
Antioxidant and Anti-Inflammatory Capacities of Fractions and Constituents from Vicia tetrasperma
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Extraction and Separation of Compounds (1–9)
2.3. UPLC-ESI-QTOF-MS Assay
2.4. Spectroscopic Data of Compounds 1–7
2.5. Cell Culture and Cell Viability
2.6. Measurement of ROS Accumulation
2.7. Measurement of NO Production
2.8. DPPH and ABTS Assay
2.9. Western Blotting
2.10. Measurement of IL-8 Production
2.11. Molecular Docking
2.12. Method Validation
2.12.1. UPLC-DAD System and Separation Condition
2.12.2. Detection of Wavelength
2.12.3. Preparation of Calibration Standard Solution
2.13. Statistical Analysis
3. Results
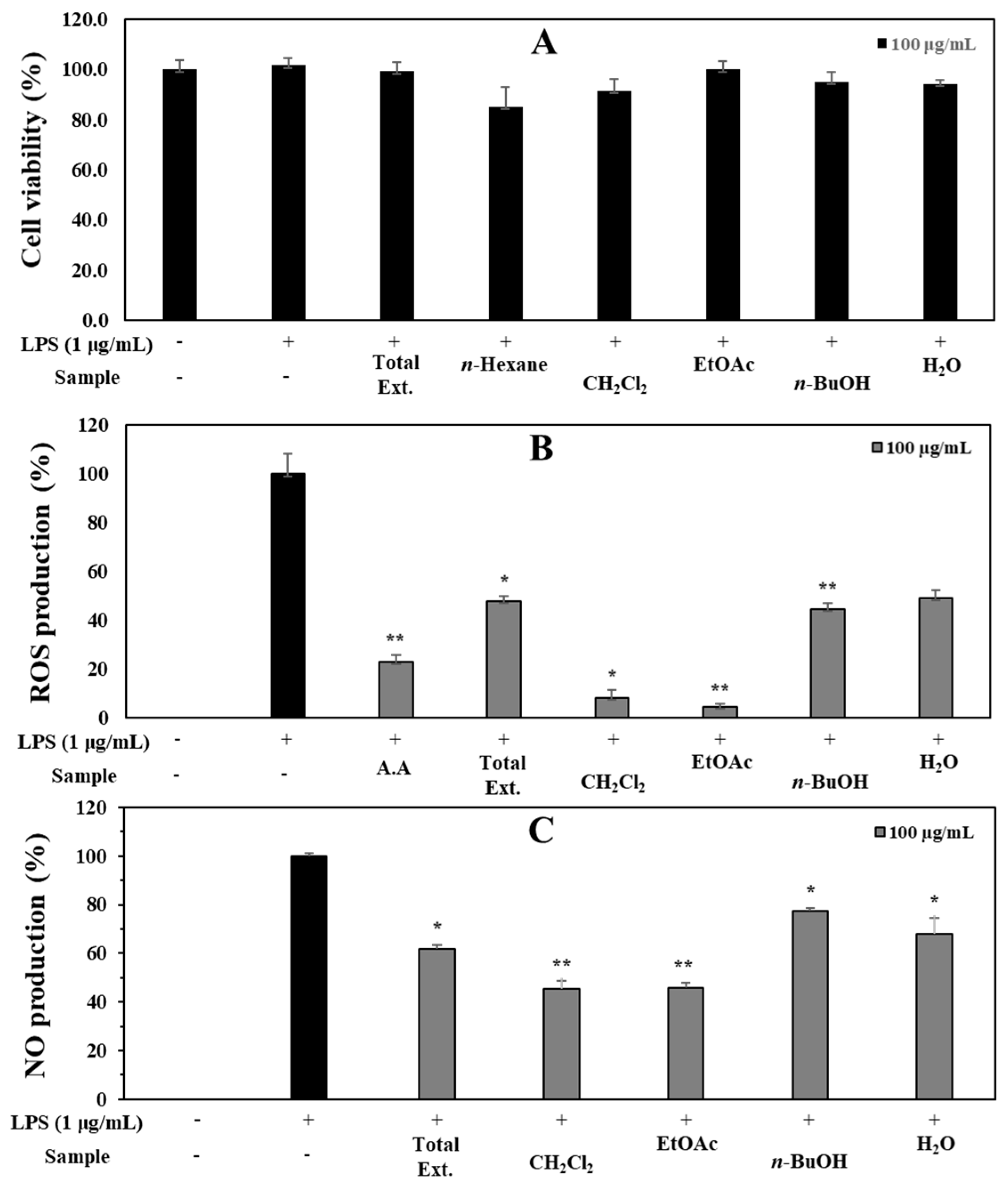
3.1. Antioxidant and Anti-Inflammatory Capacities of Total Extract and Fractions
3.2. Identification of Constituents
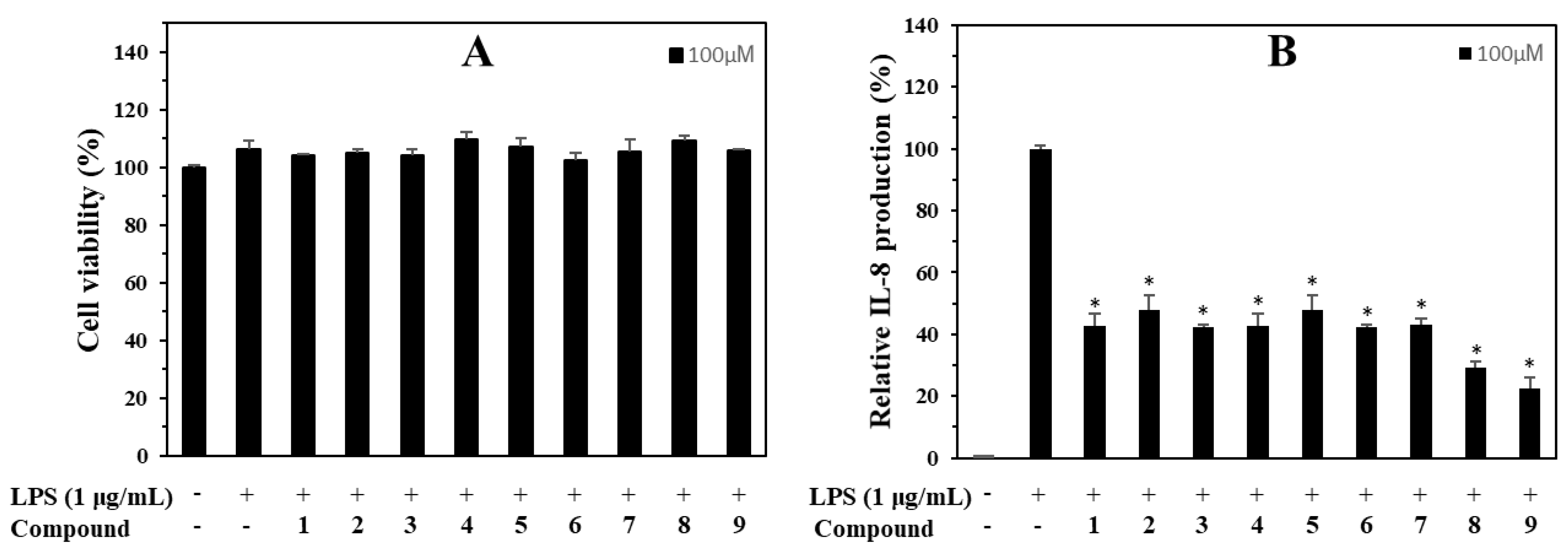
3.3. Anti-Inflammatory and Cytotoxic Capacities
3.4. Inhibitory Mechanism of Flavonoid Glycosides against iNOS and COX-2 Levels Expression
3.5. Anti-Inflammatory Capacity on IL-8 Production in LPS-Stimulated HT-29 Cells
3.6. Molecular Docking Analysis
3.7. Establishment and Validation Analysis of Marker Compounds (1 and 3–7) from the Herbal Extract of V. teterasperma
3.8. Qualification of Marker Compounds from the V. tetrasperma Extract
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jialal, I.; Devaraj, S. Low-density lipoprotein oxidation, antioxidants, and atherosclerosis: A clinical biochemistry perspective. Clin. Chem. 1996, 42, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, K.; Puri, S. Free radicals and antioxidants in health and disease: A review. East. Mediterr. Health J. 1998, 4, 350–360. [Google Scholar] [CrossRef]
- Bhatia, S.; Shukla, R.; Venkata Madhu, S.; Kaur Gambhir, J.; Madhava Prabhu, K. Antioxidant status, lipid peroxidation and nitric oxide end products in patients of type 2 diabetes mellitus with nephropathy. Clin. Biochem. 2003, 36, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cai, Y.; Sui, A.; Yao, Y.; Su, T.; Zhu, Y.; Xie, B.; Shen, X. Etanercept as a TNF-alpha inhibitor depresses experimental retinal neovascularization. Graefe’s Arch. Clin. Exp. Ophthalmol. 2021, 259, 661–671. [Google Scholar] [CrossRef]
