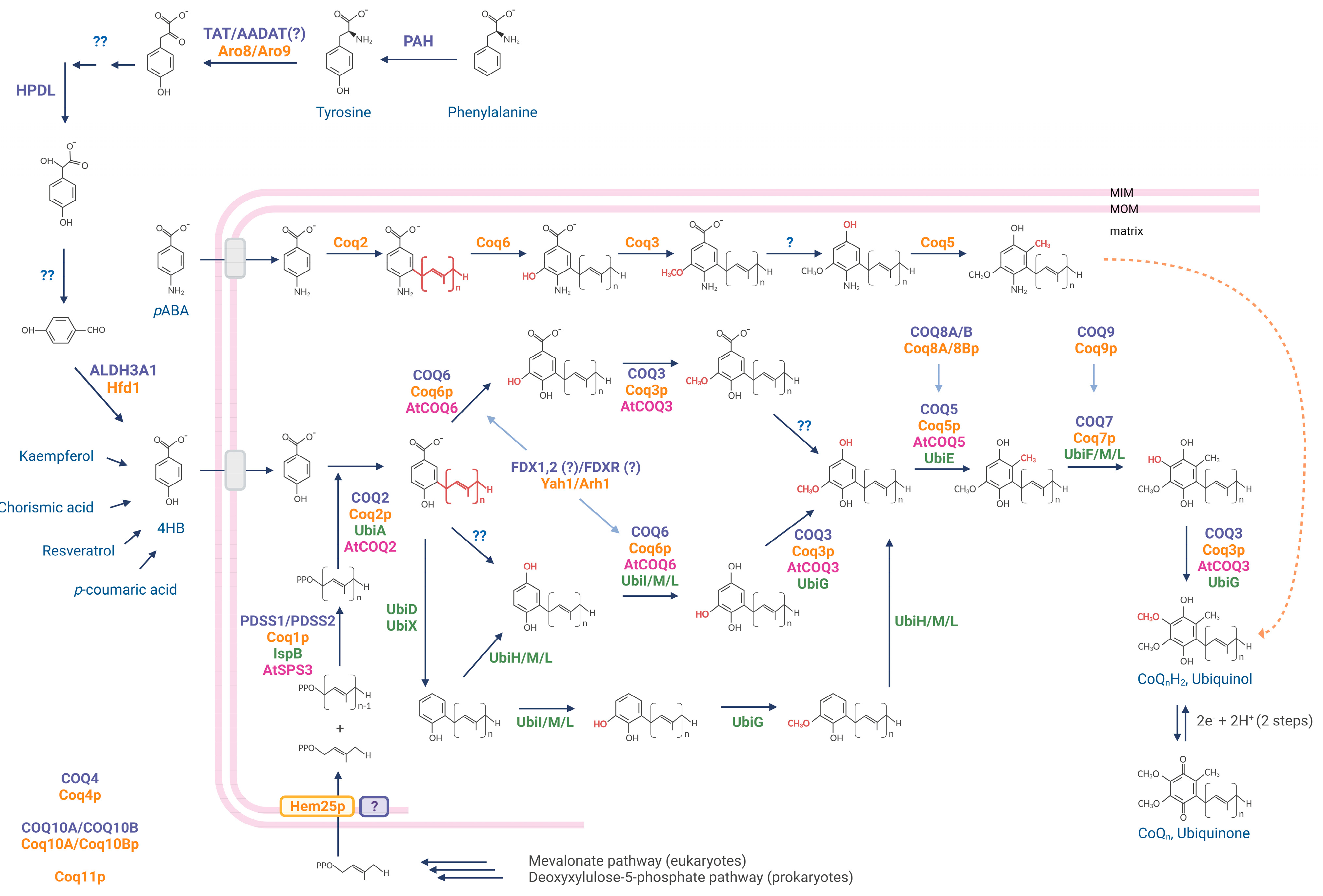{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CoQ Biosynthesis
3. Deficiency of Coenzyme Q10
4. Primary CoQ10 Deficiencies
5. Factors Causing Secondary CoQ10 Deficiency
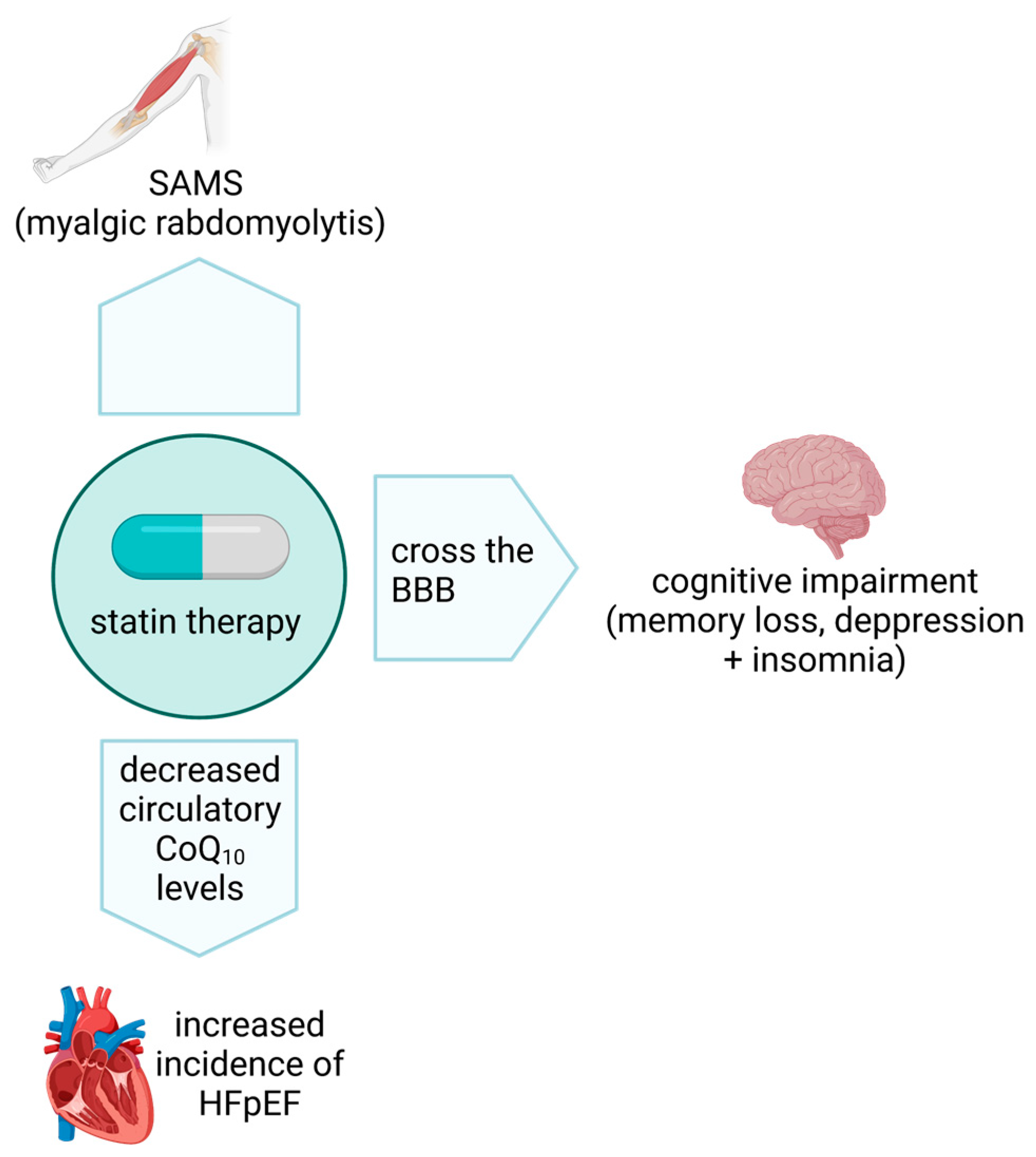
6. Statins and CoQ10
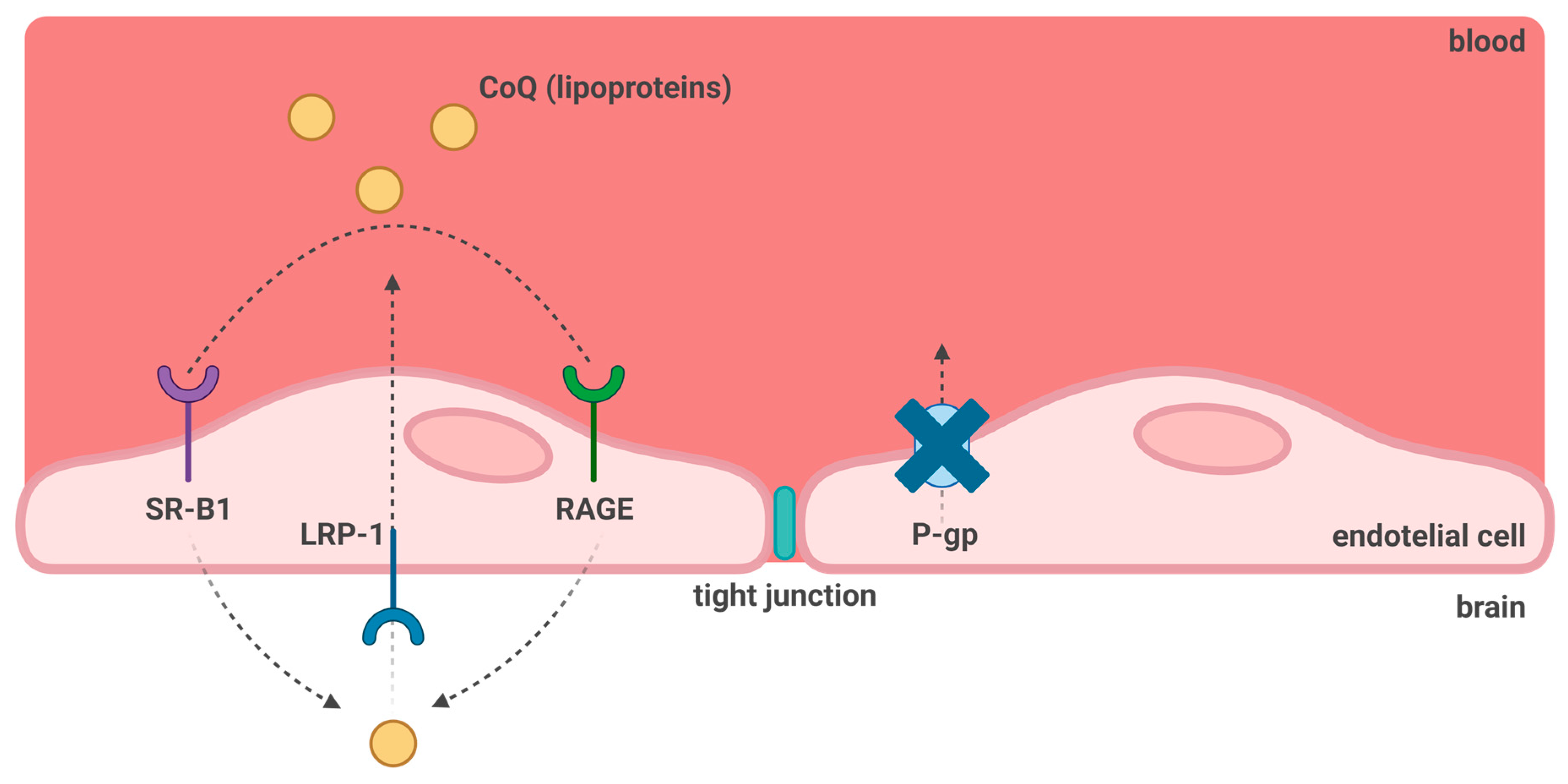
7. CoQ10 and Blood-Brain Barrier Transport
8. Bypass Treatments for CoQ10 Deficiencies
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crane, F.; Hatefi, Y.; Lester, R.; Widmer, C. Isolation of a quinone from beef heart mitochondria. Biochim. Biophys. Acta 1957, 25, 220–221. [Google Scholar] [CrossRef] [PubMed]
- Beyer, R.E. The role of ascorbate in antioxidant protection of biomembranes: Interaction with vitamin E and coenzyme Q. J. Bioenerg. Biomembr. 1994, 26, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frei, B.; Kim, M.C.; Ames, B.N. Ubiquinol-10 is an effective lipid-soluble antioxidant at physiological concentrations. Proc. Natl. Acad. Sci. USA 1990, 87, 4879–4883. [Google Scholar] [CrossRef]
- Banerjee, R.; Purhonen, J.; Kallijärvi, J. The mitochondrial coenzyme Q junction and complex III: Biochemistry and pathophysiology. FEBS J. 2021, 289, 6936–6958. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef]
- Nagel, R.; Schmidt, A.; Peters, R.J. Isoprenyl diphosphate synthases: The chain length determining step in terpene biosynthesis. Planta 2018, 249, 9–20. [Google Scholar] [CrossRef]
- Vo, C.-D.; Michaud, J.; Elsen, S.; Faivre, B.; Bouveret, E.; Barras, F.; Fontecave, M.; Pierrel, F.; Lombard, M.; Pelosi, L. The O2-independent pathway of ubiquinone biosynthesis is essential for denitrification in Pseudomonas aeruginosa. J. Biol. Chem. 2020, 295, 9021–9032. [Google Scholar] [CrossRef]
- Emekli-Alturfan, E.; Alturfan, A.A. The emerging relationship between vitamin K and neurodegenerative diseases: A review of current evidence. Mol. Biol. Rep. 2022, 50, 815–828. [Google Scholar] [CrossRef]
- Carrié, I.; Bélanger, E.; Portoukalian, J.; Rochford, J.; Ferland, G. Lifelong Low-Phylloquinone Intake Is Associated with Cognitive Impairments in Old Rats. J. Nutr. 2011, 141, 1495–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoepp-Cothenet, B.; Lieutaud, C.; Baymann, F.; Verméglio, A.; Friedrich, T.; Kramer, D.M.; Nitschke, W. Menaquinone as pool quinone in a purple bacterium. Proc. Natl. Acad. Sci. USA 2009, 106, 8549–8554. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Vo, C.D.T.; Abby, S.S.; Loiseau, L.; Rascalou, B.; Hajj Chehade, M.; Faivre, B.; Gousse, M.; Chenal, C.; Touati, N.; et al. Ubiquinone Biosynthesis over the Entire O2 Range: Characterization of a Conserved O2-Independent Pathway. MBio 2019, 10, e01319-19. [Google Scholar] [CrossRef] [Green Version]
- Manicki, M.; Aydin, H.; Abriata, L.A.; Overmyer, K.A.; Guerra, R.M.; Coon, J.J.; Peraro, M.D.; Frost, A.; Pagliarini, D.J. Structure and functionality of a multimeric human COQ7:COQ9 complex. Mol. Cell 2022, 82, 4307–4323.e10. [Google Scholar] [CrossRef]
- Fernández-Ayala, D.J.; López-Lluch, G.; García-Valdés, M.; Arroyo, A.; Navas, P. Specificity of coenzyme Q10 for a balanced function of respiratory chain and endogenous ubiquinone biosynthesis in human cells. Biochim. Biophys. Acta (BBA)-Bioenerg. 2005, 1706, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Deshwal, S.; Onishi, M.; Tatsuta, T.; Bartsch, T.; Cors, E.; Ried, K.; Lemke, K.; Nolte, H.; Giavalisco, P.; Langer, T. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nature 2023, 25, 246–257. [Google Scholar] [CrossRef]
- Chehade, M.H.; Pelosi, L.; Fyfe, C.D.; Loiseau, L.; Rascalou, B.; Brugière, S.; Kzaemzadeh, K.; Vo, C.D.T.; Ciccone, L.; Aussel, L.; et al. A Soluble Metabolon Synthesizes the Isoprenoid Lipid Ubiquinone. Cell Chem. Biol. 2019, 26, 482–492.e7. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.; Cox, G.B.; Gibson, F. Biosynthesis of Ubiquinone in Escherichia coli K-12: Biochemical and Genetic Characterization of a Mutant Unable to Convert Chorismate into 4-Hydroxybenzoate. J. Bacteriol. 1974, 118, 41–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, R.M.; Pagliarini, D.J. Coenzyme Q biochemistry and biosynthesis. Trends Biochem. Sci. 2023, 48, 463–476. [Google Scholar] [CrossRef]
- Block, A.; Widhalm, J.R.; Fatihi, A.; Cahoon, R.E.; Wamboldt, Y.; Elowsky, C.; Mackenzie, S.A.; Cahoon, E.B.; Chapple, C.; Dudareva, N.; et al. The Origin and Biosynthesis of the Benzenoid Moiety of Ubiquinone (Coenzyme Q) in Arabidopsis. Plant Cell 2014, 26, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.; Latimer, S.; Stutts, L.R.; Soubeyrand, E.; Block, A.K.; Basset, G.J. Kaempferol as a precursor for ubiquinone (coenzyme Q) biosynthesis: An atypical node between specialized metabolism and primary metabolism. Curr. Opin. Plant Biol. 2022, 66, 102165. [Google Scholar] [CrossRef]
- Fernández-Del-Río, L.; Clarke, C.F. Coenzyme Q Biosynthesis: An Update on the Origins of the Benzenoid Ring and Discovery of New Ring Precursors. Metabolites 2021, 11, 385. [Google Scholar] [CrossRef]
