
Protective Roles of Hydrogen Sulfide in Alzheimer’s Disease and Traumatic Brain Injury

Abstract
:1. Introduction
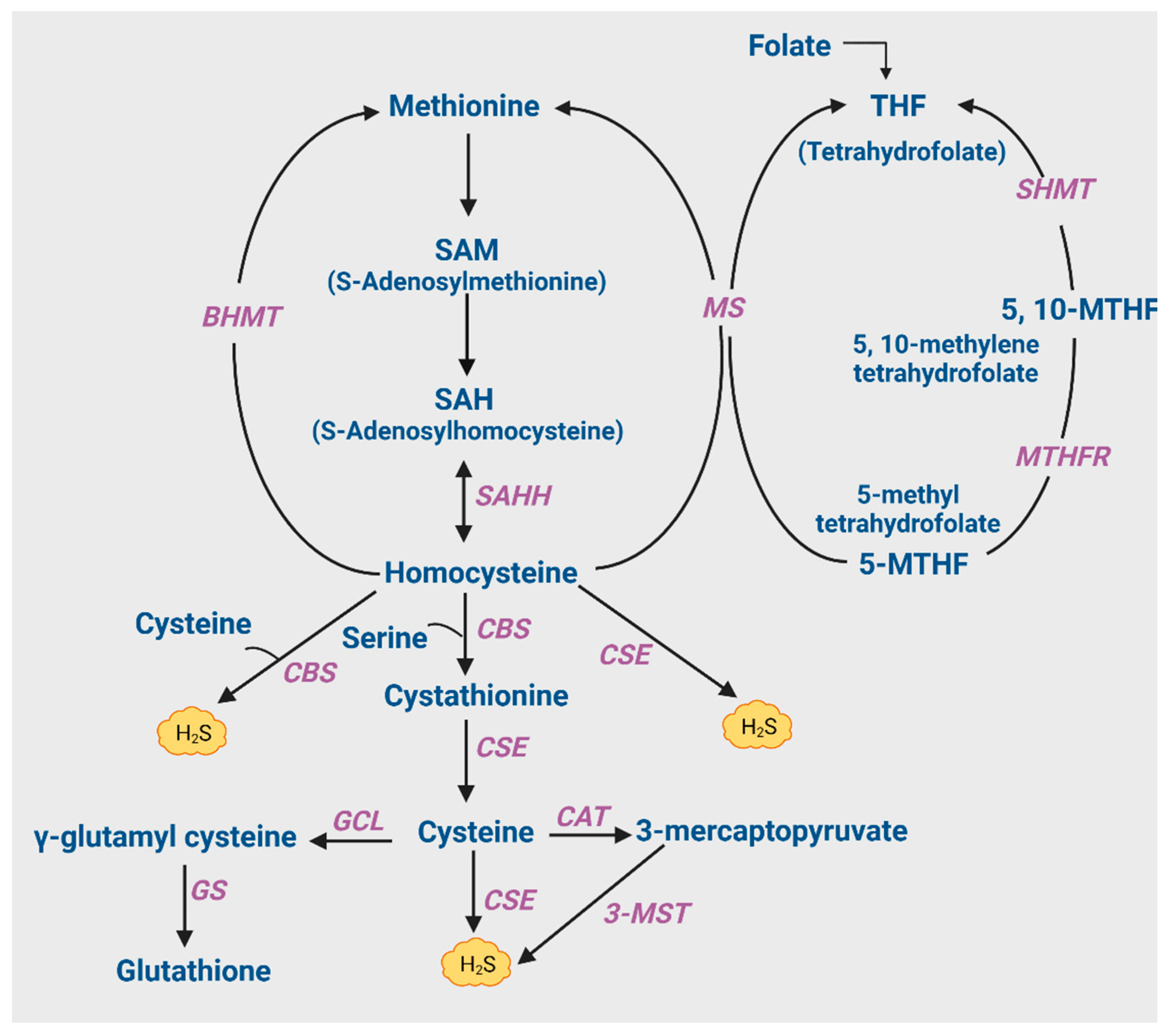
2. Biosynthesis of H2S in the Brain
3. H2S Signaling in Cognitive Functions
3.1. Alzheimer’s Disease
3.1.1. Hyperhomocysteinemia and Its Causes
3.1.2. Cysteine and GSH Metabolism
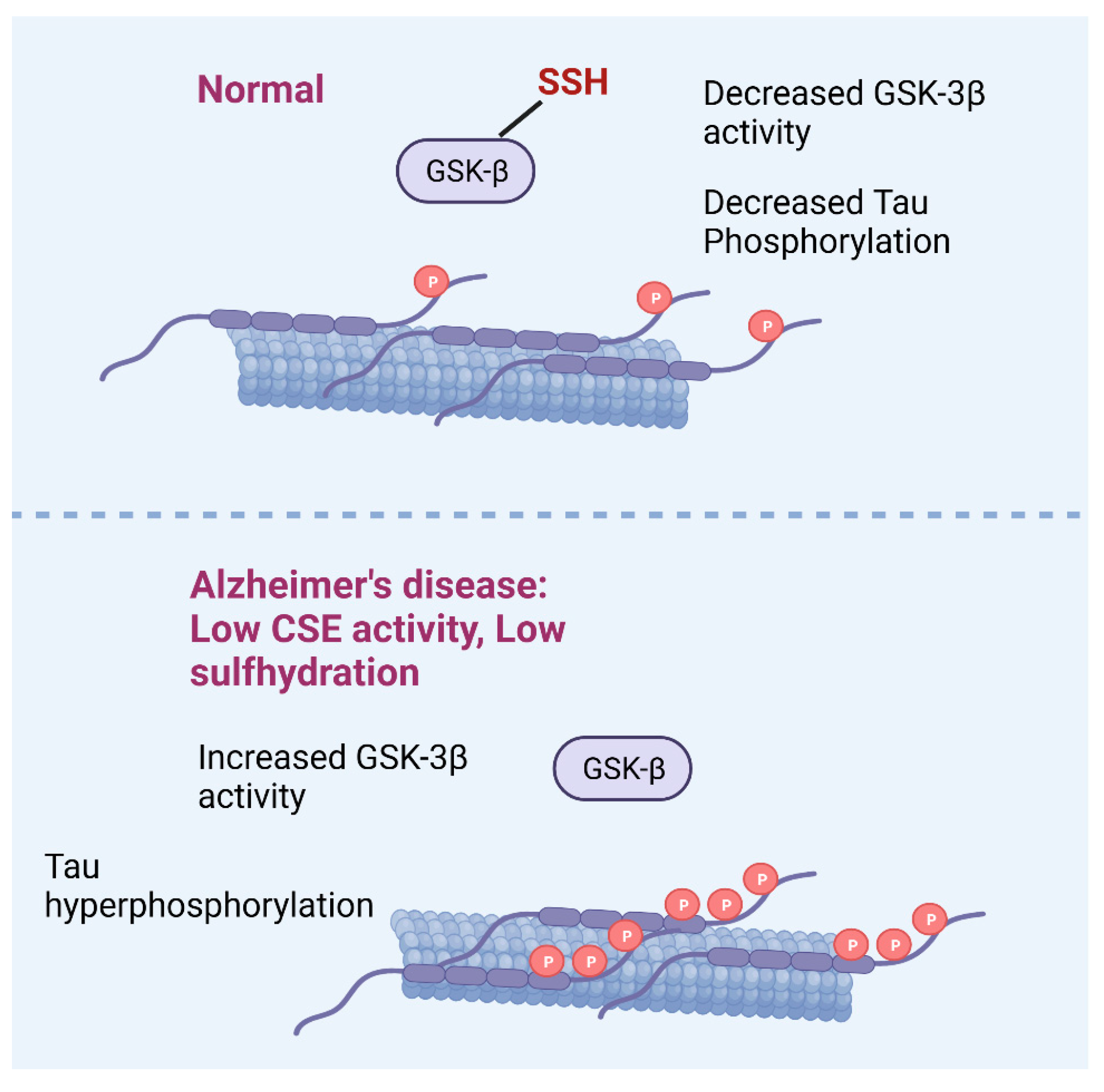
3.1.3. H2S and AD
Persulfidation, AD, and Aging
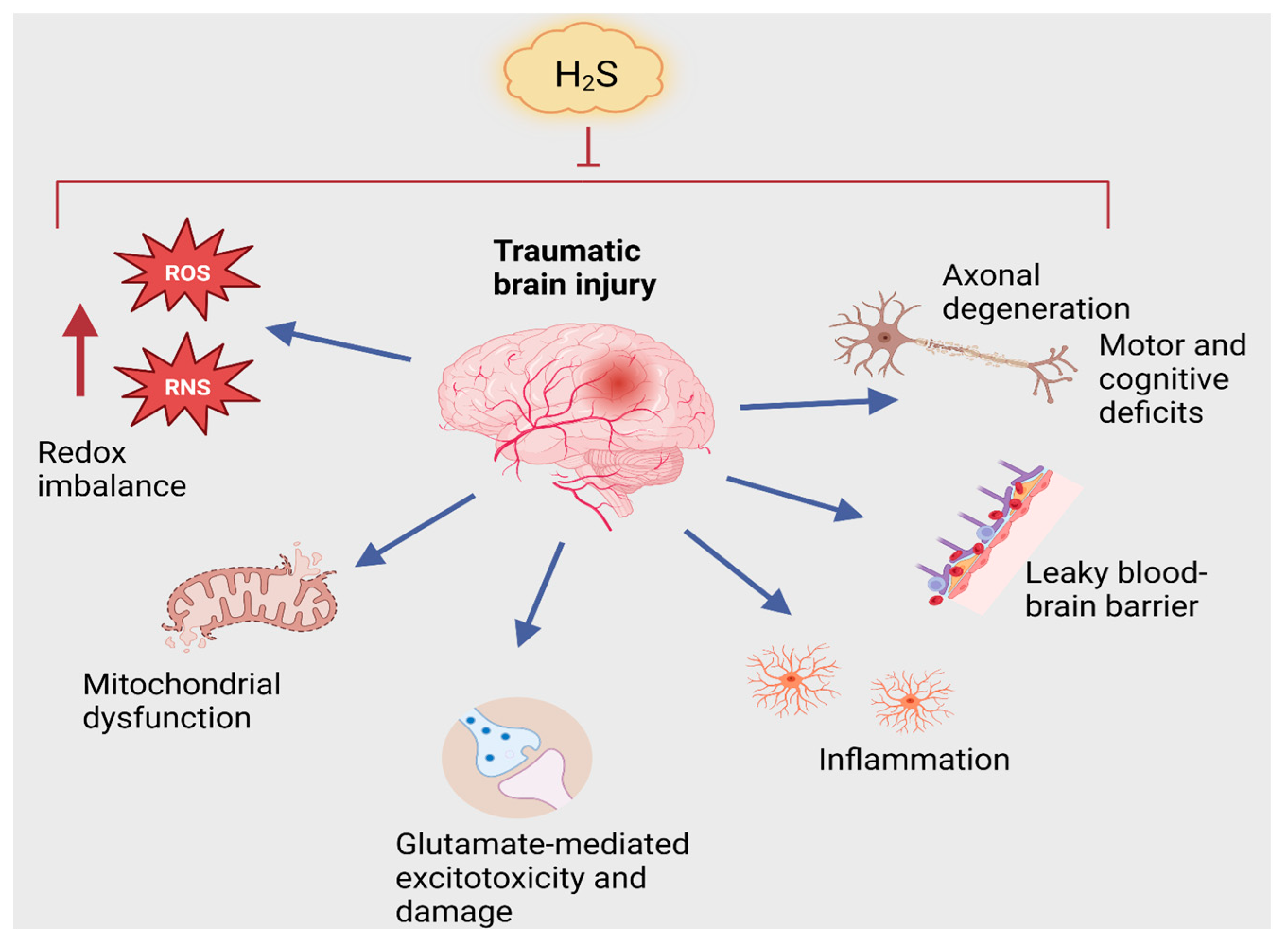
3.2. Traumatic Brain Injury
TBI and H2S
4. Dysregulation of Iron Homeostasis in AD, TBI and Intersection with H2S Signaling
5. Therapeutic Opportunities
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MST | 3-mercaptopyruvate sulfurtransferase |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| ATB346 | 2-(6-methoxynapthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester |
| ATF6 | Activating factor 6 |
| BBB | Blood–brain barrier |
| CAT | Cysteine aminotransferase |
| CBF | Cerebral blood flow |
| CBS | Cystathionine β-synthase |
| CSE | Cystathionine γ-lyase |
| CCI | Controlled cortical impact |
| COX-2 | Cyclo-oxygenase-2 |
| DADS | Diallyl disulfide |
| DATS | Diallyl trisulfide |
