
Heme: The Lord of the Iron Ring
 , , and
, , and 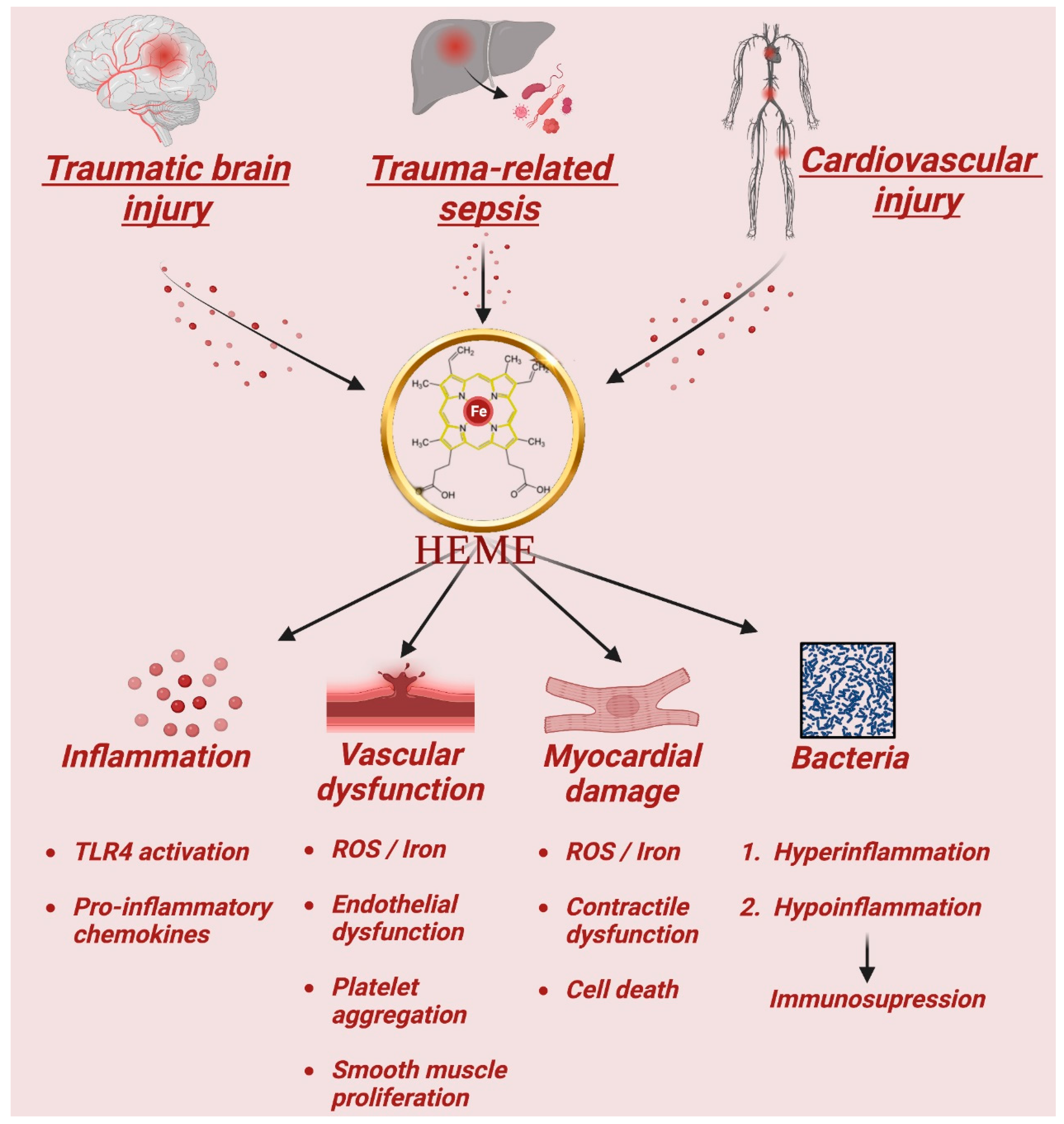{kind=link}
{kind=link}
Abstract
:1. Molecular Structure, Physiological Function, and Toxic Effects of Free Heme
1.1. Heme Structure
1.2. Heme Physiology
1.3. Heme Synthesis
1.4. Heme Scavenging
1.5. Heme Degradation
1.6. Heme Toxicity
1.7. Heme and Oxidative Stress
1.8. Hemolysis
1.9. Heme and Inflammation
1.10. Heme in Trauma and Inflammatory Diseases
1.11. Heme in Traumatic Brain Injury
1.12. Heme in Trauma-Related Sepsis
1.13. Heme-Induced Injury in the Cardiovascular System
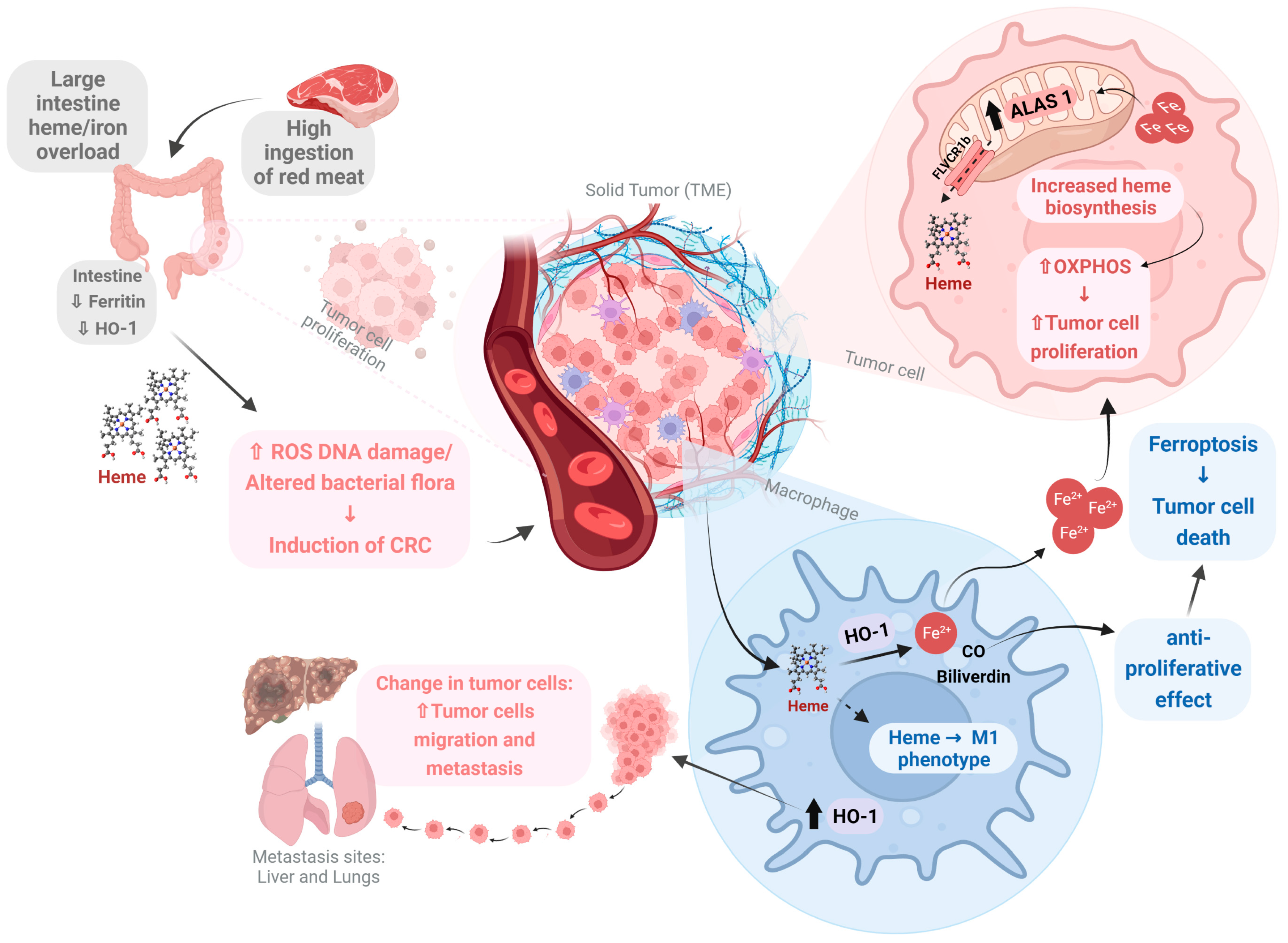
1.14. Heme in Cancer
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ogun, A.S.; Joy, N.V.; Valentine, M. Biochemistry, Heme Synthesis; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Hederstedt, L. Heme A biosynthesis. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 920–927. [Google Scholar] [CrossRef]
- Bowman, S.E.J.; Bren, K.L. The chemistry and biochemistry of heme c: Functional bases for covalent attachment. Nat. Prod. Rep. 2008, 25, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Rich, P.R.; Maréchal, A. 8.5 Electron Transfer Chains: Structures, Mechanisms and Energy Coupling. Compr. Biophys. 2012, 8, 72–93. [Google Scholar] [CrossRef]
- Gidari, A.S.; Levere, R.D. Enzymatic formation and cellular regulation of heme synthesis. Semin. Hematol. 1977, 14, 145–168. [Google Scholar] [PubMed]
- Ponka, P. Cell Biology of Heme. Am. J. Med. Sci. 1999, 318, 241. [Google Scholar] [CrossRef]
- Yamamoto, M.; Hayashi, N.; Kikuchi, G. Evidence for the transcriptional inhibition by heme of the synthesis of Delta-aminolevulinate synthase in rat liver. Biochem. Biophys. Res. Commun. 1982, 105, 985–990. [Google Scholar] [CrossRef]
- Yamamoto, M.; Hayashi, N.; Kikuchi, G. Translational inhibition by heme of the synthesis of hepatic Delta-aminolevulinate synthase in a cell-free system. Biochem. Biophys. Res. Commun. 1983, 115, 225–231. [Google Scholar] [CrossRef]
- Hamilton, J.W.; Bement, W.J.; Sinclair, P.R.; Sinclair, J.F.; Alcedo, J.A.; Wetterhahn, K.E. Heme regulates hepatic 5-aminolevulinate synthase mRNA expression by decreasing mRNA half-life and not by altering its rate of transcription. Arch. Biochem. Biophys. 1991, 289, 387–392. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Noriega, G.O.; Tomaro, M.L.; del Batlle, A.M.C. Bilirubin is highly effective in preventing in vivo δ-aminolevulinic acid-induced oxidative cell damage. Biochim. Biophys. Acta Mol. Basis Dis. 2003, 1638, 173–178. [Google Scholar] [CrossRef]
- Nishio, Y.; Fujino, M.; Zhao, M.; Ishii, T.; Ishizuka, M.; Ito, H.; Takahashi, K.; Abe, F.; Nakajima, M.; Tanaka, T.; et al. 5-Aminolevulinic acid combined with ferrous iron enhances the expression of heme oxygenase-1. Int. Immunopharmacol. 2014, 19, 300–307. [Google Scholar] [CrossRef]
- Muller-Eberhard, U.; Fraig, M. Bioactivity of heme and its containment. Am. J. Hematol. 1993, 42, 59–62. [Google Scholar] [CrossRef]
- Delanghe, J.R.; Langlois, M.R. Hemopexin: A review of biological aspects and the role in laboratory medicine. Clin. Chim. Acta 2001, 312, 13–23. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef]
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. How iron is handled in the course of heme catabolism: Integration of heme oxygenase with intracellular iron transport mechanisms mediated by poly (rC)-binding protein-2. Arch. Biochem. Biophys. 2019, 672, 108071. [Google Scholar] [CrossRef]
- Hvidberg, V.; Maniecki, M.B.; Jacobsen, C.; Højrup, P.; Møller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Maines, M.D. The Heme Oxygenase System: A Regulator of Second Messenger Gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Omura, T.; Sato, R. The Carbon Monoxide-binding Pigment of Liver Microsomes. I. EVIDENCE. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar] [CrossRef]
- Yoshinaga, T.; Sassa, S.; Kappas, A. The occurrence of molecular interactions among NADPH-cytochrome c reductase, heme oxygenase, and biliverdin reductase in heme degradation. J. Biol. Chem. 1982, 257, 7786–7793. [Google Scholar] [CrossRef]
- Kim, H.P.; Wang, X.; Galbiati, F.; Ryter, S.W.; Choi, A.M.K. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. 2004, 18, 1080–1089. [Google Scholar] [CrossRef]
- Converso, D.P.; Taillé, C.; Carreras, M.C.; Jaitovich, A.; Poderoso, J.J.; Boczkowski, J.; Converso, D.P.; Taillé, C.; Carreras, M.C.; Jaitovich, A.; et al. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J. 2006, 20, 1236–1238. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.-H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme Oxygenase-1 Protein Localizes to the Nucleus and Activates Transcription Factors Important in Oxidative Stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Uchiyama, K.; Yoshikawa, T. Heme oxygenase-1: A novel therapeutic target for gastrointestinal diseases. J. Clin. Biochem. Nutr. 2011, 48, 126. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Raupach, A. The Role of Heme Oxygenase-1 in Remote Ischemic and Anesthetic Organ Conditioning. Antioxidants 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, A.K.; Oh, J.; Otterbein, L.E. Carbon Monoxide and the Brain: Time to Rethink the Dogma. Curr. Pharm. Des. 2013, 19, 2771–2775. [Google Scholar] [CrossRef]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. A Review on Hemeoxygenase-2: Focus on Cellular Protection and Oxygen Response. Oxidative Med. Cell. Longev. 2014, 2014, 604981. [Google Scholar] [CrossRef]
