
Loss of PRDX6 Aborts Proliferative and Migratory Signaling in Hepatocarcinoma Cell Lines
 , , , and
, , , and
Abstract
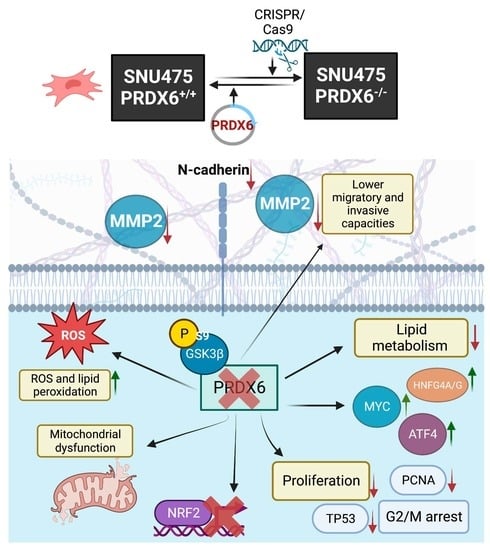
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Cell Lines
2.3. Creation of a Peroxiredoxin 6-Deficient SNU475 Hepatocarcinoma Cell Line (SNU475PRDX6−/−) Using the CRISPR/Cas9 Methodology
2.4. Plasmid Transfection
2.5. Measurement of Protein, ROS, and Lipid Peroxidation
2.6. SDS-PAGE and Western Blot
2.7. Immunofluorescence
2.8. Extracellular Flux Analysis of Mitochondrial Respiration and Glycolytic Function
2.9. Determination of the Enzymatic Activities of Mitochondrial Complexes
2.10. Cell Proliferation, Viability, and Cell Cycle Analysis
2.11. Cell Death Assay
2.12. Metalloproteinase Activity Detection Assay
2.13. Wound Healing Assay
2.14. Transwell Invasion Assay
2.15. Global Proteome Analysis
2.16. Statistics
3. Results
3.1. Creation and Characterization of SNU475 Hepatocarcinoma Cell Line Lacking PRDX6
3.2. Differential Quantitative Proteomic Analysis of SNU475PRDX6+/+ and SNU475PRDX6−/− Cells
3.3. Oxidative Stress Levels Increased in Hepatocarcinoma Cell Lines Lacking PRDX6
3.4. PRDX6 Deficiency Led to Mitochondrial Dysfunction and Metabolic Alterations in SNU475 Cells
3.5. PRDX6 Deficiency Decreased Proliferation, Altered the Cytoskeleton, Promoted Cell Cycle Arrest at G2/M Phase, and Affected the AKT/GSK3β Signaling Pathway
3.6. Peroxiredoxin 6 Deficiency Induced a More Epithelial Phenotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reczek, C.; Chandel, N. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.; Huang, B.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Benhar, M.; Engelberg, D.; Levitzki, A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 2002, 3, 420–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasauer, A.; Chandel, N. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Manevich, Y.; Sweitzer, T.; Pak, J.; Feinstein, S.; Muzykantov, V.; Fisher, A. 1-Cys peroxiredoxin overexpression protects cells against phospholipid peroxidation-mediated membrane damage. Proc. Natl. Acad. Sci. USA 2002, 99, 11599–11604. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.; Dodia, C.; Sorokina, E.; Li, H.; Zhou, S.; Raabe, T.; Feinstein, S. A novel lysophosphatidylcholine acyl transferase activity is expressed by peroxiredoxin 6. J. Lipid Res. 2016, 57, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Arevalo, J.; Vázquez-Medina, J. The role of peroxiredoxin 6 in cell signaling. Antioxidants 2018, 7, 172. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Feinstein, S.; Dodia, C.; Sorokina, E.; Lien, Y.-C.; Nguyen, S.; Debolt, K.; Speicher, D.; Fisher, A. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J. Biol. Chem. 2011, 286, 11696–11706. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Wang, A.; Burke, D.; Bodreau, H.; Lekstrom, K.; Korzeiowska, A.; Sugamata, R.; Kim, Y.-S.; Yi, L.; Ersoy, I.; et al. Peroxiredoxin 6 (Prdx6) supports NADPH oxidase1 (Nox1)-based superoxide generation and cell migration. Free Radic. Biol. Med. 2016, 96, 99–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, H.-M.; Park, K.-R.; Lee, H.; Lee, D.; Jo, M.; Shin, D.; Yoon, D.-Y.; Han, S.; Hong, J. PRDX6 promotes lung tumor progression via its GPx and iPLA2 activities. Free Radic. Biol. Med. 2014, 69, 367–376. [Google Scholar] [CrossRef]