- Banh, R.S.; Kim, E.S.; Spillier, Q.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.W.; Shi, G.; Jones, D.R.; Kimmelman, A.C.; Pacold, M.E. The polar oxy-metabolome reveals the 4-hydroxymandelate CoQ10 synthesis pathway. Nature 2021, 597, 420–425. [Google Scholar] [CrossRef]
- Fernández-Del-Río, L.; Nag, A.; Casado, E.G.; Ariza, J.; Awad, A.M.; Joseph, A.I.; Kwon, O.; Verdin, E.; de Cabo, R.; Schneider, C.; et al. Kaempferol increases levels of coenzyme Q in kidney cells and serves as a biosynthetic ring precursor. Free. Radic. Biol. Med. 2017, 110, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Tai, J.; Guerra, R.M.; Rogers, S.W.; Fang, Z.; Muehlbauer, L.K.; Shishkova, E.; Overmyer, K.A.; Coon, J.J.; Pagliarini, D.J. Hem25p is a mitochondrial IPP transporter. bioRxiv 2023. bioRxiv: 2023.03.14.532620. [Google Scholar]
- Kainou, T.; Okada, K.; Suzuki, K.; Nakagawa, T.; Matsuda, H.; Kawamukai, M. Dimer Formation of Octaprenyl-diphosphate Synthase (IspB) Is Essential for Chain Length Determination of Ubiquinone. J. Biol. Chem. 2001, 276, 7876–7883. [Google Scholar] [CrossRef] [Green Version]
- Okada, K.; Suzuki, K.; Kamiya, Y.; Zhu, X.; Fujisaki, S.; Nishimura, Y.; Nishino, T.; Nakagawad, T.; Kawamukai, M.; Matsuda, H. Polyprenyl diphosphate synthase essentially defines the length of the side chain of ubiquinone. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1996, 1302, 217–223. [Google Scholar] [CrossRef]
- Lopez, M.J.A.; Trevisson, E.; Canton, M.; Vazquez-Fonseca, L.; Morbidoni, V.; Baschiera, E.; Frasson, C.; Pelosi, L.; Rascalou, B.; Desbats, M.A.; et al. Vanillic Acid Restores Coenzyme Q Biosynthesis and ATP Production in Human Cells Lacking COQ6. Oxidative Med. Cell. Longev. 2019, 2019, 3904905. [Google Scholar] [CrossRef] [Green Version]
- Pelosi, L.; Ducluzeau, A.-L.; Loiseau, L.; Barras, F.; Schneider, D.; Junier, I.; Pierrel, F. Evolution of Ubiquinone Biosynthesis: Multiple Proteobacterial Enzymes with Various Regioselectivities to Catalyze Three Contiguous Aromatic Hydroxylation Reactions. Msystems 2016, 1, e00091-16. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hekimi, S. The Complexity of Making Ubiquinone. Trends Endocrinol. Metab. 2019, 30, 929–943. [Google Scholar] [CrossRef]
- Awad, A.M.; Bradley, M.C.; Fernández-Del-Río, L.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays Biochem. 2018, 62, 361–376. [Google Scholar]
- Hayashi, K.; Ogiyama, Y.; Yokomi, K.; Nakagawa, T.; Kaino, T.; Kawamukai, M. Functional Conservation of Coenzyme Q Biosynthetic Genes among Yeasts, Plants, and Humans. PLoS ONE 2014, 9, e99038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamukai, M. Biosynthesis of coenzyme Q in eukaryotes. Biosci. Biotechnol. Biochem. 2016, 80, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.T.; Hsu, A.Y.; Ha, H.T.; Clarke, C.F. A C-methyltransferase involved in both ubiquinone and menaquinone biosynthesis: Isolation and identification of the Escherichia coli ubiE gene. J. Bacteriol. 1997, 179, 1748–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducluzeau, A.L.; Wamboldt, Y.; Elowsky, C.G.; Mackenzie, S.A.; Schuurink, R.C.; Basset, G.J. Gene network reconstruction identifies the authentic trans-prenyl diphosphate synthase that makes the solanesyl moiety of ubiquinone-9 in Arabidopsis. Plant J. 2012, 69, 366–375. [Google Scholar] [CrossRef]
- Okada, K.; Ohara, K.; Yazaki, K.; Nozaki, K.; Uchida, N.; Kawamukai, M.; Nojiri, H.; Yamane, H. The AtPPT1 gene encoding 4-hydroxybenzoate polyprenyl diphosphate transferase in ubiquinone biosynthesis is required for embryo development in Arabidopsis thaliana. Plant Mol. Biol. 2004, 55, 567–577. [Google Scholar] [CrossRef]
- Marbois, B.; Gin, P.; Gulmezian, M.; Clarke, C.F. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2009, 1791, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Reidenbach, A.G.; Kemmerer, Z.A.; Aydin, D.; Jochem, A.; McDevitt, M.T.; Hutchins, P.D.; Stark, J.L.; Stefely, J.A.; Reddy, T.; Hebert, A.S.; et al. Conserved Lipid and Small-Molecule Modulation of COQ8 Reveals Regulation of the Ancient Kinase-like UbiB Family. Cell Chem. Biol. 2017, 25, 154–165.e11. [Google Scholar] [CrossRef] [Green Version]
- Tsui, H.S.; Pham, N.; Amer, B.R.; Bradley, M.C.; Gosschalk, J.E.; Gallagher-Jones, M.; Ibarra, H.; Clubb, R.T.; Blaby-Haas, C.E.; Clarke, C.F. Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function. J. Lipid Res. 2019, 60, 1293–1310. [Google Scholar] [CrossRef]
- Pierrel, F.; Hamelin, O.; Douki, T.; Kieffer-Jaquinod, S.; Mühlenhoff, U.; Ozeir, M.; Lill, R.; Fontecave, M. Involvement of Mitochondrial Ferredoxin and Para-Aminobenzoic Acid in Yeast Coenzyme Q Biosynthesis. Chem. Biol. 2010, 17, 449–459. [Google Scholar] [CrossRef]
- Bradley, M.C.; Yang, K.; Fernández-Del-Río, L.; Ngo, J.; Ayer, A.; Tsui, H.S.; Novales, N.A.; Stocker, R.; Shirihai, O.S.; Barros, M.H.; et al. COQ11 deletion mitigates respiratory deficiency caused by mutations in the gene encoding the coenzyme Q chaperone protein Coq10. J. Biol. Chem. 2020, 295, 6023–6042. [Google Scholar] [CrossRef] [Green Version]