- Kapturczak, M.H.; Wasserfall, C.; Brusko, T.; Campbell-Thompson, M.; Ellis, T.M.; Atkinson, M.A.; Agarwal, A. Heme Oxygenase-1 Modulates Early Inflammatory Responses: Evidence from the Heme Oxygenase-1-Deficient Mouse. Am. J. Pathol. 2004, 165, 1045–1053. [Google Scholar] [CrossRef]
- De Montellano, P.R.O. The mechanism of heme oxygenase. Curr. Opin. Chem. Biol. 2000, 4, 221–227. [Google Scholar] [CrossRef]
- Wegiel, B.; Otterbein, L.E. Go Green: The Anti-Inflammatory Effects of Biliverdin Reductase. Front. Pharmacol. 2012, 3, 47. [Google Scholar] [CrossRef]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [CrossRef]
- Regan, R.F.; Kumar, N.; Gao, F.; Guo, Y. Ferritin induction protects cortical astrocytes from heme-mediated oxidative injury. Neuroscience 2002, 113, 985–994. [Google Scholar] [CrossRef]
- Kwon, D.H.; Cha, H.-J.; Lee, H.; Hong, S.-H.; Park, C.; Park, S.-H.; Kim, G.-Y.; Kim, S.; Kim, H.-S.; Hwang, H.-J.; et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants 2019, 8, 82. [Google Scholar] [CrossRef]
- Shviro, Y.; Shaklai, N. Glutathione as a scavenger of free hemin: A mechanism of preventing red cell membrane damage. Biochem. Pharmacol. 1987, 36, 3801–3807. [Google Scholar] [CrossRef]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, Â.; Ferreira, A.; et al. A Central Role for Free Heme in the Pathogenesis of Severe Sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef]
- Kourie, J.I. Interaction of reactive oxygen species with ion transport mechanisms. Am. J. Physiol. 1998, 275, C1–C24. [Google Scholar] [CrossRef]
- Schallner, N.; Pandit, R.; Leblanc, R.; Thomas, A.J.; Ogilvy, C.S.; Zuckerbraun, B.S.; Gallo, D.; Otterbein, L.E.; Hanafy, K.A. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Investig. 2015, 125, 2609–2625. [Google Scholar] [CrossRef]
- Chang, E.F.; Claus, C.P.; Vreman, H.J.; Wong, R.J.; Noble-Haeusslein, L.J. Heme Regulation in Traumatic Brain Injury: Relevance to the Adult and Developing Brain. J. Cereb. Blood Flow Metab. 2005, 25, 1401–1417. [Google Scholar] [CrossRef]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014, 111, E4110–E4118. [Google Scholar] [CrossRef]
- Dutra, F.F.; Bozza, M.T. Heme on innate immunity and inflammation. Front. Pharmacol. 2014, 5, 115. [Google Scholar] [CrossRef]
- Lin, S.; Yin, Q.; Zhong, Q.; Lv, F.-L.; Zhou, Y.; Li, J.-Q.; Wang, J.-Z.; Su, B.-Y.; Yang, Q.-W. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflamm. 2012, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.Y.; Takeshige, K.; Cheung, B.S.; Minakami, S. Bilirubin inhibits the activation of superoxide-producing NADPH oxidase in a neutrophil cell-free system. Biochim. Biophys. Acta 1991, 1076, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Lavrovsky, Y.; Schwartzman, M.L.; Levere, R.D.; Kappas, A.; Abraham, N.G. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc. Natl. Acad. Sci. USA 1994, 91, 5987–5991. [Google Scholar] [CrossRef] [PubMed]
- Shono, T.; Ono, M.; Izumi, H.; Jimi, S.I.; Matsushima, K.; Okamoto, T.; Kohno, K.; Kuwano, M. Involvement of the transcription factor NF-kappaB in tubular morphogenesis of human microvascular endothelial cells by oxidative stress. Mol. Cell. Biol. 1996, 16, 4231–4239. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C.; Smith, A. Antioxidant protection by haemopexin of haem-stimulated lipid peroxidation. Biochem. J. 1988, 256, 861–865. [Google Scholar] [CrossRef]
- Grinberg, L.N.; O’brien, P.J.; Hrkal, Z. The effects of heme-binding proteins on the peroxidative and catalatic activities of hemin. Free Radic. Biol. Med. 1999, 27, 214–219. [Google Scholar] [CrossRef]
- Belcher, J.D.; Beckman, J.D.; Balla, G.; Balla, J.; Vercellotti, G. Heme Degradation and Vascular Injury. Antioxid. Redox Signal. 2010, 12, 233. [Google Scholar] [CrossRef]
- Bergwik, J.; Kristiansson, A.; Allhorn, M.; Gram, M.; Åkerström, B. Structure, Functions, and Physiological Roles of the Lipocalin α1-Microglobulin (A1M). Front. Physiol. 2021, 12, 645650. [Google Scholar] [CrossRef]
- Larsson, J.; Allhorn, M.; Åkerström, B. The lipocalin Alpha(1)-microglobulin binds heme in different species. Arch. Biochem. Biophys. 2004, 432, 196–204. [Google Scholar] [CrossRef]
- Xu, B.; Tian, R.; Wang, X.; Zhan, S.; Wang, R.; Guo, Y.; Ge, W. Protein profile changes in the frontotemporal lobes in human severe traumatic brain injury. Brain Res. 2016, 1642, 344–352. [Google Scholar] [CrossRef]
- Huang, L.; He, S.; Cai, Q.; Li, F.; Wang, S.; Tao, K.; Xi, Y.; Qin, H.; Gao, G.; Feng, D. Polydatin alleviates traumatic brain injury: Role of inhibiting ferroptosis. Biochem. Biophys. Res. Commun. 2021, 556, 149–155. [Google Scholar] [CrossRef]
- Abu Hamdeh, S.; Shevchenko, G.; Mi, J.; Musunuri, S.; Bergquist, J.; Marklund, N. Proteomic differences between focal and diffuse traumatic brain injury in human brain tissue. Sci. Rep. 2018, 8, 81. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Gao, C.; Chen, W.; Chen, G.; Zhang, M.; Luo, C.; Wang, T.; Chen, X.; Tao, L. TMT-based proteomics analysis to screen potential biomarkers of acute-phase TBI in rats. Life Sci. 2021, 264, 118631. [Google Scholar] [CrossRef]
- Yao, X.; Liu, S.; Ding, W.; Yue, P.; Jiang, Q.; Zhao, M.; Hu, F.; Zhang, H. TLR4 signal ablation attenuated neurological deficits by regulating microglial M1/M2 phenotype after traumatic brain injury in mice. J. Neuroimmunol. 2017, 310, 38–45. [Google Scholar] [CrossRef]