- Sebba, A. Tocilizumab: The first interleukin-6-receptor inhibitor. Am. J. Health-Syst. Pharm. 2008, 65, 1413–1418. [Google Scholar] [CrossRef]
- Oray, M.; Abu Samra, K.; Ebrahimiadib, N.; Meese, H.; Foster, C.S. Long-term side effects of glucocorticoids. Expert Opin. Drug Saf. 2016, 15, 457–465. [Google Scholar] [CrossRef]
- Oomah, B.D.; Luc, G.; Leprelle, C.; Drover, J.C.G.; Harrison, J.E.; Olson, M. Phenolics, Phytic Acid, and Phytase in Canadian-Grown Low-Tannin Faba Bean (Vicia faba L.) Genotypes. J. Agric. Food Chem. 2011, 59, 3763–3771. [Google Scholar] [CrossRef]
- Pastor-Cavada, E.; Juan, R.; Pastor, J.E.; Alaiz, M.; GirÓN-Calle, J.; Vioque, J. Antioxidative activity in the seeds of 28 vicia species from southern Spain. J. Food Biochem. 2011, 35, 1373–1380. [Google Scholar] [CrossRef]
- Amarowicz, R.; Troszyńska, A.; Pegg, R.B. Antioxidative and radical scavenging effects of phenolics from Vicia sativum. Fitoterapia 2008, 79, 121–122. [Google Scholar] [CrossRef]
- Gamal-Eldeen, A.M.; Kawashty, S.A.; Ibrahim, L.F.; Shabana, M.M.; El-Negoumy, S.I. Evaluation of antioxidant, anti-Inflammatory, and antinociceptive properties of aerial parts of Vicia sativa and its flavonoids. J. Nat. Remedies 2004, 4, 81–96. [Google Scholar] [CrossRef]
- Boudjou, S.; Oomah, B.D.; Zaidi, F.; Hosseinian, F. Phenolics content and antioxidant and anti-inflammatory activities of legume fractions. Food Chem. 2013, 138, 1543–1550. [Google Scholar] [CrossRef]
- Abdalla Hussein, M. Anti-inflammatory effect of natural heterocycle glucoside vicine obtained from Vicia faba L. its aglucone (divicine) their effect on some oxidative stress biomarkers in Albino rats. Free Radic. Antioxid. 2012, 2, 44–54. [Google Scholar] [CrossRef]
- Ryszard, A.; Fereidoon, S. Antioxidant activity of faba bean extract and fractions thereof. J. Food Bioact. 2018, 2, 112–118. [Google Scholar] [CrossRef]
- Choudhary, D.K.; Mishra, A. In vitro and in silico interaction of porcine α-amylase with Vicia faba crude seed extract and evaluation of antidiabetic activity. Bioengineered 2017, 8, 393–403. [Google Scholar] [CrossRef]