- Allan, C.M.; Awad, A.M.; Johnson, J.S.; Shirasaki, D.I.; Wang, C.; Blaby-Haas, C.E.; Merchant, S.S.; Loo, J.A.; Clarke, C.F. Identification of Coq11, a New Coenzyme Q Biosynthetic Protein in the CoQ-Synthome in Saccharomyces cerevisiae. J. Biol. Chem. 2015, 290, 7517–7534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapointe, C.P.; Stefely, J.A.; Jochem, A.; Hutchins, P.D.; Wilson, G.M.; Kwiecien, N.W.; Coon, J.J.; Wickens, M.; Pagliarini, D.J. Multi-omics Reveal Specific Targets of the RNA-Binding Protein Puf3p and Its Orchestration of Mitochondrial Biogenesis. Cell Syst. 2017, 6, 125–135.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veling, M.T.; Reidenbach, A.G.; Freiberger, E.C.; Kwiecien, N.W.; Hutchins, P.D.; Drahnak, M.J.; Jochem, A.; Ulbrich, A.; Rush, M.J.; Russell, J.D.; et al. Multi-omic Mitoprotease Profiling Defines a Role for Oct1p in Coenzyme Q Production. Mol. Cell 2017, 68, 970–977.e11. [Google Scholar] [CrossRef] [Green Version]
- Vögtle, F.N.; Prinz, C.; Kellermann, J.; Lottspeich, F.; Pfanner, N.; Meisinger, C. A Highlights from MBoC Selection: Mitochondrial protein turnover: Role of the precursor intermediate peptidase Oct1 in protein stabilization. Mol. Biol. Cell 2011, 22, 2135. [Google Scholar] [CrossRef]
- Subramanian, K.; Jochem, A.; Le Vasseur, M.; Lewis, S.; Paulson, B.R.; Reddy, T.R.; Rusell, J.D.; Coon, J.J.; Pagliarini, D.; Nunnari, J. Coenzyme Q biosynthetic proteins assemble in a substrate-dependent manner into domains at ER-mitochondria contacts. J. Cell Biol. 2019, 218, 1353–1369. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Bord, M.; Tsui, H.S.; Antunes, D.; Fernández-Del-Río, L.; Bradley, M.C.; Dunn, C.D.; Nguyen, T.P.T.; Rapaport, D.; Clarke, C.F.; Schuldiner, M. The Endoplasmic Reticulum-Mitochondria Encounter Structure Complex Coordinates Coenzyme Q Biosynthesis. Contact 2019, 2, 2515256418825409. [Google Scholar] [CrossRef] [Green Version]
- Mugoni, V.; Postel, R.; Catanzaro, V.; De Luca, E.; Turco, E.; Digilio, G.; Silengo, L.; Murphy, M.P.; Medana, C.; Stainier, D.Y.; et al. Ubiad1 Is an Antioxidant Enzyme that Regulates eNOS Activity by CoQ10 Synthesis. Cell 2013, 152, 504–518. [Google Scholar] [CrossRef] [Green Version]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [Green Version]
- Alcázar-Fabra, M.; Rodríguez-Sánchez, F.; Trevisson, E.; Brea-Calvo, G. Primary Coenzyme Q deficiencies: A literature review and online platform of clinical features to uncover genotype-phenotype correlations. Free. Radic. Biol. Med. 2021, 167, 141–180. [Google Scholar] [CrossRef]
- Navas, P.; Cascajo, M.V.; Alcázar-Fabra, M.; Hernández-Camacho, J.D.; Sánchez-Cuesta, A.; Rodríguez, A.B.C.; Ballesteros-Simarro, M.; Arroyo-Luque, A.; Rodríguez-Aguilera, J.C.; Fernández-Ayala, D.J.M.; et al. Secondary CoQ10 deficiency, bioenergetics unbalance in disease and aging. Biofactors 2021, 47, 551–569. [Google Scholar] [CrossRef]
- Yubero, D.; Montero, R.; Martín, M.A.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Rodriguez-Aguilera, J.C.; Hargreaves, I.P.; Salviati, L.; et al. Secondary coenzyme Q10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Lopez-Lluch, G.; Hargreaves, I.P. Coenzyme Q10 Metabolism: A Review of Unresolved Issues. Int. J. Mol. Sci. 2023, 24, 2585. [Google Scholar] [CrossRef]
- Alcazar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [PubMed]
- Hargreaves, I.P. Ubiquinone: Cholesterol’s reclusive cousin. Ann. Clin. Biochem. Int. J. Biochem. Lab. Med. 2003, 40, 207–218. [Google Scholar] [CrossRef]
- Berardo, A.I.; Quinzii, C.M. Redefining infantile-onset multisystem phenotypes of coenzyme Q10-deficiency in the next-generation sequencing era. J. Transl. Genet. Genom. 2020, 4, 22–35. [Google Scholar] [CrossRef]
- Malicdan, M.C.V.; Vilboux, T.; Ben-Zeev, B.; Guo, J.; Eliyahu, A.; Pode-Shakked, B.; Dori, A.; Kakani, S.; Chandrasekharappa, S.C.; Ferreira, C.R.; et al. A novel inborn error of the coenzyme Q10 biosynthesis pathway: Cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum Mutat. 2017, 39, 69–79. [Google Scholar] [CrossRef]
- Emmanuele, V.; López, L.C.; Berardo, A.; Naini, A.; Tadesse, S.; Wen, B.; D’Agostino, E.; Solomon, M.; DiMauro, S.; Quinzii, C.; et al. Heterogeneity of Coenzyme Q10 Deficiency: Patient Study and Literature Review. Arch. Neurol. 2012, 69, 978–983. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, I.; Heaton, R.A.; Mantle, D. Disorders of Human Coenzyme Q10 Metabolism: An Overview. Int. J. Mol. Sci. 2020, 21, 6695. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A. Antioxidants, Oxidative Stress, and Degenerative Neurological Disorders. Proc. Soc. Exp. Boil. Med. 1999, 222, 236–245. [Google Scholar] [CrossRef]