- Pierce, J.D.; Gupte, R.; Thimmesch, A.; Shen, Q.; Hiebert, J.B.; Brooks, W.M.; Clancy, R.L.; Diaz, F.J.; Harris, J.L. Ubiquinol treatment for TBI in male rats: Effects on mitochondrial integrity, injury severity, and neurometabolism. J. Neurosci. Res. 2018, 96, 1080–1092. [Google Scholar] [CrossRef]
- Loane, D.J.; Faden, A.I. Neuroprotection for traumatic brain injury: Translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 2010, 31, 596–604. [Google Scholar] [CrossRef]
- Alluri, H.; Shaji, C.A.; Davis, M.L.; Tharakan, B. A Mouse Controlled Cortical Impact Model of Traumatic Brain Injury for Studying Blood–Brain Barrier Dysfunctions. Methods Mol. Biol. 2018, 1717, 37–52. [Google Scholar] [CrossRef]
- Sharp, D.J.; Scott, G.; Leech, R. Network dysfunction after traumatic brain injury. Nat. Rev. Neurol. 2014, 10, 156–166. [Google Scholar] [CrossRef]
- Cauda, F.; Micon, B.M.; Sacco, K.; Duca, S.; D’Agata, F.; Geminiani, G.; Canavero, S. Disrupted intrinsic functional connectivity in the vegetative state. J. Neurol. Neurosurg. Psychiatry 2009, 80, 429–431. [Google Scholar] [CrossRef]
- Schiff, N.D.; Giacino, J.T.; Kalmar, K.; Victor, J.D.; Baker, K.; Gerber, M.; Fritz, B.; Eisenberg, B.; O’connor, J.; Kobylarz, E.J.; et al. Behavioural improvements with thalamic stimulation after severe traumatic brain injury. Nature 2007, 448, 600–603. [Google Scholar] [CrossRef]
- Lu, X.; Chen-Roetling, J.; Regan, R.F. Systemic hemin therapy attenuates blood–brain barrier disruption after intracerebral hemorrhage. Neurobiol. Dis. 2014, 70, 245. [Google Scholar] [CrossRef] [PubMed]
- Russell, N.H.; Black, R.T.; Lee, N.N.; Doperalski, A.E.; Reeves, T.M.; Phillips, L.L. Time-dependent hemeoxygenase-1, lipocalin-2 and ferritin induction after non-contusion traumatic brain injury. Brain Res. 2019, 1725, 146466. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.R.; Gallo, D.; de Souza, R.W.A.; Tiwari-Heckler, S.; Csizmadia, E.; Harbison, J.D.; Shankar, S.; Banner-Goodspeed, V.; Yaffe, M.B.; Longhi, M.S.; et al. Trauma-induced heme release increases susceptibility to bacterial infection. JCI Insight 2021, 6, e150813. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Scott Panter, S.; Sharp, F.R.; Noble, L.J. Induction of heme oxygenase-1 (HO-1) after traumatic brain injury in the rat. Neurosci. Lett. 1995, 199, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.P.; Bergeron, M.; Matz, P.; Zegna, A.; Noble, L.J.; Panter, S.S.; Sharp, F.R. Heme Oxygenase-1 is Induced in Glia Throughout Brain by Subarachnoid Hemoglobin. J. Cereb. Blood Flow Metab. 1998, 18, 257–273. [Google Scholar] [CrossRef]
- Yi, J.-H.; Hazell, A.S. N-acetylcysteine attenuates early induction of heme oxygenase-1 following traumatic brain injury. Brain Res. 2005, 1033, 13–19. [Google Scholar] [CrossRef]
- Alfieri, A.; Srivastava, S.; Siow, R.C.M.; Cash, D.; Modo, M.; Duchen, M.R.; Fraser, P.A.; Williams, S.C.R.; Mann, G.E. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 2013, 65, 1012–1022. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Z.; Luo, B.; Schluesener, H.J.; Zhang, Z. Lesional accumulation of heme oxygenase-1+ microglia/macrophages in rat traumatic brain injury. Neuroreport 2013, 24, 281–286. [Google Scholar] [CrossRef]
- Zhao, J.; Xi, G.; Wu, G.; Keep, R.F.; Hua, Y. Deferoxamine Attenuated the Upregulation of Lipocalin-2 Induced by Traumatic Brain Injury in Rats. Acta Neurochir. Suppl. 2016, 121, 291–294. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kim, Y.-M. Beneficial and Detrimental Roles of Heme Oxygenase-1 in the Neurovascular System. Int. J. Mol. Sci. 2022, 23, 7041. [Google Scholar] [CrossRef]
- Beschorner, R.; Adjodah, D.; Schwab, J.M.; Mittelbronn, M.; Pedal, I.; Mattern, R.; Schluesener, H.J.; Meyermann, R. Long-term expression of heme oxygenase-1 (HO-1, HSP-32) following focal cerebral infarctions and traumatic brain injury in humans. Acta Neuropathol. 2000, 100, 377–384. [Google Scholar] [CrossRef]
- Heidari, M.; Johnstone, D.M.; Bassett, B.; Graham, R.M.; Chua, A.C.G.; House, M.J.; Collingwood, J.F.; Bettencourt, C.; Houlden, H.; Ryten, M.; et al. Brain iron accumulation affects myelin-related molecular systems implicated in a rare neurogenetic disease family with neuropsychiatric features. Mol. Psychiatry 2016, 21, 1599–1607. [Google Scholar] [CrossRef]
- Kim, M.; Kim, J.; Moon, S.; Choi, B.Y.; Kim, S.; Jeon, H.S.; Suh, S.W.; Kim, Y.-M.; Choi, Y.K. Korean Red Ginseng Improves Astrocytic Mitochondrial Function by Upregulating HO-1-Mediated AMPKα–PGC-1α–ERRα Circuit after Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 13081. [Google Scholar] [CrossRef]
- Kim, M.; Moon, S.; Jeon, H.S.; Kim, S.; Koh, S.-H.; Chang, M.-S.; Kim, Y.-M.; Choi, Y.K. Dual Effects of Korean Red Ginseng on Astrocytes and Neural Stem Cells in Traumatic Brain Injury: The HO-1–Tom20 Axis as a Putative Target for Mitochondrial Function. Cells 2022, 11, 892. [Google Scholar] [CrossRef]
- Choi, Y.K.; Maki, T.; Mandeville, E.T.; Koh, S.-H.; Hayakawa, K.; Arai, K.; Kim, Y.-M.; Whalen, M.J.; Xing, C.; Wang, X.; et al. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat. Med. 2016, 22, 1335–1341. [Google Scholar] [CrossRef]
- Reynolds, A.; Krebs, N.F.; Stewart, P.A.; Austin, H.; Johnson, S.L.; Withrow, N.; Molloy, C.; James, S.J.; Johnson, C.; Clemons, T.; et al. Iron Status in Children with Autism Spectrum Disorder. Pediatrics 2012, 130 (Suppl. S2), S154–S159. [Google Scholar] [CrossRef]
- Feifel, D.; Young, C.W. Iron Overload Among a Psychiatric Outpatient Population. J. Clin. Psychiatry 1997, 58, 74–78. [Google Scholar] [CrossRef]