| EAAT3/EAAC1 | Excitatory amino acid transporter 3 |
| GDNF | Glial-derived neurotrophic factor |
| GLO-1,2 | Glyoxalase 1,2 |
| GSH | Glutathione |
| GSK-3β | Glycogen synthase kinase-3β |
| HD | Huntington’s disease |
| iNOS | Inducible nitric oxide synthase |
| MCI | Mild cognitive impairment |
| MRS | Magnetic resonance spectroscopy |
| MTHFR | Methylenetetrahydrofolate reductase |
| NGF | Nerve growth factor |
| Nrf2 | Nuclear factor erythroid-2-related factor 2 |
| PD | Parkinson’s disease |
| PDE5 | Phosphodiesterase 5 |
| PS1 | Presenilin-1 |
| ROS | Reactive Oxygen Species |
| RNS | Reactive Nitrogen Species |
| SAC | S-allyl cysteine |
| SAHH | S-adenosylhomocysteine hydrolase |
| SAM | S-adenosyl methionine |
| SCA | Spinocerebellar ataxia |
| α-SNAP | α-soluble NSF attachment protein |
| SNARE | Soluble NSF attachment protein receptor |
| TNF-α | Tumor necrosis factor alpha |
| VEGF | Vascular endothelial growth factor |
References
- Ramazzini, B. De Morbis Artificum Diatriba. Mutinae (Modena). Antonii Capponi. In Google Scholar; 1700. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1446785/ (accessed on 1 April 2023).