- Dangol, D.R. Traditional uses of plants of commonland hanitats in western Chitwan, Nepal. J. Inst. Agric. Anim. Sci. 2008, 29, 71–78. [Google Scholar]
- Han, S.; Sebastin, R.; Wang, X.; Lee, K.J.; Cho, G.-T.; Hyun, D.Y.; Chung, J.-W. Identification of Vicia Species Native to South Korea Using Molecular and Morphological Characteristics. Front. Plant Sci. 2021, 12, 608559. [Google Scholar] [CrossRef]
- Lahuta, L.B.; Górecki, R.J.; Horbowicz, M. High concentrations of d-pinitol or d-chiro-inositol inhibit the biosynthesis of raffinose family oligosaccharides in maturing smooth tare (Vicia tetrasperma [L.] Schreb.) seeds. Acta Physiol. Plant. 2005, 27, 505–513. [Google Scholar] [CrossRef]
- Devi, S.K.; Devi, L.I.; Singh, L.R. Purification and Characterization of a New Dimeric Mannose/Glucose-Binding Isolectin from Vicia tetrasperma (L.) Schreber. Prep. Biochem. Biotechnol. 2008, 39, 57–71. [Google Scholar] [CrossRef]
- Corsetto, P.A.; Montorfano, G.; Zava, S.; Colombo, I.; Ingadottir, B.; Jonsdottir, R.; Sveinsdottir, K.; Rizzo, A.M. Characterization of Antioxidant Potential of Seaweed Extracts for Enrichment of Convenience Food. Antioxidants 2020, 9, 249. [Google Scholar] [CrossRef]
- Le, D.; Han, S.; Ahn, J.; Yu, J.; Kim, C.-K.; Lee, M. Analysis of Antioxidant Phytochemicals and Anti-Inflammatory Effect from Vitex rotundifolia L.f. Antioxidants 2022, 11, 454. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Wei, Y.; Jiang, S.; Xu, F.; Wang, H.; Zhan, P.; Shao, X. ROS Stress and Cell Membrane Disruption are the Main Antifungal Mechanisms of 2-Phenylethanol against Botrytis cinerea. J. Agric. Food Chem. 2022, 70, 14468–14479. [Google Scholar] [CrossRef] [PubMed]
- Marín, A.; Ferreres, F.; Tomás-Barberán, F.A.; Gil, M.I. Characterization and Quantitation of Antioxidant Constituents of Sweet Pepper (Capsicum annuum L.). J. Agric. Food Chem. 2004, 52, 3861–3869. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhuang, L.; Song, D.; Lu, C.; Xu, X. Isolation, purification, and identification of the main phenolic compounds from leaves of celery (Apium graveolens L. var. dulce Mill./Pers.). J. Sep. Sci. 2017, 40, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Slimestad, R.; Fossen, T.; Brede, C. Flavonoids and other phenolics in herbs commonly used in Norwegian commercial kitchens. Food Chem. 2020, 309, 125678. [Google Scholar] [CrossRef]