- Pecorelli, A.; Leoncini, S.; De Felice, C.; Signorini, C.; Cerrone, C.; Valacchi, G.; Ciccoli, L.; Hayek, J. Non-protein-bound iron and 4-hydroxynonenal protein adducts in classic autism. Brain Dev. 2013, 35, 146–154. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Johnstone, D.; Milward, E.A. Molecular genetic approaches to understanding the roles and regulation of iron in brain health and disease. J. Neurochem. 2010, 113, 1387–1402. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.; Kurian, M.A.; Hayflick, S.J. Neurodegeneration with Brain Iron Accumulation: Genetic Diversity and Pathophysiological Mechanisms. Annu. Rev. Genom. Hum. Genet. 2015, 16, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, P. Neurodegeneration with Brain Iron Accumulation: Diagnosis and Management. J. Mov. Disord. 2015, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Daglas, M.; Adlard, P.A. The Involvement of Iron in Traumatic Brain Injury and Neurodegenerative Disease. Front. Neurosci. 2018, 12, 981. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef]
- Nisenbaum, E.J.; Novikov, D.S.; Lui, Y.W. The Presence and Role of Iron in Mild Traumatic Brain Injury: An Imaging Perspective. J. Neurotrauma 2014, 31, 301. [Google Scholar] [CrossRef]
- Loan, J.J.M.; Scott, N.W.; Jansen, J.O. Long-term survival and five year hospital resource usage following traumatic brain injury in Scotland from 1997 to 2015: A population-based retrospective cohort study. Injury 2019, 50, 82–89. [Google Scholar] [CrossRef]
- Etminan, N.; Chang, H.-S.; Hackenberg, K.; De Rooij, N.K.; Vergouwen, M.D.I.; Rinkel, G.J.E.; Algra, A. Worldwide Incidence of Aneurysmal Subarachnoid Hemorrhage According to Region, Time Period, Blood Pressure, and Smoking Prevalence in the Population: A Systematic Review and Meta-Analysis. JAMA Neurol. 2019, 76, 588–597. [Google Scholar] [CrossRef]
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abedi, V.; et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Loan, J.J.M.; Al-Shahi Salman, R.; McColl, B.W.; Hardingham, G.E. Activation of Nrf2 to Optimise Immune Responses to Intracerebral Haemorrhage. Biomolecules 2022, 12, 1438. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.I.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Liu, J.-T.; Peng, Z.-W.; Fan, H.; Yao, A.-H.; Cheng, P.; Liu, L.; Ju, G.; Kuang, F. Different TLR4 expression and microglia/macrophage activation induced by hemorrhage in the rat spinal cord after compressive injury. J. Neuroinflamm. 2013, 10, 112. [Google Scholar] [CrossRef]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef]
- Glezer, I.; Simard, A.R.; Rivest, S. Neuroprotective role of the innate immune system by microglia. Neuroscience 2007, 147, 867–883. [Google Scholar] [CrossRef]
- Strangman, G.E.; O’Neil-Pirozzi, T.M.; Supelana, C.; Goldstein, R.; Katz, D.I.; Glenn, M.B. Fractional anisotropy helps predicts memory rehabilitation outcome after traumatic brain injury. Neurorehabilitation 2012, 31, 295–310. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Yoo, W.-K.; Ko, M.-H.; Park, C.-H.; Kim, S.T.; Na, D.L. Plasticity of the Attentional Network After Brain Injury and Cognitive Rehabilitation. Neurorehabilit. Neural Repair 2009, 23, 468–477. [Google Scholar] [CrossRef]
- Sidaros, A.; Engberg, A.W.; Sidaros, K.; Liptrot, M.G.; Herning, M.; Petersen, P.; Paulson, O.B.; Jernigan, T.L.; Rostrup, E. Diffusion tensor imaging during recovery from severe traumatic brain injury and relation to clinical outcome: A longitudinal study. Brain 2008, 131, 559–572. [Google Scholar] [CrossRef]
- Graham, D.I.; McIntosh, T.K.; Maxwell, W.L.; Nicoll, J.A.R. Recent Advances in Neurotrauma. J. Neuropathol. Exp. Neurol. 2000, 59, 641–651. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of Cell Protection by Heme Oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef]
- CDC (Center for Disease Control and Prevention). Leading Causes of Death Reports; CDC: Atlanta, GA, USA, 2020. [Google Scholar]
- Dutton, R.P.; Stansbury, L.G.; Leone, S.; Kramer, E.; Hess, J.R.; Scalea, T.M. Trauma Mortality in Mature Trauma Systems: Are We Doing Better? An Analysis of Trauma Mortality Patterns, 1997–2008. J. Trauma 2010, 69, 620–626. [Google Scholar] [CrossRef]
- Fraser, D.R.; Dombrovskiy, V.Y.; Vogel, T.R. Infectious Complications after Vehicular Trauma in the United States. Surg. Infect. 2011, 12, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.S. Risk factors for infection in the trauma patient. J. Natl. Med. Assoc. 1992, 84, 1019–1023. [Google Scholar] [PubMed]
- Oyeniyi, B.T.; Fox, E.E.; Scerbo, M.; Tomasek, J.S.; Wade, C.E.; Holcomb, J.B. Trends in 1029 trauma deaths at a level 1 trauma center: Impact of a bleeding control bundle of care. Injury 2017, 48, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Hauser, C.J.; Otterbein, L.E. Heme as a danger molecule in pathogen recognition. Free Radic. Biol. Med. 2015, 89, 651–661. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Larsen, R.; Gouveia, Z.; Soares, M.P.; Gozzelino, R. Heme Cytotoxicity and the Pathogenesis of Immune-Mediated Inflammatory Diseases. Front. Pharmacol. 2012, 3, 77. [Google Scholar] [CrossRef]
- Fortes, G.B.; Alves, L.S.; De Oliveira, R.; Dutra, F.F.; Rodrigues, D.; Fernandez, P.L.; Souto-Padron, T.; DE Rosa, M.J.; Kelliher, M.; Golenbock, D.; et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 2012, 119, 2368–2375. [Google Scholar] [CrossRef]
- Wegiel, B.; Larsen, R.; Gallo, D.; Chin, B.Y.; Harris, C.; Mannam, P.; Kaczmarek, E.; Lee, P.J.; Zuckerbraun, B.S.; Flavell, R.; et al. Macrophages sense and kill bacteria through carbon monoxide–dependent inflammasome activation. J. Clin. Investig. 2014, 124, 4926–4940. [Google Scholar] [CrossRef]