- du Vigneaud, V.; Loring, G.S.; Craft, H.A. The oxidation of the sulfur of homocystine, methionine, and S-methylcysteine in the animal body. J. Biol. Chem. 1934, 105, 481–488. [Google Scholar] [CrossRef]
- Binkley, F.; Du Vigneato, V. The formation of cysteine from homocysteine and serine by liver tissue of rats. J. Biol. Chem. 1942, 144, 507–511. [Google Scholar] [CrossRef]
- Hanson, H.; Eisfeld, G. Intermediary sulfur metabolism. III. Formation of hydrogen sulfide from cystine and cysteine by the liver. Z. Gesamte Inn. Med. 1952, 7, 801–810. [Google Scholar] [PubMed]
- Chatagner, F.; Sauret-Ignazi, G. Role of transamination and pyridoxal phosphate in the enzymatic formation of hydrogen sulfide from cysteine by the rat liver under anaerobiosis. Bull. Soc. Chim. Biol. 1956, 38, 415–428. [Google Scholar]
- Ubuka, T.; Yuasa, S.; Ishimoto, Y.; Shimomura, M. Desulfuration of l-cysteine through transamination and transsulfuration in rat liver. Physiol. Chem. Phys. 1977, 9, 241–246. [Google Scholar]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef]
- Warenycia, M.W.; Goodwin, L.R.; Benishin, C.G.; Reiffenstein, R.J.; Francom, D.M.; Taylor, J.D.; Dieken, F.P. Acute hydrogen sulfide poisoning. Demonstration of selective uptake of sulfide by the brainstem by measurement of brain sulfide levels. Biochem. Pharmacol. 1989, 38, 973–981. [Google Scholar] [CrossRef]
- Goodwin, L.R.; Francom, D.; Dieken, F.P.; Taylor, J.D.; Warenycia, M.W.; Reiffenstein, R.J.; Dowling, G. Determination of sulfide in brain tissue by gas dialysis/ion chromatography: Postmortem studies and two case reports. J. Anal. Toxicol. 1989, 13, 105–109. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed]
- Haouzi, P.; Sonobe, T.; Judenherc-Haouzi, A. Hydrogen sulfide intoxication induced brain injury and methylene blue. Neurobiol. Dis. 2020, 133, 104474. [Google Scholar] [CrossRef]
- Reiffenstein, R.J.; Hulbert, W.C.; Roth, S.H. Toxicology of hydrogen sulfide. Annu. Rev. Pharmacol. Toxicol. 1992, 32, 109–134. [Google Scholar] [CrossRef]
- Nicholls, P.; Kim, J.K. Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can. J. Biochem. 1982, 60, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, E.; Morrison, M.; Roth, M.B. H2S induces a suspended animation-like state in mice. Science 2005, 308, 518. [Google Scholar] [CrossRef]
- Nicholls, P.; Marshall, D.C.; Cooper, C.E.; Wilson, M.T. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 2013, 41, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Collman, J.P.; Ghosh, S.; Dey, A.; Decreau, R.A. Using a functional enzyme model to understand the chemistry behind hydrogen sulfide induced hibernation. Proc. Natl. Acad. Sci. USA 2009, 106, 22090–22095. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H.; Kashfi, K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. 2021, 38, 101772. [Google Scholar] [CrossRef] [PubMed]
- Modis, K.; Bos, E.M.; Calzia, E.; van Goor, H.; Coletta, C.; Papapetropoulos, A.; Hellmich, M.R.; Radermacher, P.; Bouillaud, F.; Szabo, C. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part II. Pathophysiological and therapeutic aspects. Br. J. Pharmacol. 2014, 171, 2123–2146. [Google Scholar] [CrossRef]
- Szabo, C.; Ransy, C.; Modis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 2023, 103, 31–276. [Google Scholar] [CrossRef] [PubMed]
- Panagaki, T.; Randi, E.B.; Augsburger, F.; Szabo, C. Overproduction of H(2)S, generated by CBS, inhibits mitochondrial Complex IV and suppresses oxidative phosphorylation in Down syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 18769–18771. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, A.; Kanaumi, T.; Takashima, S.; Enokido, Y.; Nagai, Y.; Kimura, H. Cystathionine beta-synthase is enriched in the brains of Down’s patients. Biochem. Biophys. Res. Commun. 2005, 338, 1547–1550. [Google Scholar] [CrossRef]
- Szabo, C. The re-emerging pathophysiological role of the cystathionine-beta-synthase—Hydrogen sulfide system in Down syndrome. FEBS J. 2020, 287, 3150–3160. [Google Scholar] [CrossRef]
- Barker, S.; Paul, B.D.; Pieper, A.A. Increased risk of aging-related neurodegenerative disease after traumatic brain injury. biomedicines 2023, 11, 1154. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Modes of physiologic H2S signaling in the brain and peripheral tissues. Antioxid. Redox Signal. 2015, 22, 411–423. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br. J. Pharmacol. 2019, 176, 583–593. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018, 149, 101–109. [Google Scholar] [CrossRef]
- Paul, B.D. Neuroprotective Roles of the Reverse Transsulfuration Pathway in Alzheimer’s Disease. Front Aging Neurosci 2021, 13, 659402. [Google Scholar] [CrossRef]
- Morikawa, T.; Kajimura, M.; Nakamura, T.; Hishiki, T.; Nakanishi, T.; Yukutake, Y.; Nagahata, Y.; Ishikawa, M.; Hattori, K.; Takenouchi, T.; et al. Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Enokido, Y.; Suzuki, E.; Iwasawa, K.; Namekata, K.; Okazawa, H.; Kimura, H. Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 2005, 19, 1854–1856. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Peter, E.A.; Bir, S.; Wang, R.; Kevil, C.G. Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic. Biol. Med. 2012, 52, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, M.; Hiraki, K.; Umemura, K.; Ogasawara, Y.; Ishii, K.; Kimura, H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxid. Redox Signal. 2009, 11, 205–214. [Google Scholar] [CrossRef]
- Khaspekov, L.G.; Frumkina, L.E. Molecular Mechanisms of Astrocyte Involvement in Synaptogenesis and Brain Synaptic Plasticity. Biochemistry 2023, 88, 502–514. [Google Scholar] [CrossRef]
- Farizatto, K.L.G.; Baldwin, K.T. Astrocyte-synapse interactions during brain development. Curr. Opin. Neurobiol. 2023, 80, 102704. [Google Scholar] [CrossRef]
- Jorgacevski, J.; Potokar, M. Immune Functions of Astrocytes in Viral Neuroinfections. Int. J. Mol. Sci. 2023, 24, 3514. [Google Scholar] [CrossRef]
- Finkelstein, J.D.; Kyle, W.E.; Martin, J.L.; Pick, A.M. Activation of cystathionine synthase by adenosylmethionine and adenosylethionine. Biochem. Biophys. Res. Commun. 1975, 66, 81–87. [Google Scholar] [CrossRef]
- Morrison, L.D.; Smith, D.D.; Kish, S.J. Brain S-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J. Neurochem. 1996, 67, 1328–1331. [Google Scholar] [CrossRef]