- Mencherini, T.; Picerno, P.; Scesa, C.; Aquino, R. Triterpene, Antioxidant, and Antimicrobial Compounds from Melissa officinalis. J. Nat. Prod. 2007, 70, 1889–1894. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Uemura, T.; Shimoda, H.; Kishi, A.; Kawahara, Y.; Matsuda, H. Medicinal Foodstuffs. XVIII. Phytoestrogens from the Aerial Part of Petroselinum crispum MILL. (PARSLEY) and Structures of 6”-Acetylapiin and a New Monoterpene Glycoside, Petroside. Chem. Pharm. Bull. 2000, 48, 1039–1044. [Google Scholar] [CrossRef]
- Zhou, K.; Zhao, F.; Liu, Z.; Zhuang, Y.; Chen, L.; Qiu, F. Triterpenoids and Flavonoids from Celery (Apium graveolens). J. Nat. Prod. 2009, 72, 1563–1567. [Google Scholar] [CrossRef]
- Rinaldo, D.; Rodrigues, C.; Rodrigues, J.; Sannomiya, M.; Santos, L.; Vilegas, W. New flavone from the leaves of Neea theifera (Nyctaginaceae). J. Braz. Chem. Soc. 2007, 18, 1132–1135. [Google Scholar] [CrossRef]
- Le, D.; Han, S.; Min, K.H.; Lee, M. Anti-Inflammatory Activity of Compounds Derived from Vitex rotundifolia. Metabolites 2023, 13, 249. [Google Scholar] [CrossRef]
- Maldonado-Rojas, W.; Olivero-Verbel, J. Food-Related Compounds That Modulate Expression of Inducible Nitric Oxide Synthase May Act as Its Inhibitors. Molecules 2012, 17, 8118–8135. [Google Scholar] [CrossRef]
- Tao, Y.; Yang, S.; Xu, H.; Tao, X. Molecular docking analysis of triptoquinones from genus Tripterygium with iNOS and in silico ADMET prediction. SN Appl. Sci. 2019, 1, 1533. [Google Scholar] [CrossRef]
- Li, P.; Jia, J.; Zhang, D.; Xie, J.; Xu, X.; Wei, D. In vitro and in vivo antioxidant activities of a flavonoid isolated from celery (Apium graveolens L. var. dulce). Food Funct. 2014, 5, 50–56. [Google Scholar] [CrossRef]
- Shin, J.Y.; Che, D.N.; Cho, B.O.; Kang, H.J.; Kim, J.-S.; Jang, S.I. Anti-inflammatory effect of hydrolyzed celery leaves extract in murine primary splenocyte. J. Food Biochem. 2019, 43, e12970. [Google Scholar] [CrossRef]