- Hu, X.; Lu, E.; Pan, C.; Xu, Y.; Zhu, X. Overexpression and biological function of PRDX6 in human cervical cancer. J. Cancer 2020, 11, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.-M.; Park, K.-R.; Park, M.; Kim, D.; Jo, M.; Kim, J.; Kim, E.-C.; Yoon, D.; Han, S.; Hong, J. PRDX6 promotes tumor development via the JAK2/STAT3 pathway in a urethane-induced lung tumor model. Free Radic. Biol. Med. 2015, 80, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Pak, J.; Choi, W.; Lee, H.; Joo, W.-D.; Kim, J.-H.; Kim, Y.-T.; Kim, Y.-M.; Nam, J.-H. Peroxiredoxin 6 overexpression attenuates cisplatin-induced apoptosis in human ovarian cancer cells. Cancer Investig. 2011, 29, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-S.; Huang, C.-Y.; Hsieh, M.-C.; Kuo, Y.-H.; Tung, S.-Y.; Shen, C.-H.; Hsieh, Y.-Y.; Teng, C.-C.; Lee, K.-C.; Lee, K.-F.; et al. Expression of PRDX6 Correlates with Migration and Invasiveness of Colorectal Cancer Cells. Cell. Physiol. Biochem. 2018, 51, 2616–2630. [Google Scholar] [CrossRef]
- He, Y.; Xu, W.; Xiao, Y.; Pan, L.; Chen, G.; Tang, Y.; Zhou, J.; Wu, J.; Zhu, W.; Zhang, S.; et al. Overexpression of Peroxiredoxin 6 (PRDX6) Promotes the Aggressive Phenotypes of Esophageal Squamous Cell Carcinoma. J. Cancer 2018, 9, 3939–3949. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.-Z.; Li, D.-Q.; Hou, Y.-F.; Wu, J.; Lu, J.-S.; Di, G.-H.; Jin, W.; Ou, Z.-L.; Shen, Z.-Z.; Shao, Z.-M. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007, 9, R76. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.-N.; Lee, S.; Lee, S.-S.; Yoon, S.; Kang, G.; Hwang, S.-G.; Um, H.-D. Phospholipase A2 activity of peroxiredoxin 6 promotes invasion and metastasis of lung cancer cells. Mol. Cancer Ther. 2010, 9, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, D.; Li, B.; Zhen, H.; Chen, W.; Men, Q. PRDX6 Overexpression Promotes Proliferation, Invasion, and Migration of A549 Cells in vitro and in vivo. Cancer Manag. Res. 2021, 13, 1245–1255. [Google Scholar] [CrossRef]
- Yeung, K.T.; Yang, J. Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Grueso, M.; Tarradas, R.; Carmona-Hidalgo, B.; Lagal, D.; Peinado, J.; McDonagh, B.; Requejo-Aguilar, R.; Bárcena, J.; Padilla, C. Peroxiredoxin 6 Down-Regulation Induces Metabolic Remodeling and Cell Cycle Arrest in HepG2 Cells. Antioxidants 2019, 8, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Grueso, M.; Lagal, D.; García-Jiménez, A.; Tarradas, R.; Carmona-Hidalgo, B.; Peinado, J.; Requejo-Aguilar, R.; Bárcena, J.; Padilla, C. Knockout of PRDX6 induces mitochondrial dysfunction and cell cycle arrest at G2/M in HepG2 hepatocarcinoma cells. Redox. Biol. 2020, 37, 101737. [Google Scholar] [CrossRef] [PubMed]