- Linnebank, M.; Popp, J.; Smulders, Y.; Smith, D.; Semmler, A.; Farkas, M.; Kulic, L.; Cvetanovska, G.; Blom, H.; Stoffel-Wagner, B.; et al. S-adenosylmethionine is decreased in the cerebrospinal fluid of patients with Alzheimer’s disease. Neurodegener. Dis. 2010, 7, 373–378. [Google Scholar] [CrossRef]
- Kimura, H.; Shibuya, N.; Kimura, Y. Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid. Redox Signal. 2012, 17, 45–57. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789. [CrossRef]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278, discussion 278–284. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Snitz, B.E.; Weissfeld, L.A.; Lopez, O.L.; Kuller, L.H.; Saxton, J.; Singhabahu, D.M.; Klunk, W.E.; Mathis, C.A.; Price, J.C.; Ives, D.G.; et al. Cognitive trajectories associated with beta-amyloid deposition in the oldest-old without dementia. Neurology 2013, 80, 1378–1384. [Google Scholar] [CrossRef]
- Rentz, D.M.; Locascio, J.J.; Becker, J.A.; Moran, E.K.; Eng, E.; Buckner, R.L.; Sperling, R.A.; Johnson, K.A. Cognition, reserve, and amyloid deposition in normal aging. Ann. Neurol. 2010, 67, 353–364. [Google Scholar] [CrossRef]
- Alves, F.; Kallinowski, P.; Ayton, S. Accelerated Brain Volume Loss Caused by Anti-beta-Amyloid Drugs: A Systematic Review and Meta-analysis. Neurology 2023. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H(2)S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef]
- Chen, X.; Jhee, K.H.; Kruger, W.D. Production of the neuromodulator H2S by cystathionine beta-synthase via the condensation of cysteine and homocysteine. J. Biol. Chem. 2004, 279, 52082–52086. [Google Scholar] [CrossRef]
- Beard, R.S., Jr.; Bearden, S.E. Vascular complications of cystathionine beta-synthase deficiency: Future directions for homocysteine-to-hydrogen sulfide research. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H13–H26. [Google Scholar] [CrossRef]
- Sen, U.; Mishra, P.K.; Tyagi, N.; Tyagi, S.C. Homocysteine to hydrogen sulfide or hypertension. Cell Biochem. Biophys. 2010, 57, 49–58. [Google Scholar] [CrossRef]
- Kitzlerova, E.; Fisar, Z.; Jirak, R.; Zverova, M.; Hroudova, J.; Benakova, H.; Raboch, J. Plasma homocysteine in Alzheimer’s disease with or without co-morbid depressive symptoms. Neuro Endocrinol. Lett. 2014, 35, 42–49. [Google Scholar]
- Farina, N.; Jerneren, F.; Turner, C.; Hart, K.; Tabet, N. Homocysteine concentrations in the cognitive progression of Alzheimer’s disease. Exp. Gerontol. 2017, 99, 146–150. [Google Scholar] [CrossRef] [PubMed]
- McCaddon, A.; Davies, G.; Hudson, P.; Tandy, S.; Cattell, H. Total serum homocysteine in senile dementia of Alzheimer type. Int. J. Geriatr. Psychiatry 1998, 13, 235–239. [Google Scholar] [CrossRef]
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Miller, J.W.; Rosenberg, I.H.; Obeid, R. Homocysteine and Dementia: An International Consensus Statement. J. Alzheimers Dis. 2018, 62, 561–570. [Google Scholar] [CrossRef]
- Clarke, R.; Smith, A.D.; Jobst, K.A.; Refsum, H.; Sutton, L.; Ueland, P.M. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch. Neurol. 1998, 55, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xie, X.; Sun, Y.; Zhou, F. Blood and CSF Homocysteine Levels in Alzheimer’s Disease: A Meta-Analysis and Meta-Regression of Case-Control Studies. Neuropsychiatr. Dis. Treat. 2022, 18, 2391–2403. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.; Rivera, I.; Ravasco, P.; Jakobs, C.; Blom, H.J.; Camilo, M.E.; de Almeida, I.T. 5,10-Methylenetetrahydrofolate reductase 677C-->T and 1298A-->C mutations are genetic determinants of elevated homocysteine. QJM 2003, 96, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hegele, R.A. Genomic basis of cystathioninuria (MIM 219500) revealed by multiple mutations in cystathionine gamma-lyase (CTH). Hum. Genet. 2003, 112, 404–408. [Google Scholar] [CrossRef]
- Wang, J.; Huff, A.M.; Spence, J.D.; Hegele, R.A. Single nucleotide polymorphism in CTH associated with variation in plasma homocysteine concentration. Clin. Genet. 2004, 65, 483–486. [Google Scholar] [CrossRef]
- Wan, X.; Ma, B.; Wang, X.; Guo, C.; Sun, J.; Cui, J.; Li, L. S-Adenosylmethionine Alleviates Amyloid-beta-Induced Neural Injury by Enhancing Trans-Sulfuration Pathway Activity in Astrocytes. J. Alzheimers Dis. 2020, 76, 981–995. [Google Scholar] [CrossRef]
- Li, Q.; Cui, J.; Fang, C.; Liu, M.; Min, G.; Li, L. S-Adenosylmethionine Attenuates Oxidative Stress and Neuroinflammation Induced by Amyloid-beta Through Modulation of Glutathione Metabolism. J. Alzheimers Dis. 2017, 58, 549–558. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Paul, B.D.; Parker, G.M.; Hester, L.D.; Snowman, A.M.; Taniguchi, Y.; Kamiya, A.; Snyder, S.H.; Sawa, A. The glutathione cycle shapes synaptic glutamate activity. Proc. Natl. Acad. Sci. USA 2019, 116, 2701–2706. [Google Scholar] [CrossRef]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free. Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Oxidative stress and ageing: Is ageing a cysteine deficiency syndrome? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2355–2372. [Google Scholar] [CrossRef]
- Han, H.; Wang, F.; Chen, J.; Li, X.; Fu, G.; Zhou, J.; Zhou, D.; Wu, W.; Chen, H. Changes in Biothiol Levels Are Closely Associated with Alzheimer’s Disease. J. Alzheimers Dis. 2021, 82, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3beta and inhibiting Tau hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid-beta cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers Dis. 2013, 36, 197–209. [Google Scholar] [CrossRef]
- Duerson, K.; Woltjer, R.L.; Mookherjee, P.; Leverenz, J.B.; Montine, T.J.; Bird, T.D.; Pow, D.V.; Rauen, T.; Cook, D.G. Detergent-insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of Alzheimer’s disease patients. Brain Pathol. 2009, 19, 267–278. [Google Scholar] [CrossRef]
- Pontrello, C.G.; McWhirt, J.M.; Glabe, C.G.; Brewer, G.J. Age-Related Oxidative Redox and Metabolic Changes Precede Intraneuronal Amyloid-beta Accumulation and Plaque Deposition in a Transgenic Alzheimer’s Disease Mouse Model. J. Alzheimers Dis. 2022, 90, 1501–1521. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef]