- Manzar, H.; Abdulhussein, D.; Yap, T.E.; Cordeiro, M.F. Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain. Int. J. Mol. Sci. 2020, 21, 9299. [Google Scholar] [CrossRef]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; de Lonlay, P.; Munnich, A.; Rötig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Sondheimer, N.; Hewson, S.; Cameron, J.M.; Somers, G.R.; Broadbent, J.D.; Ziosi, M.; Quinzii, C.M.; Naini, A.B. Novel recessive mutations in COQ4 cause severe infantile cardiomyopathy and encephalopathy associated with CoQ 10 deficiency. Mol. Genet. Metab. Rep. 2017, 12, 23–27. [Google Scholar] [CrossRef]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; López, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.I.; Hardy, J.; et al. A Nonsense Mutation in COQ9 Causes Autosomal-Recessive Neonatal-Onset Primary Coenzyme Q10 Deficiency: A Potentially Treatable Form of Mitochondrial Disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brea-Calvo, G.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Bürgi, S.; et al. COQ4 Mutations Cause a Broad Spectrum of Mitochondrial Disorders Associated with CoQ10 Deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Rötig, A. CABC1 Gene Mutations Cause Ubiquinone Deficiency with Cerebellar Ataxia and Seizures. Am. J. Hum. Genet. 2008, 82, 623. [Google Scholar] [CrossRef] [Green Version]
- Olgac, A.; Öztoprak, U.; Kasapkara, S.; Kılıç, M.; Yüksel, D.; Derinkuyu, E.B.; Yıldız, Y.T.; Ceylaner, S.; Ezgu, F.S. A rare case of primary coenzyme Q10 deficiency due to COQ9 mutation. J. Pediatr. Endocrinol. Metab. 2019, 33, 165–170. [Google Scholar] [CrossRef]
- Iványi, B.; Rácz, G.Z.; Gál, P.; Brinyiczki, K.; Bódi, I.; Kalmár, T.; Maróti, Z.; Bereczki, C. Diffuse mesangial sclerosis in a PDSS2 mutation-induced coenzyme Q10 deficiency. Pediatr. Nephrol. 2017, 33, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Danhauser, K.; Herebian, D.; Haack, T.B.; Rodenburg, R.J.; Strom, T.M.; Meitinger, T.; Klee, D.; Mayatepek, E.; Prokisch, H.; Distelmaier, F. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur. J. Hum. Genet. 2015, 24, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Nair, P.; Lama, M.; El-Hayek, S.; Abou Sleymane, G.; Stora, S.; Obeid, M.; Al-Ali, M.T.; Delague, V.; Megarbane, A. COQ8A and MED25 Mutations in a Child with Intellectual Disability, Microcephaly, Seizures, and Spastic Ataxia: Synergistic Effect of Digenic Variants? Mol. Syndromol. 2019, 9, 319–323. [Google Scholar] [CrossRef]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J. Med. Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardecchia, F.; De Giorgi, A.; Palombo, F.; Fiorini, C.; De Negri, A.M.; Carelli, V.; Caporali, L.; Leuzzi, V. Missense PDSS1 mutations in CoenzymeQ10 synthesis cause optic atrophy and sensorineural deafness. Ann. Clin. Transl. Neurol. 2020, 8, 247–251. [Google Scholar] [CrossRef]
- Caglayan, A.O.; Gumus, H.; Sandford, E.; Kubisiak, T.L.; Ma, Q.; Ozel, A.B.; Per, H.; Li, J.Z.; Shakkottai, V.G.; Burmeister, M. COQ4 Mutation Leads to Childhood-Onset Ataxia Improved by CoQ10 Administration. Cerebellum 2019, 18, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.M.; Kamsteeg, E.-J.; Rodenburg, R.J.; van Deutekom, A.W.; Buis, D.R.; Engelen, M.; Cobben, J.-M. Coenzyme Q10 deficiency due to a COQ4 gene defect causes childhood-onset spinocerebellar ataxia and stroke-like episodes. Mol. Genet. Metab. Rep. 2018, 17, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Mero, S.; Salviati, L.; Leuzzi, V.; Rubegni, A.; Calderan, C.; Nardecchia, F.; Galatolo, D.; Desbats, M.A.; Naef, V.; Gemignani, F.; et al. New pathogenic variants in COQ4 cause ataxia and neurodevelopmental disorder without detectable CoQ10 deficiency in muscle or skin fibroblasts. J. Neurol. 2021, 268, 3381–3389. [Google Scholar] [CrossRef]
- Cordts, I.; Semmler, L.; Prasuhn, J.; Seibt, A.; Herebian, D.; Navaratnarajah, T.; Park, J.; Deininger, N.; Laugwita, L.; Goricke, S.L.; et al. Bi-Allelic COQ4 Variants Cause Adult-Onset Ataxia-Spasticity Spectrum Disease. Mov. Disord. 2022, 37, 2147–2253. [Google Scholar] [CrossRef] [PubMed]
- Laugwitz, L.; Seibt, A.; Herebian, D.; Peralta, S.; Kienzle, I.; Buchert, R.; Falb, R.; Gauck, D.; Müller, A.; Grimmel, M.; et al. Human COQ4 deficiency: Delineating the clinical, metabolic and neuroimaging phenotypes. J. Med. Genet. 2021, 59, 878–887. [Google Scholar] [CrossRef]
- Naini, A.; Lewis, V.J.; Hirano, M.; DiMauro, S. Primary coenzyme Q10 deficiency and the brain. BioFactors 2003, 18, 145–152. [Google Scholar] [CrossRef]
- Hargreaves, I.P.; Lane, A.; Sleiman, P.M. The coenzyme Q10 status of the brain regions of Parkinson’s disease patients. Neurosci. Lett. 2008, 447, 17–19. [Google Scholar] [CrossRef]
- Fecher, C.; Trovò, L.; Müller, S.A.; Snaidero, N.; Wettmarshausen, J.; Heink, S.; Ortiz, O.; Wagner, I.; Kuhn, R.; Hartmann, J.; et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 2019, 22, 1731–1742. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, Q.; Peiffer, L.B.; Hicks, J.L.; Haffner, M.C.; Rosenberg, A.Z.; Levi, M.; Wang, X.X.; Ozbek, B.; Valle, J.B.-D.; et al. An in Situ Atlas of Mitochondrial DNA in Mammalian Tissues Reveals High Content in Stem and Proliferative Compartments. Am. J. Pathol. 2020, 190, 1565–1579. [Google Scholar] [CrossRef]
- Scalais, E.; Chafai, R.; Van Coster, R.; Bindl, L.; Nuttin, C.; Panagiotaraki, C.; Seneca, S.; Lissens, W.; Ribes, A.; Geers, C.; et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur. J. Paediatr. Neurol. 2013, 17, 625–630. [Google Scholar] [CrossRef]