- Hatefi, Y.; Rieske, J. Preparation and properties of DPNH-coenzyme Q reductase (complex I of the respiratory chain). Methods Enzymol. 1967, 10, 235–239. [Google Scholar]
- Tajhya, R.; Patel, R.; Beeton, C. Detection of Matrix Metalloproteinases by Zymography. Methods Mol. Biol. 2017, 1579, 231–244. [Google Scholar] [CrossRef] [Green Version]
- López-Grueso, M.; González-Ojeda, R.; Requejo-Aguilar, R.; McDonagh, B.; Fuentes-Almagro, C.; Muntané, J.; Bárcena, J.; Padilla, C. Thioredoxin and glutaredoxin regulate metabolism through different multiplex thiol switches. Redox. Biol. 2019, 21, 101049. [Google Scholar] [CrossRef]
- Huang, D.; Sherman, B.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.; Lane, H.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.; Lin, Y.-H.; Wita, A.; Xu, K.; Correia, M.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1–DELE1–HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef]
- Jin, G.-Z.; Zhang, Y.; Cong, W.-M.; Wu, X.; Wang, X.; Wu, S.; Wang, S.; Zhou, W.; Yuan, S.; Gao, H.; et al. Phosphoglucomutase 1 inhibits hepatocellular carcinoma progression by regulating glucose trafficking. PLoS Biol. 2018, 16, e2006483. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, L.; Fang, P.; Sun, Y.; Jiang, X.; Wang, H.; Yang, X.-F. Lysophospholipids induce innate immune transdifferentiation of endothelial cells, resulting in prolonged endothelial activation. J. Biol. Chem. 2018, 293, 11033–11045. [Google Scholar] [CrossRef] [Green Version]
- Vishwanatha, J.; Jindal, H.; Davis, R. The role of primer recognition proteins in DNA replication: Association with nuclear matrix in HeLa cells. J. Cell Sci. 1992, 101, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, L.; Hu, B.; Chen, L.; Jv, M.; Wang, L.; Zhou, C.; Wei, M.; Zhao, L. Advances in cancer treatment: A new therapeutic target, Annexin A2. J. Cancer 2021, 12, 3587–3596. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.; Qi, Y.; Hann, S. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [Green Version]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen synthase kinases: Moonlighting proteins with theranostic potential in cancer. Semin. Cancer Biol. 2019, 56, 25–36. [Google Scholar] [CrossRef]
- Dubois, V.; Staels, B.; Lefebvre, P.; Verzi, M.; Eeckhoute, J. Control of Cell Identity by the Nuclear Receptor HNF4 in Organ Pathophysiology. Cells 2020, 9, 2185. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, W.; Tang, J.; Ma, X.-K.; Dai, J.-Y.; Li, Y.; Jiang, J.-L.; Zhang, S.-H.; Chen, Z.-N. Annexin II promotes invasion and migration of human hepatocellular carcinoma cells in vitro via its interaction with HAb18G/CD147. Cancer Sci. 2010, 101, 387–395. [Google Scholar] [CrossRef]