- Zabel, M.; Nackenoff, A.; Kirsch, W.M.; Harrison, F.E.; Perry, G.; Schrag, M. Markers of oxidative damage to lipids, nucleic acids and proteins and antioxidant enzymes activities in Alzheimer’s disease brain: A meta-analysis in human pathological specimens. Free. Radic. Biol. Med. 2018, 115, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain glutathione levels--a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Mandal, P.K.; Mishra, R.; Punjabi, K.; Dwivedi, D.; Tripathi, M.; Badhautia, V. Hippocampal Glutathione Depletion and pH Increment in Alzheimer’s Disease: An in vivo MRS Study. J. Alzheimers Dis. 2021, 84, 1139–1152. [Google Scholar] [CrossRef]
- Chen, J.J.; Thiyagarajah, M.; Song, J.; Chen, C.; Herrmann, N.; Gallagher, D.; Rapoport, M.J.; Black, S.E.; Ramirez, J.; Andreazza, A.C.; et al. Altered central and blood glutathione in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Alzheimers Res. Ther. 2022, 14, 23. [Google Scholar] [CrossRef]
- Dwivedi, D.; Megha, K.; Mishra, R.; Mandal, P.K. Glutathione in Brain: Overview of Its Conformations, Functions, Biochemical Characteristics, Quantitation and Potential Therapeutic Role in Brain Disorders. Neurochem. Res. 2020, 45, 1461–1480. [Google Scholar] [CrossRef]
- Navarro, J.F.; Croteau, D.L.; Jurek, A.; Andrusivova, Z.; Yang, B.; Wang, Y.; Ogedegbe, B.; Riaz, T.; Stoen, M.; Desler, C.; et al. Spatial Transcriptomics Reveals Genes Associated with Dysregulated Mitochondrial Functions and Stress Signaling in Alzheimer Disease. iScience 2020, 23, 101556. [Google Scholar] [CrossRef]
- Thornalley, P.J. Glyoxalase I--structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef]
- Eto, K.; Asada, T.; Arima, K.; Makifuchi, T.; Kimura, H. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 293, 1485–1488. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Liu, X.Q.; Jiang, P.; Huang, H.; Yan, Y. Plasma levels of endogenous hydrogen sulfide and homocysteine in patients with Alzheimer’s disease and vascular dementia and the significance thereof. Zhonghua Yi Xue Za Zhi 2008, 88, 2246–2249. [Google Scholar]
- Sun, P.; Chen, H.C.; Lu, S.; Hai, J.; Guo, W.; Jing, Y.H.; Wang, B. Simultaneous Sensing of H(2)S and ATP with a Two-Photon Fluorescent Probe in Alzheimer’s Disease: Toward Understanding Why H(2)S Regulates Glutamate-Induced ATP Dysregulation. Anal. Chem. 2022, 94, 11573–11581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Ma, S.; Liu, X.; Du, Y.; Zhu, X.; Liu, Y.; Wu, X. Activating transcription factor 6 regulates cystathionine to increase autophagy and restore memory in Alzheimer’s disease model mice. Biochem. Biophys. Res. Commun. 2022, 615, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Guo, Y.; Liang, X.; Yuan, Y.; Qi, X.; Wang, M.; Ma, J.; Zhou, H. Hydrogen sulfide protects against amyloid beta-peptide induced neuronal injury via attenuating inflammatory responses in a rat model. J. Biomed. Res. 2013, 27, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Bian, J.S. Hydrogen sulfide protects amyloid-beta induced cell toxicity in microglia. J. Alzheimers Dis. 2010, 22, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.Q.; Yang, C.T.; Chen, J.; Yin, W.L.; Tian, S.W.; Hu, B.; Feng, J.Q.; Li, Y.J. Effect of hydrogen sulphide on beta-amyloid-induced damage in PC12 cells. Clin. Exp. Pharmacol. Physiol. 2008, 35, 180–186. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Zhao, F.; Dai, Z.; Meng, T.; Tu, S.; Yan, Y. Hydrogen sulfide reduces mRNA and protein levels of beta-site amyloid precursor protein cleaving enzyme 1 in PC12 cells. Neurochem. Int. 2011, 58, 169–175. [Google Scholar] [CrossRef]
- Xuan, A.; Long, D.; Li, J.; Ji, W.; Zhang, M.; Hong, L.; Liu, J. Hydrogen sulfide attenuates spatial memory impairment and hippocampal neuroinflammation in beta-amyloid rat model of Alzheimer’s disease. J. Neuroinflammation 2012, 9, 202. [Google Scholar] [CrossRef]
- Giuliani, D.; Ottani, A.; Zaffe, D.; Galantucci, M.; Strinati, F.; Lodi, R.; Guarini, S. Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms. Neurobiol. Learn. Mem. 2013, 104, 82–91. [Google Scholar] [CrossRef]
- Ali, R.; Hameed, R.; Chauhan, D.; Sen, S.; Wahajuddin, M.; Nazir, A.; Verma, S. Multiple Actions of H(2)S-Releasing Peptides in Human beta-Amyloid Expressing C. elegans. ACS Chem. Neurosci. 2022, 13, 3378–3388. [Google Scholar] [CrossRef]
- Xi, Y.; Zhang, Y.; Zhou, Y.; Liu, Q.; Chen, X.; Liu, X.; Grune, T.; Shi, L.; Hou, M.; Liu, Z. Effects of methionine intake on cognitive function in mild cognitive impairment patients and APP/PS1 Alzheimer’s Disease model mice: Role of the cystathionine-beta-synthase/H(2)S pathway. Redox Biol. 2023, 59, 102595. [Google Scholar] [CrossRef]
- Rose, P.; Moore, P.K.; Zhu, Y.Z. Garlic and Gaseous Mediators. Trends Pharmacol. Sci. 2018, 39, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Benavides, G.A.; Squadrito, G.L.; Mills, R.W.; Patel, H.D.; Isbell, T.S.; Patel, R.P.; Darley-Usmar, V.M.; Doeller, J.E.; Kraus, D.W. Hydrogen sulfide mediates the vasoactivity of garlic. Proc. Natl. Acad. Sci. USA 2007, 104, 17977–17982. [Google Scholar] [CrossRef]
- Liang, D.; Wu, H.; Wong, M.W.; Huang, D. Diallyl Trisulfide Is a Fast H2S Donor, but Diallyl Disulfide Is a Slow One: The Reaction Pathways and Intermediates of Glutathione with Polysulfides. Org. Lett. 2015, 17, 4196–4199. [Google Scholar] [CrossRef]
- Chuah, S.C.; Moore, P.K.; Zhu, Y.Z. S-allylcysteine mediates cardioprotection in an acute myocardial infarction rat model via a hydrogen sulfide-mediated pathway. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2693–H2701. [Google Scholar] [CrossRef] [PubMed]
- Ghazimoradi, M.M.; Ghoushi, E.; Ghobadi Pour, M.; Karimi Ahmadabadi, H.; Rafieian-Kopaei, M. A review on garlic as a supplement for Alzheimer’s disease: A mechanistic insight in its direct and indirect effects. Curr. Pharm. Des. 2023, 29, 519–526. [Google Scholar] [CrossRef]
- Ray, B.; Chauhan, N.B.; Lahiri, D.K. The “aged garlic extract:” (AGE) and one of its active ingredients S-allyl-L-cysteine (SAC) as potential preventive and therapeutic agents for Alzheimer’s disease (AD). Curr. Med. Chem. 2011, 18, 3306–3313. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.B. Effect of aged garlic extract on APP processing and tau phosphorylation in Alzheimer’s transgenic model Tg2576. J. Ethnopharmacol. 2006, 108, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, P.; Nigro, M.; Travagli, A.; Catani, M.; Cavazzini, A.; Merighi, S.; Gessi, S. Therapeutic Potential of Allicin and Aged Garlic Extract in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 6950. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.; Selassie, M.; Gwebu, E.T. Effect of Aged Garlic Extract on the Cytotoxicity of Alzheimer beta-Amyloid Peptide in Neuronal PC12 Cells. Nutr. Neurosci. 2000, 3, 139–142. [Google Scholar] [CrossRef]