- Atmaca, M.; Gulhan, B.; Korkmaz, E.; Inozu, M.; Soylemezoglu, O.; Candan, C.; Bayazıt, A.K.; Elmacı, A.M.; Parmaksiz, G.; Duzova, A.; et al. Follow-up results of patients with ADCK4 mutations and the efficacy of CoQ10 treatment. Pediatr. Nephrol. 2017, 32, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, E.; Lipska-Ziętkiewicz, B.S.; Boyer, O.; Gribouval, O.; Fourrage, C.; Tabatabaei, M.; Schnaidt, S.; Gucer, S.; Kaymaz, F.; Arici, M.; et al. ADCK4-Associated Glomerulopathy Causes Adolescence-Onset FSGS. J. Am. Soc. Nephrol. 2016, 27, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevisson, E.; DiMauro, S.; Navas, P.; Salviati, L. Coenzyme Q deficiency in muscle. Curr. Opin. Neurol. 2011, 24, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Diomedi-Camassei, F.; Di Giandomenico, S.; Santorelli, F.M.; Caridi, G.; Piemonte, F.; Montini, G.; Ghiggeri, G.M.; Murer, L.; Barisoni, L.; Pastore, A.; et al. COQ2 nephropathy: A newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Ahn, Y.H.; Kang, H.G.; Yoo, K.H.; Won, N.H.; Lee, K.B.; Moon, K.C.; Seong, M.-W.; Gwon, T.R.; Park, S.S.; et al. COQ6 Mutations in Children with Steroid-Resistant Focal Segmental Glomerulosclerosis and Sensorineural Hearing Loss. Am. J. Kidney Dis. 2017, 70, 139–144. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Malgireddy, K.; Thompson, R.; Torres-Russotto, D. A novel CABC1/ADCK3 mutation in adult-onset cerebellar ataxia. Park Relat, Disord. 2016, 33, 151–152. [Google Scholar] [CrossRef]
- Hikmat, O.; Tzoulis, C.; Knappskog, P.M.; Johansson, S.; Boman, H.; Sztromwasser, P.; Lien, E.; Broodtkorb, E.; Ghezzi, D.; Bindoff, L.A. ADCK3 mutations with epilepsy, stroke-like episodes and ataxia: A POLG mimic? Eur. J. Neurol. 2016, 23, 1188–1194. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, F.; Liu, X.; Zhong, X.; Yao, Y.; Xiao, H. Steroid-resistant nephrotic syndrome caused by co-inheritance of mutations at NPHS1 and ADCK4 genes in two Chinese siblings. Intractable Rare Dis. Res. 2017, 6, 299–303. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-T.; Hersheson, J.; Plagnol, V.; Fawcett, K.; Duberley, K.E.C.; Preza, E.; Hargreaves, I.P.; Chalasani, A.; Laura, M.; Wood, N.W.; et al. Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: Clinical, genetic and biochemical characterisation. J. Neurol. Neurosurg. Psychiatry 2013, 85, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Desbats, M.A.; Lunardi, G.; Doimo, M.; Trevisson, E.; Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ10) deficiency. J. Inherit. Metab. Dis. 2014, 38, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Gigante, M.; Diella, S.; Santangelo, L.; Trevisson, E.; Acosta, M.; Amatruda, M.; Finzi, G.; Caridi, G.; Murer, L.; Accetturo, M.; et al. Further phenotypic heterogeneity of CoQ10 deficiency associated with steroid resistant nephrotic syndrome and novel COQ2 and COQ6 variants. Clin. Genet. 2017, 92, 224–226. [Google Scholar] [CrossRef] [Green Version]
- Montini, G.; Malaventura, C.; Salviati, L. Early Coenzyme Q10 Supplementation in Primary Coenzyme Q10 Deficiency. N. Engl. J. Med. 2008, 358, 2849–2850. [Google Scholar] [CrossRef]
- Montero, R.; Grazina, M.; López-Gallardo, E.; Montoya, J.; Briones, P.; Navarro-Sastre, A.; Land, J.M.; Hargreaves, I.P.; Artuch, R.; O’Callaghan, M.D.M.; et al. Coenzyme Q10 deficiency in mitochondrial DNA depletion syndromes. Mitochondrion 2013, 13, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Maeda, H.; Goto, Y.-I.; Nonaka, I. Muscle coenzyme Q10 in mitochondrial encephalomyopathies. Neuromuscul. Disord. 1991, 1, 443–447. [Google Scholar] [CrossRef]
- Sacconi, S.; Trevisson, E.; Salviati, L.; Aymé, S.; Rigal, O.; Redondo, A.G.; Mancuso, M.; Siciliano, G.; Tonin, P.; Angelini, C.; et al. Coenzyme Q10 is frequently reduced in muscle of patients with mitochondrial myopathy. Neuromuscul. Disord. 2010, 20, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Kattah, A.G.; Naini, A.; Akman, H.O.; Mootha, V.K.; DiMauro, S.; Hirano, M. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology 2005, 64, 539–541. [Google Scholar] [CrossRef]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.H.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef]
- Aeby, A.; Sznajer, Y.; Cavé, H.; Rebuffat, E.; Van Coster, R.; Rigal, O.; Van Bogaert, P. Cardiofaciocutaneous (CFC) syndrome associated with muscular coenzyme Q10 deficiency. J. Inherit. Metab. Dis. 2007, 30, 827. [Google Scholar] [CrossRef]
- Buján, N.; Arias, A.; Montero, R.; García-Villoria, J.; Lissens, W.; Seneca, S.; Espinós, C.; Navas, P.; De Meirleir, L.; Artuch, R.; et al. Characterization of CoQ10 biosynthesis in fibroblasts of patients with primary and secondary CoQ10 deficiency. J. Inherit. Metab. Dis. 2013, 37, 53–62. [Google Scholar] [CrossRef]
- Fatehi, F.; Okhovat, A.A.; Nilipour, Y.; Mroczek, M.; Straub, V.; Töpf, A.; Palibrk, A.; Peric, S.; Stojanovic, V.R.; Najmabadi, H.; et al. Adult-onset very-long-chain acyl-CoA dehydrogenase deficiency (VLCADD). Eur. J. Neurol. 2020, 27, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Yanjanin, N.M.; Bianconi, S.; Pavan, W.J.; Porter, F.D. Oxidative stress in Niemann-Pick disease, type C. Mol. Genet Metab. 2010, 101, 214–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazakerley, D.J.; Chaudhuri, R.; Yang, P.; Maghzal, G.J.; Thomas, K.C.; Krycer, J.R.; Humphrey, S.J.; Oarker, B.L.; Fisher-Wellman, K.H.; Meoli, C.C.; et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife 2018, 7, e32111. [Google Scholar] [CrossRef]