- Kitano, A.; Shimasaki, T.; Chikano, Y.; Nakada, M.; Hirose, M.; Higashi, T.; Ishigaki, Y.; Endo, Y.; Takino, T.; Sato, H.; et al. Aberrant Glycogen Synthase Kinase 3bIs Involved inPancreatic Cancer Cell Invasion and Resistance toTherapy. PLoS ONE 2013, 8, e55289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikano, Y.; Domoto, T.; Furuta, T.; Sabit, H.; Kitano-Tamura, A.; Pyko, I.; Takino, T.; Sai, Y.; Hayashi, Y.; Sato, H.; et al. Glycogen Synthase Kinase 3β Sustains Invasion of Glioblastoma via the Focal Adhesion Kinase, Rac1, and c-Jun N-Terminal Kinase-Mediated Pathway. Mol. Cancer Ther. 2015, 14, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Paluchova, V.; Cajka, T.; Duran, T.; Vigor, C.; Dodia, C.; Chatterjee, S.; Fisher, A.; Kuda, O. The role of peroxiredoxin 6 in biosynthesis of FAHFAs. Free Radic. Biol. Med. 2022, 193, 787–794. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Reyes, I.; Cardona, L.R.; Kong, H.; Vasan, K.; McElroy, G.S.; Werner, M.; Kihshen, H.; Reczek, C.R.; Weinberg, S.E.; Gao, P.; et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 2020, 585, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.; Prado, M.; Zamboni, N.; D’Amico, D.; Williams, R.; Finley, D.; Gygi, S.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.M.; Seo, Y.H.; Park, C.B.; Yoon, S.H.; Yoon, G. Roles of GSK3 in metabolic shift toward abnormal anabolism in cell senescence. Ann. N. Y. Acad. Sci. 2010, 1201, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, S.; Minguzzi, M.; Platano, D.; Santi, S.; Trisolino, G.; Filardo, G.; Mariani, E.; Borzi, R.M. Glycogen Synthase Kinase-3beta Inhibition Links Mitochondrial Dysfunction, Extracellular Matrix Remodelling and Terminal Differentiation in Chondrocytes. Sci. Rep. 2017, 7, 12059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Wong, R.; Rajapakse, N.; Murphy, E.; Steenbergen, C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ. Res. 2008, 103, 983–991. [Google Scholar] [CrossRef]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of Human Plasma Lysophospholipase D, a Lysophosphatidic Acid-producing Enzyme, as Autotaxin, a Multifunctional Phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef] [Green Version]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: Implications for targeting mTOR during malignancy. Oncogene 2012, 31, 1949–1962. [Google Scholar] [CrossRef] [Green Version]
- Augello, G.; Puleio, R.; Emma, M.R.; Cusimano, A.; Loria, G.R.; McCubrey, J.A.; Montalto, G.; Cervello, M. A PTEN inhibitor displays preclinical activity against hepatocarcinoma cells. Cell Cycle 2016, 15, 573–583. [Google Scholar] [CrossRef]
- Abe, K.; Yamamoto, N.; Domoto, T.; Bolidong, D.; Hayashi, K.; Takeuchi, A.; Miwa, S.; Igarashi, K.; Inatani, H.; Aoki, Y.; et al. Glycogen synthase kinase 3beta as a potential therapeutic target in synovial sarcoma and fibrosarcoma. Cancer Sci. 2020, 111, 429–440. [Google Scholar] [CrossRef]
- Abe, K.; Shimozaki, S.; Domoto, T.; Yamamoto, N.; Tsuchiya, H.; Minamoto, T. Glycogen synthase kinase 3β biology in bone and soft tissue sarcomas. J. Cancer Metastasis Treat. 2020, 6, 429–440. [Google Scholar] [CrossRef]
- DeGregori, J.; Kowalik, T.; Nevins, J. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell Biol. 1995, 15, 4215–4224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrozik, K.; Blaschuk, O.; Cheong, C.; Zannettino, A.; Vandyke, K. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajka, D.; Budziak, B.; Pietras, L.; Duda, P.; McCubrey, J.; Gizak, A. GSK3 as a Regulator of Cytoskeleton Architecture: Consequences for Health and Disease. Cells 2021, 10, 2092. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) Category | Expression Trend | Term | Count | % | p-Value |