- Martins, R.; Maier, J.; Gorki, A.-D.; Huber, K.V.M.; Sharif, O.; Starkl, P.; Saluzzo, S.; Quattrone, F.; Gawish, R.; Lakovits, K.; et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat. Immunol. 2016, 17, 1361–1372. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Su, W.C.; Liu, X.; Macias, A.A.; Baron, R.M.; Perrella, M.A. Heme oxygenase-1–derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J. Clin. Investig. 2008, 118, 239–247. [Google Scholar] [CrossRef]
- de Souza, R.W.A.; Gallo, D.; Lee, G.R.; Katsuyama, E.; Schaufler, A.; Weber, J.; Csizmadia, E.; Tsokos, G.C.; Koch, L.G.; Britton, S.L.; et al. Skeletal muscle heme oxygenase-1 activity regulates aerobic capacity. Cell Rep. 2021, 35, 109018. [Google Scholar] [CrossRef]
- Skinner, N.A.; MacIsaac, C.M.; Hamilton, J.A.; Visvanathan, K. Regulation of Toll-like receptor (TLR)2 and TLR4 on CD14dimCD16+ monocytes in response to sepsis-related antigens. Clin. Exp. Immunol. 2005, 141, 270–278. [Google Scholar] [CrossRef]
- Gillrie, M.R.; Zbytnuik, L.; McAvoy, E.; Kapadia, R.; Lee, K.; Waterhouse, C.C.M.; Davis, S.P.; Muruve, D.A.; Kubes, P.; Ho, M. Divergent roles of Toll-like receptor 2 in response to lipoteichoic acid and Staphylococcus aureus in vivo. Eur. J. Immunol. 2010, 40, 1639–1650. [Google Scholar] [CrossRef]
- Takeuchi, O.; Hoshino, K.; Akira, S. Cutting Edge: TLR2-Deficient and MyD88-Deficient Mice Are Highly Susceptible to Staphylococcus aureus Infection. J. Immunol. 2000, 165, 5392–5396. [Google Scholar] [CrossRef]
- Echchannaoui, H.; Frei, K.; Schnell, C.; Leib, S.L.; Zimmerli, W.; Landmann, R. Toll-Like Receptor 2–Deficient Mice Are Highly Susceptible to Streptococcus pneumoniae Meningitis because of Reduced Bacterial Clearing and Enhanced Inflammation. J. Infect. Dis. 2002, 186, 798–806. [Google Scholar] [CrossRef]
- Yimin; Kohanawa, M.; Zhao, S.; Ozaki, M.; Haga, S.; Nan, G.; Kuge, Y.; Tamaki, N. Contribution of Toll-Like Receptor 2 to the Innate Response against Staphylococcus aureus Infection in Mice. PLoS ONE 2013, 8, e74287. [Google Scholar] [CrossRef]
- Fournier, A.; Voirol, P.; Krähenbühl, M.; Bonnemain, C.-L.; Fournier, C.; Dupuis-Lozeron, E.; Pantet, O.; Pagani, J.-L.; Revelly, J.-P.; Sadeghipour, F.; et al. Staphylococcus aureus carriage at admission predicts early-onset pneumonia after burn trauma. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 523–528. [Google Scholar] [CrossRef]
- Wahl, S.; Vichinsky, E. Pulmonary hypertension in hemolytic anemias. F1000 Med. Rep. 2010, 2, 10. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Balla, J.; Vercellotti, G.M.; Nath, K.; Yachie, A.; Nagy, E.; Eaton, J.W.; Balla, G. Haem, haem oxygenase and ferritin in vascular endothelial cell injury. Nephrol. Dial. Transplant. 2003, 18 (Suppl. S5), 8v–12. [Google Scholar] [CrossRef] [PubMed]
- Olgun, A.; Akman, S.; Erbil, M.K. The role of RBC destruction in vascular regions with high turbulence on atherosclerosis. Med. Hypotheses 2004, 63, 283–284. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef]
- Moraes, J.A.; Barcellos-De-Souza, P.; Rodrigues, G.; Nascimento-Silva, V.; Silva, S.V.; Assreuy, J.; Arruda, M.A.; Barja-Fidalgo, C. Heme modulates smooth muscle cell proliferation and migration via NADPH oxidase: A counter-regulatory role for heme oxygenase system. Atherosclerosis 2012, 224, 394–400. [Google Scholar] [CrossRef]
- Duckles, H.; Boycott, H.E.; Al-Owais, M.M.; Elies, J.; Johnson, E.; Dallas, M.L.; Porter, K.E.; Giuntini, F.; Boyle, J.P.; Scragg, J.L.; et al. Heme oxygenase-1 regulates cell proliferation via carbon monoxide-mediated inhibition of T-type Ca2+ channels. Pflugers Arch. 2015, 467, 415–427. [Google Scholar] [CrossRef]
- Bhoite-Solomon, V.; Kessler-Icekson, G.; Shaklai, N. Myocyte injury by hemin. Vitr. Cell. Dev. Biol. Anim. 1993, 29A, 636–642. [Google Scholar] [CrossRef]
- Alvarado, G.; Jeney, V.; Tóth, A.; Csősz, É.; Kalló, G.; Huynh, A.T.; Hajnal, C.; Kalász, J.; Pásztor, E.T.; Édes, I.; et al. Heme-induced contractile dysfunction in Human cardiomyocytes caused by oxidant damage to thick filament proteins. Free Radic. Biol. Med. 2015, 89, 248–262. [Google Scholar] [CrossRef]
- Jeney, V.; Balla, G.; Balla, J. Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front. Physiol. 2014, 5, 379. [Google Scholar] [CrossRef]
- Bunn, H.F.; Jandl, J.H. Exchange of Heme among Hemoglobins and between Hemoglobin and Albumin. J. Biol. Chem. 1968, 243, 465–475. [Google Scholar] [CrossRef]
- Ingoglia, G.; Sag, C.M.; Rex, N.; De Franceschi, L.; Vinchi, F.; Cimino, J.; Petrillo, S.; Wagner, S.; Kreitmeier, K.; Silengo, L.; et al. Hemopexin counteracts systolic dysfunction induced by heme-driven oxidative stress. Free Radic. Biol. Med. 2017, 108, 452–464. [Google Scholar] [CrossRef]
- de Back, D.Z.; Kostova, E.B.; van Kraaij, M.; van den Berg, T.K.; van Bruggen, R. Of Macrophages and Red Blood Cells; A Complex Love Story. Front. Physiol. 2014, 5, 9. [Google Scholar] [CrossRef]
- Köhler, A.C.; Sag, C.M.; Maier, L.S. Reactive oxygen species and excitation–contraction coupling in the context of cardiac pathology. J. Mol. Cell. Cardiol. 2014, 73, 92–102. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.-L.A.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A Dynamic Pathway for Calcium-Independent Activation of CaMKII by Methionine Oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef]