- Hayasaka, N.; Shimizu, N.; Komoda, T.; Mohri, S.; Tsushida, T.; Eitsuka, T.; Miyazawa, T.; Nakagawa, K. Absorption and Metabolism of Luteolin in Rats and Humans in Relation to in Vitro Anti-inflammatory Effects. J. Agric. Food Chem. 2018, 66, 11320–11329. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, M.; Jiang, B.; Yu, J.; Hao, Q.; Liu, W.; Hu, Z.; Zhang, Y.; Song, C. Extraction optimization, structural characterization and potential alleviation of hyperuricemia by flavone glycosides from celery seeds. Food Funct. 2022, 13, 9832–9846. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Antioxidant Activity (μmol TEAC/g Sample) | |
|---|---|---|
| DPPH | ABTS | |
| Total extract | 0.089 ± 0.011 | 0.186 ± 0.013 |
| n-Hexane | 0.131 ± 0.013 | 0.020 ± 0.001 |
| CH2Cl2 | 0.152 ± 0.011 | 0.254 ± 0.008 |
| EtOAc | 0.281 ± 0.015 | 0.482 ± 0.025 |
| n-BuOH | 0.172 ± 0.016 | 0.226 ± 0.018 |
| H2O | 0.077 ± 0.011 | 0.090 ± 0.003 |
| Compounds | Formula | Molecular Weight | Adduct | Found | Error | MS/MS Product Ions |
|---|---|---|---|---|---|---|
| (m/z) | (ppm) | |||||
| Luteolin 7-O-[2-β-D-apiofuranosyl)-β-D-glucopyranoside (1) | C26H28O15 | 580.1428 | [M − H]− | 579.1371 | 1.6 | 579.1371, 447.0919, 285.0403 |
| Luteolin 7-O-glucoside (2) | C21H20O11 | 448.1006 | [M − H]− | 447.0917 | −2.9 | 447.0917, 286.0432 |
| Apiin (3) | C26H28O14 | 564.14790 | [M − H]− | 563.1428 | 2.7 | 563.1428, 431.0976, 269.0469 |
| Luteolin 7-O-(2-apiosyl-6-malonyl)glucoside (4) | C29H30O18 | 666.1432 | [M − H]− | 665.1373 | 1.1 | 665.1373, 621.1457, 489.1041, 285.0403 |
| Chrysoeriol 7-O-(2-apiosyl)glucoside (5) | C27H30O15 | 594.1585 | [M − H]− | 593.1509 | −2.5 | 593.1509, 299.0576 |
| 6″-Acetylapiin (6) | C28H30O15 | 606.1585 | [M − H]− | 605.1528 | 2.9 | 605.1528, 563.1426, 269.0471 |
| Chrysoeriol 7-O-(2-apiosyl-6-acetyl)glucoside (7) | C29H32O16 | 636.1690 | [M − H]− | 635.1634 | 2.8 | 635.1634, 593.1504, 299.0574 |
| Apigenin-7-O-[2″-O-(5‴-O-feruloyl)-D-apiofuranosyl]-D-glucopyranoside (8) | C36H36O17 | 740.1952 | [M − H]− | 739.1878 | 0.4 | 739.1878, 545.1303, 269.0456 |
| Luteolin-[2″-O-(5‴-O-feruloyl)-D-apiofuranosyl]-D-glucopyranoside (9) | C36H36O18 | 756.1902 | [M − H]− | 755.1857 | 2.6 | 755.1857, 579.1351, 447.0849, 285.0406 |