- Kühl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.-G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 2017, 6, e30952. [Google Scholar] [CrossRef]
- Yen, H.C.; Yeh, W.Y.; Lee, S.H.; Feng, Y.H.; Yang, S.L. Characterization of human mitochondrial PDSS and COQ proteins and their roles in maintaining coenzyme Q10 levels and each other’s stability. Biochim. Biophys. Acta-Bioenerg. 2020, 1861, 148192. [Google Scholar] [CrossRef] [PubMed]
- Diquigiovanni, C.; Rizzardi, N.; Kampmeier, A.; Liparulo, I.; Bianco, F.; De Nicolo, B.; Cataldi-Stagetti, E.; Cuna, E.; Severi, G.; Seri, M.; et al. Mutant SPART causes defects in mitochondrial protein import and bioenergetics reversed by Coenzyme Q. Open Biol. 2023, 13, 230040. [Google Scholar] [CrossRef]
- Cornelius, N.; Byron, C.; Hargreaves, I.; Guerra, P.F.; Furdek, A.K.; Land, J.; Radford, W.W.; Frerman, F.; Corydon, T.J.; Gregersen, N.; et al. Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple Acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2013, 22, 3819–3827. [Google Scholar] [CrossRef] [PubMed]
- Prasun, P. Multiple Acyl-CoA Dehydrogenase Deficiency; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews: Seattle, WA, USA, 1993. [Google Scholar]
- Gueguen, N.; Baris, O.; Lenaers, G.; Reynier, P.; Spinazzi, M. Secondary coenzyme Q deficiency in neurological disorders. Free. Radic. Biol. Med. 2021, 165, 203–218. [Google Scholar] [CrossRef]
- Kalén, A.; Appelkvist, E.-L.; Dallner, G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 1989, 24, 579–584. [Google Scholar] [CrossRef]
- Söderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Lipid Compositions of Different Regions of the Human Brain During Aging. J. Neurochem. 1990, 54, 415–423. [Google Scholar] [CrossRef]
- Hoppe, U.; Bergemann, J.; Diembeck, W.; Ennen, J.; Gohla, S.; Harris, I.; Jacob, J.; Kielholz, J.; Mei, W.; Pollet, D.; et al. Coenzyme Q10, a cutaneous antioxidant and energizer. Biofactors 1999, 9, 371–378. [Google Scholar] [CrossRef]
- Passi, S.; De Pità, O.; Puddu, P.; Littarru, G.P. Lipophilic Antioxidants in Human Sebum and Aging. Free. Radic. Res. 2002, 36, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Kuehne, A.; Hildebrand, J.; Soehle, J.; Wenck, H.; Terstegen, L.; Gallinat, S.; Knott, A.; Winnefeld, M.; Zamboni, N. An integrative metabolomics and transcriptomics study to identify metabolic alterations in aged skin of humans in vivo. BMC Genom. 2017, 18, 169. [Google Scholar] [CrossRef] [Green Version]
- Alf, D.; Schmidt, M.E.; Siebrecht, S.C. Ubiquinol supplementation enhances peak power production in trained athletes: A double-blind, placebo controlled study. J. Int. Soc. Sports Nutr. 2013, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Granata, C.; Caruana, N.J.; Botella, J.; Jamnick, N.A.; Huynh, K.; Kuang, J.; Janssen, H.A.; Reljic, B.; Mellett, N.A.; Laskowski, A.; et al. High-intensity training induces non-stoichiometric changes in the mitochondrial proteome of human skeletal muscle without reorganisation of respiratory chain content. Nat. Commun. 2021, 12, 7056. [Google Scholar] [CrossRef] [PubMed]
- Rundek, T.; Naini, A.; Sacco, R.; Coates, K.; DiMauro, S. Atorvastatin Decreases the Coenzyme Q10 Level in the Blood of Patients at Risk for Cardiovascular Disease and Stroke. Arch. Neurol. 2004, 61, 889–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberg, F.; Appelkvist, E.L.; Bröijersén, A.; Eriksson, M.; Angelin, B.; Hjemdahl, P.; Dallner, G. Gemfibrozil-induced decrease in serum ubiquinone and a-and g-tocopherol levels in men with combined hyperlipidaemia. Eur. J. Clin. Investig. 1998, 28, 235–242. [Google Scholar] [CrossRef]
- Moreno-Fernández, A.M.; Cordero, M.D.; Garrido-Maraver, J.; Alcocer-Gómez, E.; Casas-Barquero, N.; Carmona-López, M.I.; Sánchez-Alcázar, J.A.; de Miguel, M. Oral treatment with amitriptyline induces coenzyme Q deficiency and oxidative stress in psychiatric patients. J. Psychiatr. Res. 2012, 46, 341–345. [Google Scholar] [CrossRef]
- Ortiz, T.; Villanueva-Paz, M.; Díaz-Parrado, E.; Illanes, M.; Fernández-Rodríguez, A.; Sánchez-Alcázar, J.A.; de Miguel, M. Amitriptyline down-regulates coenzyme Q10 biosynthesis in lung cancer cells. Eur. J. Pharmacol. 2017, 797, 75–82. [Google Scholar] [CrossRef]
- Palan, P.R.; Magneson, A.T.; Castillo, M.; Dunne, J.; Mikhail, M.S. Effects of menstrual cycle and oral contraceptive use on serum levels of lipid-soluble antioxidants. Am. J. Obstet. Gynecol. 2006, 194, e35–e38. [Google Scholar] [CrossRef]
- Palan, P.R.; Strube, F.; Letko, J.; Sadikovic, A.; Mikhail, M.S. Effects of Oral, Vaginal, and Transdermal Hormonal Contraception on Serum Levels of Coenzyme Q 10, Vitamin E, and Total Antioxidant Activity. Obstet. Gynecol. Int. 2010, 2010, 925635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palan, P.R.; Connell, K.; Ramirez, E.; Inegbenijie, C.; Gavara, R.Y.; Ouseph, J.A.; Mikhail, M.S. Effects of menopause and hormone replacement therapy on serum levels of coenzyme Q10and other lipid-soluble antioxidants. Biofactors 2005, 25, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Kalyan, S.; Huebbe, P.; Esatbeyoglu, T.; Niklowitz, P.; Côté, H.C.F.; Rimbach, G.; Kabelitz, D. Nitrogen-Bisphosphonate Therapy Is Linked to Compromised Coenzyme Q10 and Vitamin E Status in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2014, 99, 1307–1313. [Google Scholar] [CrossRef] [Green Version]