- Bayeva, M.; Gheorghiade, M.; Ardehali, H. Mitochondria as a Therapeutic Target in Heart Failure. J. Am. Coll. Cardiol. 2013, 61, 599–610. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Lüscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. Ferric Carboxymaltose in Patients with Heart Failure and Iron Deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef]
- Ponikowski, P.; Van Veldhuisen, D.J.; Comin-Colet, J.; Ertl, G.; Komajda, M.; Mareev, V.; McDonagh, T.; Parkhomenko, A.; Tavazzi, L.; Levesque, V.; et al. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur. Heart J. 2015, 36, 657–668. [Google Scholar] [CrossRef]
- Gozzelino, R.; Arosio, P. Iron Homeostasis in Health and Disease. Int. J. Mol. Sci. 2016, 17, 130. [Google Scholar] [CrossRef]
- Murphy, C.J.; Oudit, G.Y. Iron-Overload Cardiomyopathy: Pathophysiology, Diagnosis, and Treatment. J. Card. Fail. 2010, 16, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Epidemic of Iron Overload in Dialysis Population Caused by Intravenous Iron Products: A Plea for Moderation. Am. J. Med. 2012, 125, 951–952. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.W.; Seckington, R.C.; Deugnier, Y. Haemochromatosis. Lancet 2016, 388, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Batey, R.G.; Fong, P.L.C.; Shamir, S.; Sherlock, S. A non-transferrin-bound serum iron in idiopathic hemochromatosis. Dig. Dis. Sci. 1980, 25, 340–346. [Google Scholar] [CrossRef]
- Das, S.K.; Wang, W.; Zhabyeyev, P.; Basu, R.; McLean, B.; Fan, D.; Parajuli, N.; DesAulniers, J.; Patel, V.B.; Hajjar, R.J.; et al. Iron-overload injury and cardiomyopathy in acquired and genetic models is attenuated by resveratrol therapy. Sci. Rep. 2015, 5, 18132. [Google Scholar] [CrossRef]
- Gordan, R.; Wongjaikam, S.; Gwathmey, J.K.; Chattipakorn, N.; Chattipakorn, S.C.; Xie, L.-H. Involvement of cytosolic and mitochondrial iron in iron overload cardiomyopathy: An update. Heart Fail. Rev. 2018, 23, 801–816. [Google Scholar] [CrossRef]
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an Intestinal Heme Transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef]
- Menon, A.V.; Liu, J.; Tsai, H.P.; Zeng, L.; Yang, S.; Asnani, A.; Kim, J. Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood 2022, 139, 936–941. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Park, T.-J.; Park, J.H.; Lee, G.S.; Lee, J.-Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.-Y.; Oh, K.-J.; Han, B.-S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef]
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 2020, 160, 92–102. [Google Scholar] [CrossRef]
- Wang, C.; Yuan, W.; Hu, A.; Lin, J.; Xia, Z.; Yang, C.F.; Li, Y.; Zhang, Z. Dexmedetomidine alleviated sepsis-induced myocardial ferroptosis and septic heart injury. Mol. Med. Rep. 2020, 22, 175. [Google Scholar] [CrossRef]
- Liu, J.; Lane, S.; Lall, R.; Russo, M.; Farrell, L.; Coskun, M.D.; Curtin, C.; Araujo-Gutierrez, R.; Scherrer-Crosbie, M.; Trachtenberg, B.H.; et al. Circulating hemopexin modulates anthracycline cardiac toxicity in patients and in mice. Sci. Adv. 2022, 8, 51. [Google Scholar] [CrossRef]
- Mariotto, A.B.; Robin Yabroff, K.; Shao, Y.; Feuer, E.J.; Brown, M.L. Projections of the Cost of Cancer Care in the United States: 2010–2020. J. Natl. Cancer Inst. 2011, 103, 117–128. [Google Scholar] [CrossRef]
- Jemal, A.; Vineis, P.; Bray, F.; Torre, L.; Forman, D. The Cancer Atlas, 3rd ed.; Daniel, J.M., Ed.; American Cancer Society: Atlanta, GA, USA, 2019; ISBN 978-1-60443-265-7. [Google Scholar]
- Fiorito, V.; Chiabrando, D.; Petrillo, S.; Bertino, F.; Tolosano, E. The Multifaceted Role of Heme in Cancer. Front. Oncol. 2020, 9, 1540. [Google Scholar] [CrossRef]
- Gu, K.J.; Li, G. An Overview of Cancer Prevention: Chemoprevention and Immunoprevention. J. Cancer Prev. 2020, 25, 127. [Google Scholar] [CrossRef]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, L.; Ding, J.; Chen, Y. Iron Metabolism in Cancer. Int. J. Mol. Sci. 2018, 20, 95. [Google Scholar] [CrossRef]
- Jung, M.; Mertens, C.; Tomat, E.; Brüne, B. Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci. 2019, 20, 273. [Google Scholar] [CrossRef]
- Willett, W.C.; Stampfer, M.J.; Colditz, G.A.; Rosner, B.A.; Speizer, F.E. Relation of Meat, Fat, and Fiber Intake to the Risk of Colon Cancer in a Prospective Study among Women. N. Engl. J. Med. 1990, 323, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Sesink, A.L.A.; Termont, D.S.M.L.; Kleibeuker, J.H.; Van Der Meer, R. Red meat and colon cancer: Dietary haem-induced colonic cytotoxicity and epithelial hyperproliferation are inhibited by calcium. Carcinogenesis 2001, 22, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Loomis, D.; Guyton, K.Z.; Grosse, Y.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K.; Stewart, B.W.; et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015, 16, 1599–1600. [Google Scholar] [CrossRef] [PubMed]
- Tram, K.L.; Cross, A.J.; Consonni, D.; Randi, G.; Bagnardi, V.; Bertazzi, P.A.; Caporaso, N.E.; Sinha, R.; Subar, A.F.; Landi, M.T. Intakes of Red Meat, Processed Meat, and Meat Mutagens Increase Lung Cancer Risk. Cancer Res. 2009, 69, 932–939. [Google Scholar] [CrossRef]
- Cross, A.J.; Freedman, N.D.; Ren, J.; Ward, M.H.; Hollenbeck, A.R.; Schatzkin, A.; Sinha, R.; Abnet, C.C. Meat Consumption and Risk of Esophageal and Gastric Cancer in a Large Prospective Study. Am. J. Gastroenterol. 2011, 106, 432–442. [Google Scholar] [CrossRef]