| Ligand | Protein | Affinity Score (Kcal/mol) | Conventional Hydrogen Bond | Hydrophobic Interactions |
|---|---|---|---|---|
| 6″-Acetylapiin (6) | PDB ID: 3E7G | −9.50 | ARG199, ARG381, ASP382, ILE201, GLU377, GLY202, TRO463 | VAL352 |
| 6″-Acetylapiin (6) | PDB ID: 5IKQ | −3.28 | ASP157, GLN457, HIS214 | ALA132, ALA219, LYS459 |
| Apigenin-7-O-[2″-O-(5‴-O-feruloyl)-D-apiofuranosyl]-D-glucopyranoside (8) | PDB ID: 5D14 | −4.32 | GLN6, LYS9, CYS48 | CYS7, ILE8, THR10, LYS13, TYR11, ILE38, GLU46, ARG45, LEU47 |
| Luteolin-[2″-O-(5‴-O-feruloyl)-D-apiofuranosyl]-D-glucopyranoside (9) | PDB ID: 5D14 | −7.75 | ARG4, CYS48 | LEU3, CYS5, GLN6, CYS7, ILE8, THR10, TYR11, ILE38, GLN46, LEU47 |
| Marker Compound | Concentration Range (µg/mL) | Regression Equation | Correlation Coefficient (r2) | LOD (µg/mL) | LOQ (µg/mL) | Content (w/w) |
|---|---|---|---|---|---|---|
| Luteolin 7-O-[2-β-D-apiofuranosyl)-β-D-glucopyranoside (1) | 1.95~250 | Y = 6342.7x + 9788.7 | 0.9997 | 0.60 | 1.84 | 2.18 |
| Apiin (3) | 1.95~250 | Y = 12,674x + 20,920 | 0.9997 | 0.14 | 0.42 | 3.02 |
| Luteolin 7-O-(2-apiosyl-6-malonylglucoside) (4) | 0.39~50 | Y = 13,457x + 4379.3 | 0.9994 | 0.12 | 0.39 | 0.39 |
| Chrysoeriol 7-O-(2-apiosyl)-glucoside (5) | 1.95~250 | Y = 11,866x + 25,034 | 0.9997 | 0.53 | 1.62 | 1.94 |
| 6″-Acetylapiin (6) | 0.31~20 | Y = 15,520x + 1345.3 | 0.9995 | 0.28 | 0.85 | 0.09 |
| Chrysoeriol 7-O-(2-apiosyl-6-acetyl)-glucoside (7) | 0.31~20 | Y = 18,736x − 2737.6 | 0.9990 | 0.52 | 1.59 | 0.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, D.D.; Min, K.H.; Lee, M. Antioxidant and Anti-Inflammatory Capacities of Fractions and Constituents from Vicia tetrasperma. Antioxidants 2023, 12, 1044. https://doi.org/10.3390/antiox12051044
Le DD, Min KH, Lee M. Antioxidant and Anti-Inflammatory Capacities of Fractions and Constituents from Vicia tetrasperma. Antioxidants. 2023; 12(5):1044. https://doi.org/10.3390/antiox12051044
Chicago/Turabian StyleLe, Duc Dat, Kyung Hyun Min, and Mina Lee. 2023. "Antioxidant and Anti-Inflammatory Capacities of Fractions and Constituents from Vicia tetrasperma" Antioxidants 12, no. 5: 1044. https://doi.org/10.3390/antiox12051044