- Tricarico, P.M.; Girardelli, M.; Kleiner, G.; Knowles, A.; Valencic, E.; Crovella, S.; Marcuzzi, A. Alendronate, a double-edged sword acting in the mevalonate pathway. Mol. Med. Rep. 2015, 12, 4238–4242. [Google Scholar] [CrossRef] [Green Version]
- Mantle, D.; Turton, N.; Hargreaves, I.P. Depletion and Supplementation of Coenzyme Q10 in Secondary Deficiency Disorders. Front. Biosci. 2022, 27, 322. [Google Scholar] [CrossRef] [PubMed]
- Folkers, K.; Vadhanavikit, S.; Mortensen, S.A. Biochemical rationale and myocardial tissue data on the effective therapy of cardiomyopathy with coenzyme Q10. Proc. Natl. Acad. Sci. USA 1985, 82, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W.; Haas, R.H.; Passov, D.; Beal, M.F. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann. Neurol. 1997, 42, 261–264. [Google Scholar] [CrossRef]
- Sumbalova, Z.; Kucharska, J.; Palacka, P.; Rausova, Z.; Langsjoen, P.H.; Langsjoen, A.M.; Gvozdjakova, A. Platelet mitochondrial function and endogenous coenzyme Q10 levels are reduced in patients after COVID-19. Bratisl. Med. J. 2021, 123, 9–15. [Google Scholar] [CrossRef]
- Kelekçi, S.; Evliyaoğlu, O.; Sen, V.; Yolbaş, I.; Uluca, U.; Tan, I.; Gürkan, M.F. The relationships between clinical outcome and the levels of total antioxidant capacity (TAC) and coenzyme Q (CoQ 10) in children with pandemic influenza (H 1 N1) and seasonal flu. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1033–1038. [Google Scholar]
- Chase, M.; Cocchi, M.N.; Liu, X.; Andersen, L.W.; Holmberg, M.J.; Donnino, M.W. Coenzyme Q10 in acute influenza. Influ. Other Respir. Viruses 2018, 13, 64–70. [Google Scholar] [CrossRef]
- Hargreaves, I.P.; Duncan, A.J.; Heales, S.J.R.; Land, J.M. The effect of HMG-CoA reductase inhibitors on coenzyme Q10: Possible biochemical/clinical implications. Drug Saf. 2005, 28, 659–676. [Google Scholar] [CrossRef]
- Langsjoen, P.; Langsjoen, A. Statin-induced coenzyme Q10 depletion: Clinical consequences and therapeutic implications in skeletal and myocardial myopathies. In Coenzyme Q10: From Fact to Fiction; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2015; pp. 237–251. [Google Scholar]
- Teive, H.A.; Moro, A.; Moscovich, M.; Arruda, W.O.; Munhoz, R.P. Statin-associated cerebellar ataxia. A Brazilian case series. Park. Relat. Disord. 2016, 25, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Cholesterol Level and Statin Use in Alzheimer Disease. Arch. Neurol. 2011, 68, 1385–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langsjoen, P.H.; Langsjoen, J.O.; Langsjoen, A.M.; Rosenfeldt, F. Statin-Associated Cardiomyopathy Responds to Statin Withdrawal and Administration of Coenzyme Q 10. Perm. J. 2019, 23, 18–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goli, A.K.; Goli, S.A.; Byrd, R.P.; Roy, T.M. Simvastatin-induced lactic acidosis: A rare adverse reaction? Clin. Pharmacol. Ther. 2002, 72, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Päivä, H.; Thelen, K.M.; Coster, R.; Smet, J.; Paepe, B.; Mattila, K.M.; Laakso, J.; Lehtimäki, T.; Vonbergmann, K.; Lütjohann, D.; et al. High-dose statins and skeletal muscle metabolism in humans: A randomized, controlled trial. Clin. Pharmacol. Ther. 2005, 78, 60–68. [Google Scholar] [CrossRef]
- Berner, J.E. Statins can produce ataxia in bipolar disorder: Two case reports. J. Clin. Psychiatry 2010, 71, 359. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, A.L.; Rosenfeldt, F.; Filipiak, K.J. Effect of coenzyme Q10 in Europeans with chronic heart failure: A sub-group analysis of the Q-SYMBIO randomized double-blind trial. Cardiol. J. 2013, 26, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Pierce, J.D.; Mahoney, D.E.; Hiebert, J.B.; Thimmesch, A.R.; Diaz, F.J.; Smith, C.; Shen, Q.; Mudaranthakam, D.P.; Clancy, R.L. Study protocol, randomized controlled trial: Reducing symptom burden in patients with heart failure with preserved ejection fraction using ubiquinol and/or D-ribose. BMC Cardiovasc. Disord. 2018, 18, 57. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, I. Coenzyme Q10 as a therapy for mitochondrial disease. Int. J. Biochem. Cell Biol. 2014, 49, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M.; Matson, S.; Matson, W.R.; Cormier, K.; Del Signore, S.J.; Hagerty, S.W.; Stack, E.C.; Ryu, H.; Ferrante, R.J. Dose ranging and efficacy study of high-dose coenzyme Q10 formulations in Huntington’s disease mice. Biochim. Biophys. Acta 2006, 1762, 616–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentinger, M.; Dallner, G.; Chojnacki, T.; Swiezewska, E. Distribution and breakdown of labeled coenzyme Q10 in rat. Free. Radic. Biol. Med. 2003, 34, 563–575. [Google Scholar] [CrossRef]
- Wainwright, L.; Hargreaves, I.P.; Georgian, A.R.; Turner, C.; Dalton, R.N.; Abbott, N.J.; Heales, S.J.R.; Preston, J.E. CoQ10 Deficient Endothelial Cell Culture Model for the Investigation of CoQ10 Blood-Brain Barrier Transport. J. Clin. Med. 2020, 9, 3236. [Google Scholar] [CrossRef]