- Taunk, P.; Hecht, E.; Stolzenbergsolomon, R. Are meat and heme iron intake associated with pancreatic cancer? Results from the NIH-AARP diet and health cohort. Int. J. Cancer 2016, 138, 2172–2189. [Google Scholar] [CrossRef]
- Chang, V.C.; Cotterchio, M.; Khoo, E. Iron intake, body iron status, and risk of breast cancer: A systematic review and meta-analysis. BMC Cancer 2019, 19, 543. [Google Scholar] [CrossRef]
- Inoue-Choi, M.; Sinha, R.; Gierach, G.L.; Ward, M.H. Red and processed meat, nitrite, and heme iron intakes and postmenopausal breast cancer risk in the NIH-AARPDiet and Health Study. Int. J. Cancer 2016, 138, 1609–1618. [Google Scholar] [CrossRef]
- Pierre, F.; Taché, S.; Corpet, D.E.; Freeman, A.; Van der Meer, R. Beef Meat and Blood Sausage Promote the Formation of Azoxymethane-Induced Mucin-Depleted Foci and Aberrant Crypt Foci in Rat Colons. J. Nutr. 2004, 134, 2711–2716. [Google Scholar] [CrossRef]
- Bastide, N.M.; Chenni, F.; Audebert, M.; Santarelli, R.L.; Taché, S.; Naud, N.; Baradat, M.; Jouanin, I.; Surya, R.; Hobbs, D.A.; et al. A Central Role for Heme Iron in Colon Carcinogenesis Associated with Red Meat Intake. Cancer Res. 2015, 75, 870–879. [Google Scholar] [CrossRef]
- Sesink, A.L.A.; Termont, D.S.M.L.; Kleibeuker, J.H.; Van Der Meer, R. Red meat and colon cancer: Dietary haem, but not fat, has cytotoxic and hyperproliferative effects on rat colonic epithelium. Carcinogenesis 2000, 21, 1909–1915. [Google Scholar] [CrossRef]
- Massey, R.C.; Key, P.E.; Mallett, A.K.; Rowland, I.R. An investigation of the endogenous formation of apparent total N-nitroso compounds in conventional microflora and germ-free rats. Food Chem. Toxicol. 1988, 26, 595–600. [Google Scholar] [CrossRef]
- Gamage, S.M.K.; Dissabandara, L.; Lam, A.K.Y.; Gopalan, V. The role of heme iron molecules derived from red and processed meat in the pathogenesis of colorectal carcinoma. Crit. Rev. Oncol. Hematol. 2018, 126, 121–128. [Google Scholar] [CrossRef]
- Ijssennagger, N.; Rijnierse, A.; De Wit, N.J.W.; Boekschoten, M.V.; Dekker, J.; Schonewille, A.; Muller, M.; Van Der Meer, R. Dietary heme induces acute oxidative stress, but delayed cytotoxicity and compensatory hyperproliferation in mouse colon. Carcinogenesis 2013, 34, 1628–1635. [Google Scholar] [CrossRef]
- Ijssennagger, N.; Rijnierse, A.; De Wit, N.; Jonker-Termont, D.; Dekker, J.; Muller, M.; Van Der Meer, R. Dietary haem stimulates epithelial cell turnover by downregulating feedback inhibitors of proliferation in murine colon. Gut 2012, 61, 1041–1049. [Google Scholar] [CrossRef]
- Sasso, A.; Latella, G. Role of Heme Iron in the Association Between Red Meat Consumption and Colorectal Cancer. Nutr. Cancer 2018, 70, 1173–1183. [Google Scholar] [CrossRef]
- Ijssennagger, N.; Derrien, M.; Van Doorn, G.M.; Rijnierse, A.; van den Bogert, B.; Muller, M.; Dekker, J.; Kleerebezem, M.; Van Der Meer, R. Dietary Heme Alters Microbiota and Mucosa of Mouse Colon without Functional Changes in Host-Microbe Cross-Talk. PLoS ONE 2012, 7, e49868. [Google Scholar] [CrossRef]
- Constante, M.; Fragoso, G.; Calvé, A.; Samba-Mondonga, M.; Santos, M.M. Dietary Heme Induces Gut Dysbiosis, Aggravates Colitis, and Potentiates the Development of Adenomas in Mice. Front. Microbiol. 2017, 8, 1809. [Google Scholar] [CrossRef]
- Ijssennagger, N.; Belzer, C.; Hooiveld, G.J.; Dekker, J.; van Mil, S.W.C.; Müller, M.; Kleerebezem, M.; van der Meer, R.; Klaenhammer, T.R. Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc. Natl. Acad. Sci. USA 2015, 112, 10038–10043. [Google Scholar] [CrossRef]
- Yang, X.; Palasuberniam, P.; Kraus, D.; Chen, B. Aminolevulinic Acid-Based Tumor Detection and Therapy: Molecular Mechanisms and Strategies for Enhancement. Int. J. Mol. Sci. 2015, 16, 25865–25880. [Google Scholar] [CrossRef]
- Hooda, J.; Cadinu, D.; Alam, M.M.; Shah, A.; Cao, T.M.; Sullivan, L.A.; Brekken, R.; Zhang, L. Enhanced Heme Function and Mitochondrial Respiration Promote the Progression of Lung Cancer Cells. PLoS ONE 2013, 8, e63402. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Wang, Y.; Lian, S.; Lynch, J.; Nagai, S.; Fanshawe, B.; Kandilci, A.; Janke, L.J.; Neale, G.; Fan, Y.; et al. Upregulated heme biosynthesis, an exploitable vulnerability in MYCN-driven leukemogenesis. JCI Insight 2017, 2, e92409. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Nakada, M.; Zhao, S.-G.; Endo, Y.; Furuyama, N.; Nambu, E.; Pyko, I.V.; Hayashi, Y.; Hamada, J.-I. Silencing of ferrochelatase enhances 5-aminolevulinic acid-based fluorescence and photodynamic therapy efficacy. Br. J. Cancer 2011, 104, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Lal, S.; FitzGerald, K.E.; Zhang, L. A holistic view of cancer bioenergetics: Mitochondrial function and respiration play fundamental roles in the development and progression of diverse tumors. Clin. Transl. Med. 2016, 5, 3. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Hagiya, Y.; Nakajima, M.; Ishizuka, M.; Tanaka, T.; Ogura, S.-I. The heme precursor 5-aminolevulinic acid disrupts the Warburg effect in tumor cells and induces caspase-dependent apoptosis. Oncol. Rep. 2014, 31, 1282–1286. [Google Scholar] [CrossRef]
- Laplagne, C.; Domagala, M.; Le Naour, A.; Quemerais, C.; Hamel, D.; Fournié, J.-J.; Couderc, B.; Bousquet, C.; Ferrand, A.; Poupot, M. Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression. Int. J. Mol. Sci. 2019, 20, 4719. [Google Scholar] [CrossRef]
- Smith, A.G.; Raven, E.L.; Chernova, T. The regulatory role of heme in neurons. Metallomics 2011, 3, 955–962. [Google Scholar] [CrossRef]