- Gupta, V.B.; Indi, S.S.; Rao, K.S. Garlic extract exhibits antiamyloidogenic activity on amyloid-beta fibrillogenesis: Relevance to Alzheimer’s disease. Phytother. Res. 2009, 23, 111–115. [Google Scholar] [CrossRef]
- Luo, J.F.; Dong, Y.; Chen, J.Y.; Lu, J.H. The effect and underlying mechanisms of garlic extract against cognitive impairment and Alzheimer’s disease: A systematic review and meta-analysis of experimental animal studies. J. Ethnopharmacol. 2021, 280, 114423. [Google Scholar] [CrossRef]
- He, X.L.; Yan, N.; Zhang, H.; Qi, Y.W.; Zhu, L.J.; Liu, M.J.; Yan, Y. Hydrogen sulfide improves spatial memory impairment and decreases production of Abeta in APP/PS1 transgenic mice. Neurochem. Int. 2014, 67, 1–8. [Google Scholar] [CrossRef]
- Yang, Y.J.; Zhao, Y.; Yu, B.; Xu, G.G.; Wang, W.; Zhan, J.Q.; Tang, Z.Y.; Wang, T.; Wei, B. GluN2B-containing NMDA receptors contribute to the beneficial effects of hydrogen sulfide on cognitive and synaptic plasticity deficits in APP/PS1 transgenic mice. Neuroscience 2016, 335, 170–183. [Google Scholar] [CrossRef]
- Zhao, F.L.; Qiao, P.F.; Yan, N.; Gao, D.; Liu, M.J.; Yan, Y. Hydrogen Sulfide Selectively Inhibits gamma-Secretase Activity and Decreases Mitochondrial Abeta Production in Neurons from APP/PS1 Transgenic Mice. Neurochem. Res. 2016, 41, 1145–1159. [Google Scholar] [CrossRef]
- Li, X.H.; Deng, Y.Y.; Li, F.; Shi, J.S.; Gong, Q.H. Neuroprotective effects of sodium hydrosulfide against beta-amyloid-induced neurotoxicity. Int. J. Mol. Med. 2016, 38, 1152–1160. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, Y.; Yang, C.; Sun, Y.; Zhang, M.; Wang, S. Hydrogen sulfide prevents Abeta-induced neuronal apoptosis by attenuating mitochondrial translocation of PTEN. Neuroscience 2016, 325, 165–174. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, Y.; Liu, H.; Yin, C.; Li, X.; Gong, Q. Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: A novel mechanism mediated by the activation of Nrf2. Pharmacol. Biochem. Behav. 2016, 150–151, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Sestito, S.; Daniele, S.; Pietrobono, D.; Citi, V.; Bellusci, L.; Chiellini, G.; Calderone, V.; Martini, C.; Rapposelli, S. Memantine prodrug as a new agent for Alzheimer’s Disease. Sci. Rep. 2019, 9, 4612. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.P.; Xie, W.; Christopher Kwon, Y.I.; Juckel, N.; Xie, J.; Dronamraju, V.R.; Vince, R.; Lee, M.K.; More, S.S. Sulfanegen stimulates 3-mercaptopyruvate sulfurtransferase activity and ameliorates Alzheimer’s disease pathology and oxidative stress in vivo. Redox Biol. 2022, 57, 102484. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, M.; Ashabi, G.; Kashef, M.; Marashi, E.S.; Ghasemi, T. Aerobic Training with Naringin Supplementation Improved Spatial Cognition via H(2)S Signaling Pathway in Alzheimer’s Disease Model Rats. Exp. Aging Res. 2022, 1–14. [Google Scholar] [CrossRef]
- Aboulhoda, B.E.; Rashed, L.A.; Ahmed, H.; Obaya, E.M.M.; Ibrahim, W.; Alkafass, M.A.L.; Abd El-Aal, S.A.; ShamsEldeen, A.M. Hydrogen sulfide and mesenchymal stem cells-extracted microvesicles attenuate LPS-induced Alzheimer’s disease. J. Cell. Physiol. 2021, 236, 5994–6010. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kyles, P.; Kalani, A.; Tyagi, N. Hydrogen Sulfide Ameliorates Homocysteine-Induced Alzheimer’s Disease-Like Pathology, Blood-Brain Barrier Disruption, and Synaptic Disorder. Mol. Neurobiol. 2016, 53, 2451–2467. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.J.; Chen, S.L.; Hsieh-Li, H.M. Administration of NaHS Attenuates Footshock-Induced Pathologies and Emotional and Cognitive Dysfunction in Triple Transgenic Alzheimer’s Mice. Front. Behav. Neurosci. 2015, 9, 312. [Google Scholar] [CrossRef]
- Vandini, E.; Ottani, A.; Zaffe, D.; Calevro, A.; Canalini, F.; Cavallini, G.M.; Rossi, R.; Guarini, S.; Giuliani, D. Mechanisms of Hydrogen Sulfide against the Progression of Severe Alzheimer’s Disease in Transgenic Mice at Different Ages. Pharmacology 2019, 103, 50–60. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef]
- Petrovic, D.; Kouroussis, E.; Vignane, T.; Filipovic, M.R. The Role of Protein Persulfidation in Brain Aging and Neurodegeneration. Front. Aging Neurosci. 2021, 13, 674135. [Google Scholar] [CrossRef] [PubMed]
- Doka, E.; Pader, I.; Biro, A.; Johansson, K.; Cheng, Q.; Ballago, K.; Prigge, J.R.; Pastor-Flores, D.; Dick, T.P.; Schmidt, E.E.; et al. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2016, 2, e1500968. [Google Scholar] [CrossRef]
- Krishnan, N.; Fu, C.; Pappin, D.J.; Tonks, N.K. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal. 2011, 4, ra86. [Google Scholar] [CrossRef]
- Wedmann, R.; Onderka, C.; Wei, S.; Szijarto, I.A.; Miljkovic, J.L.; Mitrovic, A.; Lange, M.; Savitsky, S.; Yadav, P.K.; Torregrossa, R.; et al. Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 2016, 7, 3414–3426. [Google Scholar] [CrossRef]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Ko, H.S.; Lee, Y.I.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef]
- Paul, B.D.; Sbodio, J.I.; Snyder, S.H. Mutant Huntingtin Derails Cysteine Metabolism in Huntington’s Disease at Both Transcriptional and Post-Translational Levels. Antioxidants 2022, 11, 1470. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Golgi stress response reprograms cysteine metabolism to confer cytoprotection in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 780–785. [Google Scholar] [CrossRef]
- Snijder, P.M.; Baratashvili, M.; Grzeschik, N.A.; Leuvenink, H.G.D.; Kuijpers, L.; Huitema, S.; Schaap, O.; Giepmans, B.N.G.; Kuipers, J.; Miljkovic, J.L.; et al. Overexpression of Cystathionine gamma-Lyase Suppresses Detrimental Effects of Spinocerebellar Ataxia Type 3. Mol. Med. 2016, 21, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170.e1113. [Google Scholar] [CrossRef]