- Duberley, K.E.C.; Abramov, A.Y.; Chalasani, A.; Heales, S.J.; Rahman, S.; Hargreaves, I.P. Human neuronal coenzyme Q10 deficiency results in global loss of mitochondrial respiratory chain activity, increased mitochondrial oxidative stress and reversal of ATP synthase activity: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2012, 36, 63–73. [Google Scholar] [CrossRef]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Flint Beal, M.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of Coenzyme Q10 in Early Parkinson Disease: Evidence of Slowing of the Functional Decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef]
- Flint Beal, M.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Lumina, L.P. A Randomized Clinical Trial of High-Dosage Coenzyme Q10 in Early Parkinson Disease: No Evidence of Benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Lee, P.; Ulatowski, L.M. Vitamin E: Mechanism of transport and regulation in the CNS. IUBMB Life 2018, 71, 424–429. [Google Scholar] [CrossRef]
- Muthukumaran, K.; Leahy, S.; Harrison, K.; Sikorska, M.; Sandhu, J.K.; Cohen, J.; Pandey, S. Orally delivered water soluble coenzyme Q10 (Ubisol-Q10) blocks on-going neurodegeneration in rats exposed to paraquat: Potential for therapeutic application in Parkinson’s disease. BMC Neurosci. 2014, 15, 21, Erratum in BMC Neurosci. 2021, 22, 79.. [Google Scholar] [CrossRef]
- Herebian, D.; López, L.C.; Distelmaier, F. Bypassing human CoQ 10 deficiency. Mol. Genet. Metab. 2018, 123, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Pierrel, F. Impact of Chemical Analogs of 4-Hydroxybenzoic Acid on Coenzyme Q Biosynthesis: From Inhibition to Bypass of Coenzyme Q Deficiency. Front. Physiol. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.X.; Ozeir, M.; Tang, J.Y.; Chen, J.Y.; Jaquinod, S.-K.; Fontecave, M.; Clarke, C.F.; Pierrel, F. Overexpression of the Coq8 Kinase in Saccharomyces cerevisiae coq Null Mutants Allows for Accumulation of Diagnostic Intermediates of the Coenzyme Q6 Biosynthetic Pathway. J. Biol. Chem. 2012, 287, 23571–23581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herebian, D.; Seibt, A.; Smits, S.H.J.; Rodenburg, R.J.; Mayatepek, E.; Distelmaier, F. 4-Hydroxybenzoic acid restores CoQ10biosynthesis in human COQ2 deficiency. Ann. Clin. Transl. Neurol. 2017, 4, 902–908. [Google Scholar] [CrossRef]
- Wang, Y.; Oxer, D.; Hekimi, S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 2015, 6, 6393. [Google Scholar] [CrossRef] [Green Version]
- Luna-Sanchez, M.; Diaz-Casado, E.; Barca, E.; Tejada, M.A.; Montilla-Garcia, A.; Cobos, E.J.; Escames, G.; Acuna-Castroviejo, D.; Quinzii, C.M.; Lopez, L.C. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar] [CrossRef]
- Hidalgo-Gutiérrez, A.; Barriocanal-Casado, E.; Bakkali, M.; Díaz-Casado, M.E.; Sánchez-Maldonado, L.; Romero, M.; López, L.C. β-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9R239X mice. EMBO Mol. Med. 2019, 11, e9466. [Google Scholar] [CrossRef]
- Widmeier, E.; Airik, M.; Hugo, H.; Schapiro, D.; Wedel, J.; Ghosh, C.C.; Nakayama, M.; Schneider, R.; Awad, A.M.; Nag, A.; et al. Treatment with 2,4-Dihydroxybenzoic Acid Prevents FSGS Progression and Renal Fibrosis in Podocyte-Specific Coq6 Knockout Mice. J. Am. Soc. Nephrol. 2019, 30, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernández-Del-Río, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.-I.; et al. ADCK4 Deficiency Destabilizes the Coenzyme Q Complex, Which Is Rescued by 2,4-Dihydroxybenzoic Acid Treatment. J. Am. Soc. Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef]
- Ozeir, M.; Mühlenhoff, U.; Webert, H.; Lill, R.; Fontecave, M.; Pierrel, F. Coenzyme Q Biosynthesis: Coq6 Is Required for the C5-Hydroxylation Reaction and Substrate Analogs Rescue Coq6 Deficiency. Chem. Biol. 2011, 18, 1134–1142. [Google Scholar] [CrossRef]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1842, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-García, P.; Díaz-Casado, M.E.; Hidalgo-Gutiérrez, A.; Jiménez-Sánchez, L.; Bakkali, M.; Barriocanal-Casado, E.; Escames, G.; Chiozzi, R.Z.; Völlmy, F.; Zaal, E.A.; et al. The Q-junction and the inflammatory response are critical pathological and therapeutic factors in CoQ deficiency. Redox Biol. 2022, 55, 102403. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staiano, C.; García-Corzo, L.; Mantle, D.; Turton, N.; Millichap, L.E.; Brea-Calvo, G.; Hargreaves, I. Biosynthesis, Deficiency, and Supplementation of Coenzyme Q. Antioxidants 2023, 12, 1469. https://doi.org/10.3390/antiox12071469
Staiano C, García-Corzo L, Mantle D, Turton N, Millichap LE, Brea-Calvo G, Hargreaves I. Biosynthesis, Deficiency, and Supplementation of Coenzyme Q. Antioxidants. 2023; 12(7):1469. https://doi.org/10.3390/antiox12071469
Chicago/Turabian StyleStaiano, Carmine, Laura García-Corzo, David Mantle, Nadia Turton, Lauren E. Millichap, Gloria Brea-Calvo, and Iain Hargreaves. 2023. "Biosynthesis, Deficiency, and Supplementation of Coenzyme Q" Antioxidants 12, no. 7: 1469. https://doi.org/10.3390/antiox12071469