- Chiabrando, D.; Fiorito, V.; Petrillo, S.; Tolosano, E. Unraveling the Role of Heme in Neurodegeneration. Front. Neurosci. 2018, 12, 712. [Google Scholar] [CrossRef]
- Gozzelino, R. The Pathophysiology of Heme in the Brain. Curr. Alzheimer Res. 2016, 13, 174–184. [Google Scholar] [CrossRef]
- Mravec, B.; Horvathova, L.; Hunakova, L. Neurobiology of Cancer: The Role of β-Adrenergic Receptor Signaling in Various Tumor Environments. Int. J. Mol. Sci. 2020, 21, 7958. [Google Scholar] [CrossRef]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef]
- Chiabrando, D.; Castori, M.; di Rocco, M.; Ungelenk, M.; Gießelmann, S.; Di Capua, M.; Madeo, A.; Grammatico, P.; Bartsch, S.; Hübner, C.A.; et al. Mutations in the Heme Exporter FLVCR1 Cause Sensory Neurodegeneration with Loss of Pain Perception. PLoS Genet. 2016, 12, e1006461. [Google Scholar] [CrossRef]
- Lamkin, D.M.; Ho, H.-Y.; Ong, T.H.; Kawanishi, C.K.; Stoffers, V.L.; Ahlawat, N.; Ma, J.C.Y.; Arevalo, J.M.G.; Cole, S.W.; Sloan, E.K. β-Adrenergic-stimulated macrophages: Comprehensive localization in the M1-M2 spectrum. Brain Behav. Immun. 2016, 57, 338. [Google Scholar] [CrossRef]
- Sun, J.; Kim, S.J.; Park, M.K.; Kim, H.J.; Tsoy, I.; Kang, Y.J.; Lee, Y.S.; Seo, H.G.; Lee, J.H.; Chang, K.C. Selective activation of adrenergic β1receptors induces heme oxygenase 1 production in RAW264.7 cells. FEBS Lett. 2005, 579, 5494–5500. [Google Scholar] [CrossRef]
- Hull, T.D.; Boddu, R.; Guo, L.; Tisher, C.C.; Traylor, A.M.; Patel, B.; Joseph, R.; Prabhu, S.D.; Suliman, H.B.; Piantadosi, C.A.; et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 2016, 1, e85817. [Google Scholar] [CrossRef]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme Oxygenase-1, Oxidation, Inflammation, and Atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef]
- Wegiel, B.; Nemeth, Z.; Correa-Costa, M.; Bulmer, A.C.; Otterbein, L.E. Heme Oxygenase-1: A Metabolic Nike. Antioxid. Redox Signal. 2014, 20, 1709–1722. [Google Scholar] [CrossRef]
- Chiang, S.-K.; Chen, S.-E.; Chang, L.-C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 39. [Google Scholar] [CrossRef]
- Alaluf, E.; Vokaer, B.; Detavernier, A.; Azouz, A.; Splittgerber, M.; Carrette, A.; Boon, L.; Libert, F.; Soares, M.P.; Le Moine, A.; et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight 2020, 5, e133929. [Google Scholar] [CrossRef]
- Wegiel, B.; Gallo, D.; Csizmadia, E.; Harris, C.; Belcher, J.; Vercellotti, G.M.; Penacho, N.; Seth, P.; Sukhatme, V.; Ahmed, A.; et al. Carbon Monoxide Expedites Metabolic Exhaustion to Inhibit Tumor Growth. Cancer Res. 2013, 73, 7009–7021. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme Oxygenase-1/Carbon Monoxide: From Basic Science to Therapeutic Applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO System in Tumor Growth, Angiogenesis and Metabolism—Targeting HO-1 as an Anti-Tumor Therapy. Vascul. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.-W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef]
- Burt, T.D.; Seu, L.; Mold, J.E.; Kappas, A.; McCune, J.M. Naive Human T Cells Are Activated and Proliferate in Response to the Heme Oxygenase-1 Inhibitor Tin Mesoporphyrin. J. Immunol. 2010, 185, 5279–5288. [Google Scholar] [CrossRef]
- Muliaditan, T.; Caron, J.; Okesola, M.; Opzoomer, J.W.; Kosti, P.; Georgouli, M.; Gordon, P.; Lall, S.; Kuzeva, D.M.; Pedro, L.; et al. Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nat. Commun. 2018, 9, 2951. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef]
- da Silva, M.C.; Breckwoldt, M.O.; Vinchi, F.; Correia, M.P.; Stojanovic, A.; Thielmann, C.M.; Meister, M.; Muley, T.; Warth, A.; Platten, M.; et al. Iron Induces Anti-tumor Activity in Tumor-Associated Macrophages. Front. Immunol. 2017, 8, 1479. [Google Scholar] [CrossRef]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between Defective Endothelial Cells Explain Tumor Vessel Leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Vandekeere, S.; Dubois, C.; Kalucka, J.; Sullivan, M.R.; García-Caballero, M.; Goveia, J.; Chen, R.; Diehl, F.F.; Bar-Lev, L.; Souffreau, J.; et al. Serine Synthesis via PHGDH Is Essential for Heme Production in Endothelial Cells. Cell Metab. 2018, 28, 573–587.e13. [Google Scholar] [CrossRef]
- Petrillo, S.; Chiabrando, D.; Genova, T.; Fiorito, V.; Ingoglia, G.; Vinchi, F.; Mussano, F.; Carossa, S.; Silengo, L.; Altruda, F.; et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018, 25, 573–588. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Torii, C.; Hida, Y. Tumor angiogenesis—Characteristics of tumor endothelial cells. Int. J. Clin. Oncol. 2016, 21, 206–212. [Google Scholar] [CrossRef]
- Maishi, N.; Hida, K. Tumor endothelial cells accelerate tumor metastasis. Cancer Sci. 2017, 108, 1921–1926. [Google Scholar] [CrossRef]
- Choi, H.; Moon, A. Crosstalk between cancer cells and endothelial cells: Implications for tumor progression and intervention. Arch. Pharmacal Res. 2018, 41, 711–724. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voltarelli, V.A.; Alves de Souza, R.W.; Miyauchi, K.; Hauser, C.J.; Otterbein, L.E. Heme: The Lord of the Iron Ring. Antioxidants 2023, 12, 1074. https://doi.org/10.3390/antiox12051074
Voltarelli VA, Alves de Souza RW, Miyauchi K, Hauser CJ, Otterbein LE. Heme: The Lord of the Iron Ring. Antioxidants. 2023; 12(5):1074. https://doi.org/10.3390/antiox12051074
Chicago/Turabian StyleVoltarelli, Vanessa Azevedo, Rodrigo W. Alves de Souza, Kenji Miyauchi, Carl J. Hauser, and Leo Edmond Otterbein. 2023. "Heme: The Lord of the Iron Ring" Antioxidants 12, no. 5: 1074. https://doi.org/10.3390/antiox12051074