- Ji, D.; Luo, C.; Liu, J.; Cao, Y.; Wu, J.; Yan, W.; Xue, K.; Chai, J.; Zhu, X.; Wu, Y.; et al. Insufficient S-Sulfhydration of Methylenetetrahydrofolate Reductase Contributes to the Progress of Hyperhomocysteinemia. Antioxid. Redox Signal. 2022, 36, 1–14. [Google Scholar] [CrossRef]
- Yin, W.L.; He, J.Q.; Hu, B.; Jiang, Z.S.; Tang, X.Q. Hydrogen sulfide inhibits MPP+-induced apoptosis in PC12 cells. Life Sci. 2009, 85, 269–275. [Google Scholar] [CrossRef]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shan, H.; Wang, T.; Liu, W.; Wang, Y.; Wang, L.; Zhang, L.; Chang, P.; Dong, W.; Chen, X.; et al. Dynamic change of hydrogen sulfide after traumatic brain injury and its effect in mice. Neurochem. Res. 2013, 38, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Huang, Y.; Lin, W.; Gao, D.; Fei, Z. Protective effects of hydrogen sulfide in a rat model of traumatic brain injury via activation of mitochondrial adenosine triphosphate-sensitive potassium channels and reduction of oxidative stress. J. Surg. Res. 2013, 184, e27–e35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shan, H.; Chang, P.; Wang, T.; Dong, W.; Chen, X.; Tao, L. Hydrogen sulfide offers neuroprotection on traumatic brain injury in parallel with reduced apoptosis and autophagy in mice. PLoS ONE 2014, 9, e87241. [Google Scholar] [CrossRef]
- Karimi, S.A.; Hosseinmardi, N.; Janahmadi, M.; Sayyah, M.; Hajisoltani, R. The protective effect of hydrogen sulfide (H(2)S) on traumatic brain injury (TBI) induced memory deficits in rats. Brain Res. Bull. 2017, 134, 177–182. [Google Scholar] [CrossRef]
- Xu, K.; Wu, F.; Xu, K.; Li, Z.; Wei, X.; Lu, Q.; Jiang, T.; Wu, F.; Xu, X.; Xiao, J.; et al. NaHS restores mitochondrial function and inhibits autophagy by activating the PI3K/Akt/mTOR signalling pathway to improve functional recovery after traumatic brain injury. Chem. Biol. Interact. 2018, 286, 96–105. [Google Scholar] [CrossRef]
- Campolo, M.; Esposito, E.; Ahmad, A.; Di Paola, R.; Paterniti, I.; Cordaro, M.; Bruschetta, G.; Wallace, J.L.; Cuzzocrea, S. Hydrogen sulfide-releasing cyclooxygenase inhibitor ATB-346 enhances motor function and reduces cortical lesion volume following traumatic brain injury in mice. J. Neuroinflamm. 2014, 11, 196. [Google Scholar] [CrossRef]
- Zhang, M.; Shan, H.; Chang, P.; Ma, L.; Chu, Y.; Shen, X.; Wu, Q.; Wang, Z.; Luo, C.; Wang, T.; et al. Upregulation of 3-MST Relates to Neuronal Autophagy After Traumatic Brain Injury in Mice. Cell. Mol. Neurobiol. 2017, 37, 291–302. [Google Scholar] [CrossRef]
- Sun, J.; Li, X.; Gu, X.; Du, H.; Zhang, G.; Wu, J.; Wang, F. Neuroprotective effect of hydrogen sulfide against glutamate-induced oxidative stress is mediated via the p53/glutaminase 2 pathway after traumatic brain injury. Aging 2021, 13, 7180–7189. [Google Scholar] [CrossRef]
- Huerta de la Cruz, S.; Rocha, L.; Santiago-Castaneda, C.; Sanchez-Lopez, A.; Pinedo-Rodriguez, A.D.; Medina-Terol, G.J.; Centurion, D. Hydrogen Sulfide Subchronic Treatment Improves Hypertension Induced by Traumatic Brain Injury in Rats through Vasopressor Sympathetic Outflow Inhibition. J. Neurotrauma 2022, 39, 181–195. [Google Scholar] [CrossRef]
- Lopez-Preza, F.I.; Huerta de la Cruz, S.; Santiago-Castaneda, C.; Silva-Velasco, D.L.; Beltran-Ornelas, J.H.; Tapia-Martinez, J.; Sanchez-Lopez, A.; Rocha, L.; Centurion, D. Hydrogen sulfide prevents the vascular dysfunction induced by severe traumatic brain injury in rats by reducing reactive oxygen species and modulating eNOS and H(2)S-synthesizing enzyme expression. Life Sci. 2023, 312, 121218. [Google Scholar] [CrossRef]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Lin, Q.; Shahid, S.; Hone-Blanchet, A.; Huang, S.; Wu, J.; Bisht, A.; Loring, D.; Goldstein, F.; Levey, A.; Crosson, B.; et al. Magnetic resonance evidence of increased iron content in subcortical brain regions in asymptomatic Alzheimer’s disease. Hum. Brain Mapp. 2023. [Google Scholar] [CrossRef] [PubMed]
- Kenkhuis, B.; Bush, A.I.; Ayton, S. How iron can drive neurodegeneration. Trends Neurosci. 2023, 46, 333–335. [Google Scholar] [CrossRef]
- Mahoney-Sanchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.C. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef]
- Robicsek, S.A.; Bhattacharya, A.; Rabai, F.; Shukla, K.; Dore, S. Blood-Related Toxicity after Traumatic Brain Injury: Potential Targets for Neuroprotection. Mol. Neurobiol. 2020, 57, 159–178. [Google Scholar] [CrossRef]
- Nisenbaum, E.J.; Novikov, D.S.; Lui, Y.W. The presence and role of iron in mild traumatic brain injury: An imaging perspective. J. Neurotrauma 2014, 31, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Braughler, J.M.; Duncan, L.A.; Chase, R.L. The involvement of iron in lipid peroxidation. Importance of ferric to ferrous ratios in initiation. J. Biol. Chem. 1986, 261, 10282–10289. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Ashraf, A.; Jeandriens, J.; Parkes, H.G.; So, P.W. Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: Evidence of ferroptosis. Redox Biol. 2020, 32, 101494. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Torabi, S.F.; Lake, R.J.; Hong, S.; Yu, Z.; Wu, P.; Yang, Z.; Nelson, K.; Guo, W.; Pawel, G.T.; et al. Simultaneous Fe2+/Fe3+ imaging shows Fe3+ over Fe2+ enrichment in Alzheimer’s disease mouse brain. Sci. Adv. 2023, 9, eade7622. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Wang, Y.; Ying, X.; Wang, Y.; Zou, Z.; Yuan, A.; Xiao, Z.; Geng, N.; Qiao, Z.; Li, W.; Lu, X.; et al. Hydrogen sulfide alleviates mitochondrial damage and ferroptosis by regulating OPA3-NFS1 axis in doxorubicin-induced cardiotoxicity. Cell. Signal. 2023, 107, 110655. [Google Scholar] [CrossRef]
- Yu, Y.; Li, X.; Wu, X.; Li, X.; Wei, J.; Chen, X.; Sun, Z.; Zhang, Q. Sodium hydrosulfide inhibits hemin-induced ferroptosis and lipid peroxidation in BV2 cells via the CBS/H(2)S system. Cell. Signal. 2023, 104, 110594. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, W.; Dang, J.; Liu, B.; Xu, J.; Li, J.; Liu, Y.; He, L.; Ying, Y.; Cai, J.; et al. Hydrogen sulfide protects retinal pigment epithelium cells against ferroptosis through the AMPK- and p62-dependent non-canonical NRF2-KEAP1 pathway. Exp. Cell Res. 2023, 422, 113436. [Google Scholar] [CrossRef]