| Biological Process | Up | Protein Biosynthesis | 12 | 9.0 | 1.1 × 10−7 |
| Down | Lipid Metabolism | 20 | 14.3 | 3.1 × 10−6 | |
| Cellular component | Up | Cytoplasm | 83 | 62.4 | 3.1 × 10−19 |
| Nucleus | 52 | 39.1 | 3.2 × 10−3 | ||
| Down | Cytoplasm | 93 | 66.4 | 7.6 × 10−25 | |
| Cytoskeleton | 23 | 16.4 | 1.1 × 10−4 | ||
| Reactome Pathway | Up | tRNA-Aminoacylation | 8 | 6.0 | 1.7 × 10−9 |
| Down | The Role of GTSE1 in G2/M Progression After G2 checkpoint | 11 | 7.9 | 3.5 × 10−9 | |
| KEGG Pathway | Up | Aminoacyl-tRNA Biosynthesis | 8 | 6.0 | 7.8 × 10−6 |
| Nucleo-cytoplasmic Transport | 9 | 6.8 | 2.5 × 10−5 | ||
| Down | Metabolic Pathways | 42 | 30.0 | 2.2 × 10−7 | |
| (B) Transcrip. Regulator | Activation Score | p-Value | (C) Molecular and Cellular Function | Overlap (%) | p-Value |
| TP53 | −0.943 | 2.16 × 10−19 | tRNA Charging | 23.1 | 5.23 × 10−10 |
| MYC | 2.574 | 1.54 × 10−11 | BAG2 Signaling Pathway | 13.1 | 3.69 × 10−9 |
| HNF4A | 1.213 | 5.53 × 10−11 | Coronavirus Replication Pathway | 17.18 | 4.42 × 10−8 |
| NRF2 | −0.944 | 3.89 × 10−10 | Remodeling of Epithelial Adherens Junctions | 13.2 | 9.07 × 10−8 |
| ATF4 | 2.451 | 1.34 × 10−9 | Protein Ubiquitination Pathway | 5.8 | 1.49 × 10−7 |
| HNF4G | 2.828 | 4.33 × 10−8 |
| Protein IDs | Protein Description | Gene Names | Log FC | p-Value |
|---|---|---|---|---|
| P0DPH8 | Tubulin alpha-3D chain | TUBA3D | −2 | 4.18 × 10−6 |
| P08133 | Annexin A6 | ANXA6 | −0.509 | 0.00032 |
| P09525 | Annexin A4 | ANXA4 | −0.402 | 0.00155 |
| P50995 | Annexin A11 | ANXA11 | −0.832 | 0.00315 |
| P07355 | Annexin A2 | ANXA2 | −0.375 | 0.00924 |
| Q8WUP2 | Filamin-binding LIM protein 1 | FBLIM1 | −0.578 | 0.00014 |
| P21333 | Filamin-A | FLNA | −0.283 | 0.01528 |
| O75369 | Filamin-B | FLNB | −0.207 | 0.03843 |
| P11047 | Laminin subunit gamma-1 | LAMC1 | −1.704 | 0.00053 |
| G3XAI2 | Laminin subunit beta-1 | LAMB1 | −1.668 | 0.02289 |
| O43707 | Alpha-actinin-4 | ACTN4 | −0.140 | 0.01652 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagal, D.J.; López-Grueso, M.J.; Pedrajas, J.R.; Leto, T.L.; Bárcena, J.A.; Requejo-Aguilar, R.; Padilla, C.A. Loss of PRDX6 Aborts Proliferative and Migratory Signaling in Hepatocarcinoma Cell Lines. Antioxidants 2023, 12, 1153. https://doi.org/10.3390/antiox12061153
Lagal DJ, López-Grueso MJ, Pedrajas JR, Leto TL, Bárcena JA, Requejo-Aguilar R, Padilla CA. Loss of PRDX6 Aborts Proliferative and Migratory Signaling in Hepatocarcinoma Cell Lines. Antioxidants. 2023; 12(6):1153. https://doi.org/10.3390/antiox12061153
Chicago/Turabian StyleLagal, Daniel J., María J. López-Grueso, José R. Pedrajas, Thomas L. Leto, J. Antonio Bárcena, Raquel Requejo-Aguilar, and C. Alicia Padilla. 2023. "Loss of PRDX6 Aborts Proliferative and Migratory Signaling in Hepatocarcinoma Cell Lines" Antioxidants 12, no. 6: 1153. https://doi.org/10.3390/antiox12061153