- Wallace, J.L.; Nagy, P.; Feener, T.D.; Allain, T.; Ditroi, T.; Vaughan, D.J.; Muscara, M.N.; de Nucci, G.; Buret, A.G. A proof-of-concept, Phase 2 clinical trial of the gastrointestinal safety of a hydrogen sulfide-releasing anti-inflammatory drug. Br. J. Pharmacol. 2020, 177, 769–777. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| H2S Donor/ Boosting Agent | System | Effect | Ref. |
|---|---|---|---|
| NaHS | β (Aβ 25-35)-treated PC12 cells | Prevented β (Aβ 25-35)-induced cytotoxicity and apoptosis by reducing the loss of MMP and attenuating the increase in intracellular ROS. | [90] |
| NaHS | PC12 cells | Decreased beta secretase-1 (BACE-1) levels and Aβ1-42 release. Increased phosphorylation of Akt. | [91] |
| NaHS | Aβ1-40-induced toxicity in BV-2 microglial cells | Attenuated Aβ1-40-induced lactate dehydrogenase (LDH) release and expression of growth arrest DNA damage (GADD 153). Inhibited release of nitric oxide, induction of inducible nitric oxide synthase (iNOS), and expression of tumor necrosis factor α (TNF-α) and cyclooxygenase 2 (COX-2). | [89] |
| NaHS | Rat Amyloid β (Aβ 1-40)-injection model | Ameliorated learning and memory deficits. Suppressed Aβ(1-40)-induced apoptosis in the hippocampal CA1 region. Diminished hippocampal inflammation, astrogliosis, and microgliosis. | [92] |
| NaHS | APP/PS1 mouse model of AD | Improved spatial memory. Decreased expression of BACE1, PS1, and amyloidogenic C99 fragment. Increased expression of ADAM17 and nonamyloidogenic C83 fragment. | [107] |
| NaHS | APP/PS1 mouse model of AD | Increased hippocampal H2S levels, intracellular ATP, and mitochondrial COX IV activity. Improved hippocampus-dependent contextual fear memory and novel object recognition. Decreased Aβ accumulation in mitochondria. | [108] |
| NaHS | APP/PS1 mouse model of AD | Inhibited γ-secretase activity and decreases mitochondrial Aβ production in neurons. | [109] |
| NaHS | Rat amyloid β (Aβ 25-35)-injection model | Prevented neuronal loss, apoptosis, and activation of pro-caspase-3. Decreased expression of phosphodiesterase 5 (PDE-5) in the hippocampus. Upregulated expression of peroxisome proliferator-activated receptor (PPAR)-α and PPAR-γ. Prevented Aβ 25-35)-decrease in IκB-α degradation and increase in nuclear factor-κB (NF-κB) p65 phosphorylation levels. | [110] |
| NaHS | Primary hippocampal neuronal culture from C57/BL6 mice at P0-P1 | Reduced Aβ-induced apoptosis. Decreased release of cytochrome C into the cytosol and caspase-3 activity. Decreased translocation of the phosphatase and tensin homologs deleted on chromosome 10 (PTEN) from the cytosol to the mitochondria. Increased p-AKT/AKT levels in PI3K-dependent manner. | [111] |
| NaHS | APP/PS1 mouse model of AD | Mitigated cognitive deficits. Decreased the number of senile plaques, Aβ1-40 and Aβ1-42 levels, neuronal loss, beta-amyloid precursor (APP), and BACE1. Increased CBS and 3MST. Augmented antioxidant effects through induction of nuclear factor erythroid-2-related factor 2 (Nrf2), heme oxygenase-1(HO-1), and glutathione S-transferase (GST). | [112] |
|
Memit (Derived from memantine by replacing the free amine group with an isothiocyanate moiety) | Aβ oligomers-induced damage in human neurons and rat | Decreased Aβ(1-42)-induced aggregation and Aβ oligomer-induced damage in human neurons and rat microglial cells. Increased neuroprotective autophagy. | [113] |
| H2S releasing peptide conjugates | C. elegans | Reduced Aβ1–42 amyloid deposits and ROS. Increased acetylcholine levels. | [94] |
| Sulfanagen | APP/PS1 mouse model of AD | Stimulated 3-MST and prevented neuropathology. | [114] |
| Methionine restriction | APP/PS1 mouse model of AD | Decreased Aβ accumulation. Improved cognitive function. Restored synapse ultrastructure. Alleviated mitochondrial dysfunction by enhancing mitochondrial biogenesis in male mice. Balanced the redox status and activated cystathionine-β-synthase (CBS)/ H2S pathway. | [95] |
| Naringin | Rat Amyloid β (Aβ)-injection model | Improved spatial memory. Increased H2S production. | [115] |
| NaHS | LPS-induced AD-like cognitive deficits in Wistar albino rats | Reduced inflammation. Decreased oxidative stress, apoptosis, and histopathological alterations. | [116] |
| NaHS in combination with the NMDA-receptor antagonist, MK801 | Homocysteine (intracerebral injection)-induced AD-like neurodegeneration | Ameliorated BBB disruption, impaired cerebral blood flow (CBF), and diminished synaptic function. | [117] |
| NaHS | Mouse primary hippocampal neurons (E16–18), 3xTg-AD mouse model of AD | Reduced neuronal death in Aβ1-42 treated primary hippocampal neuronal culture and increased neurite length. Increased plasma H2S and decreased anxiety-like behavior and cognitive deficits in 3xTg-AD mice. Reduced amyloid deposits and hyperphosphorylation of Tau. Decreased inflammatory responses, oxidative stress, and gliosis in 3×Tg-AD mice. | [118] |
| NaHS, sulfurous water | 3xTg-AD mouse model of AD | Prevented learning and memory deficits. Reduced size of amyloid β plaques. Decreased activity of c-jun N-terminal kinases, extracellular signal-regulated kinases, and p38. | [119] |
| NaHS, Tabiano’s spa-water | Rat Aβ1–40 injection model of AD, Streptozotocin-induced AD, 3xTg-AD mouse model of AD | Improved learning and memory deficits in all three models. Decreased amyloid deposits in the rat models of AD. The spa-water decreased oxidative and nitrosative stress, MAPK activation, inflammation, and apoptosis in 3xTg-AD mice. | [93] |
| Na-GYY4137 | 3xTg-AD mouse model of AD | Improved motor and cognitive function in 3xTg-AD mice. Increased overall sulfhydration. Inhibited Tau phosphorylation by sulfhydrating glycogen synthase kinase (GSK3β) and inhibiting its activity. | [72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, B.D.; Pieper, A.A. Protective Roles of Hydrogen Sulfide in Alzheimer’s Disease and Traumatic Brain Injury. Antioxidants 2023, 12, 1095. https://doi.org/10.3390/antiox12051095
Paul BD, Pieper AA. Protective Roles of Hydrogen Sulfide in Alzheimer’s Disease and Traumatic Brain Injury. Antioxidants. 2023; 12(5):1095. https://doi.org/10.3390/antiox12051095
Chicago/Turabian StylePaul, Bindu D., and Andrew A. Pieper. 2023. "Protective Roles of Hydrogen Sulfide in Alzheimer’s Disease and Traumatic Brain Injury" Antioxidants 12, no. 5: 1095. https://doi.org/10.3390/antiox12051095