
Oxidative Stress and Inflammation in B-Cell Lymphomas
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
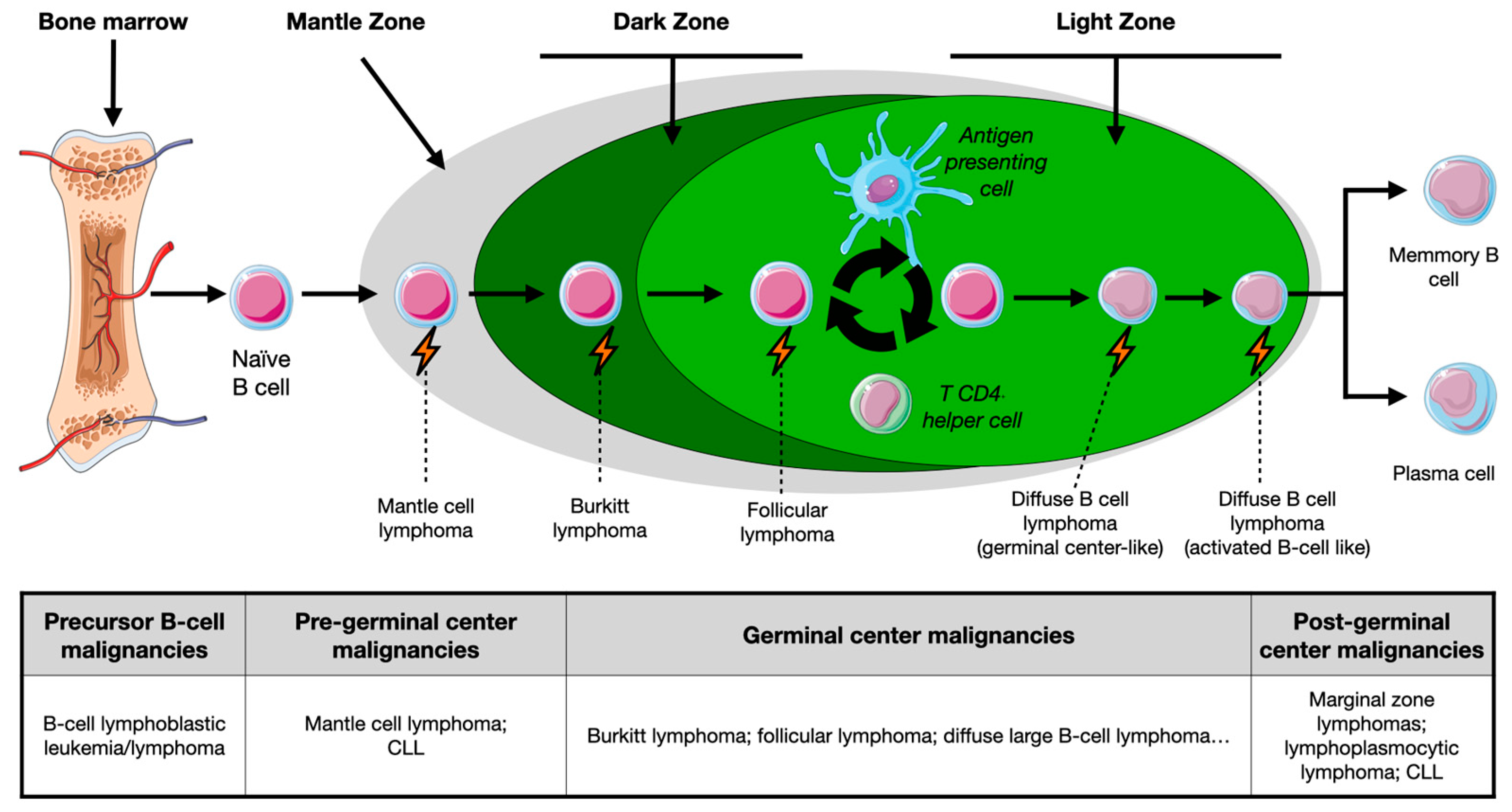
1.1. Overview of B-Cell Maturation
1.2. B-Cell Malignancies: Nature- or Nurture-Induced Disruption?
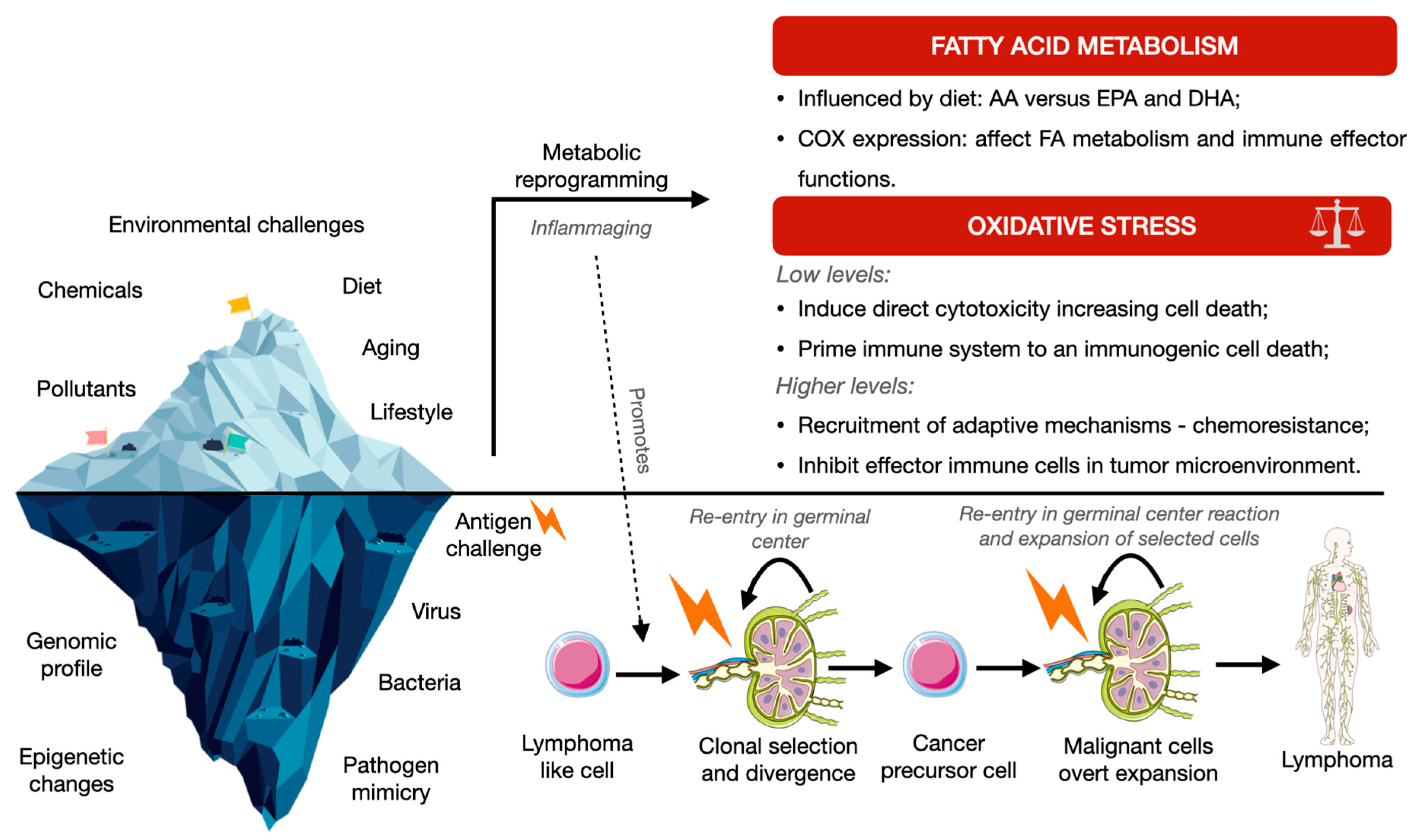
2. Oxidative Stress and Inflammation in Lymphomagenesis
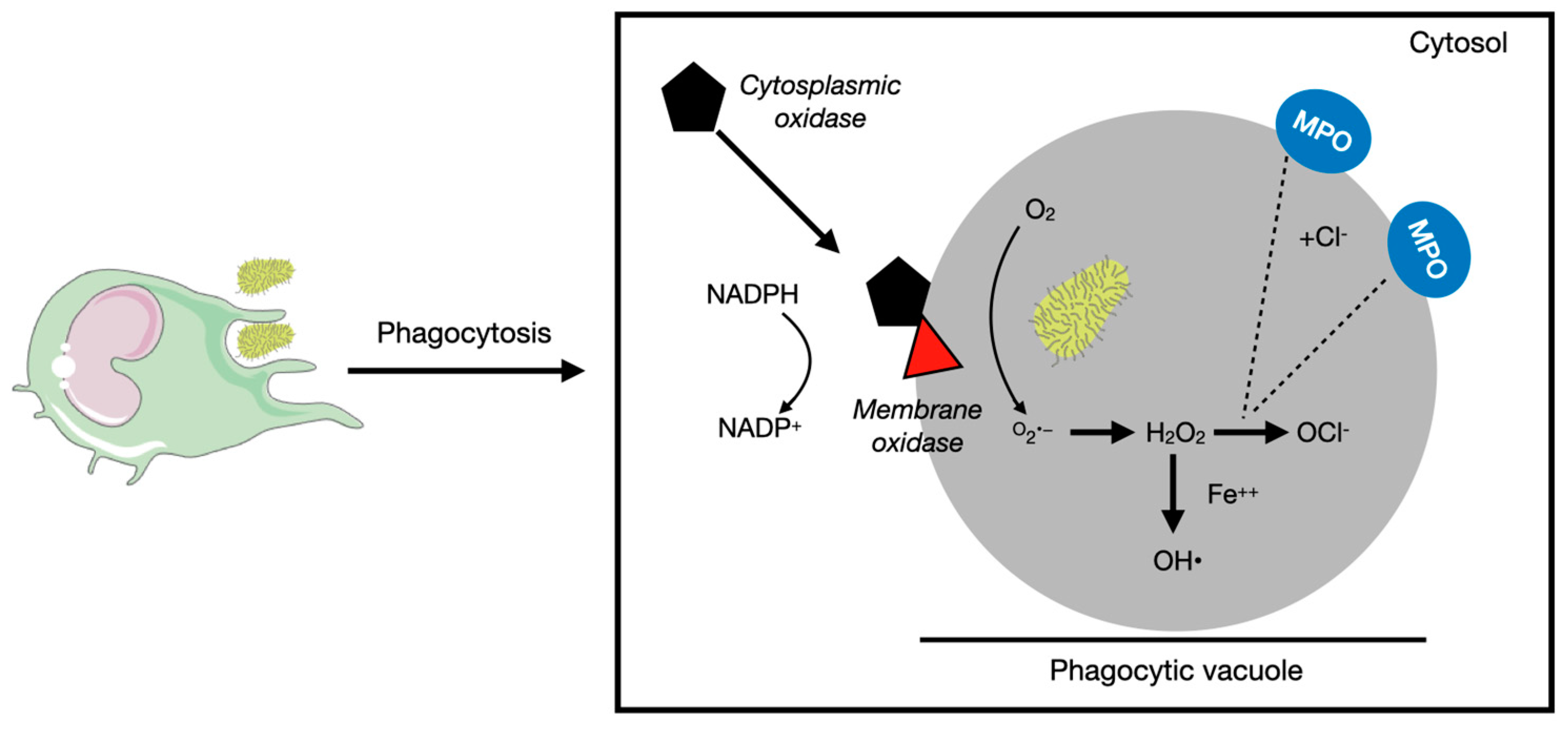
2.1. Reactive Oxygen Species
2.1.1. Oxidative Stress as a Modulator of Lymphomagenesis
2.1.2. Oxidative Stress as a Therapeutical Target and Modulator of Treatment Response
2.2. Fatty Acids
2.2.1. Fatty Acids as Environmental Modulators of Lymphomagenesis
2.2.2. Fatty Acids as a Modulator of Treatment Response and Therapeutical Target
2.3. Fatty Acids and ROS Cross-Regulation as a Result of Environmental Reprogramming
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jones, D.S.; Podolsky, S.H.; Greene, J.A. The burden of disease and the changing task of medicine. N. Engl. J. Med. 2012, 366, 2333–2338. [Google Scholar] [CrossRef] [Green Version]
- Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Non-Hodgkin Lymphoma, Long-Term Trends in SEER Age-Adjusted Incidence Rates, 1975–2019, 8 Registries. Available online: https://seer.cancer.gov/statistics-network/explorer/application.html (accessed on 27 February 2023).
- Hallek, M.; Al-Sawaf, O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am. J. Hematol. 2021, 96, 1679–1705. [Google Scholar] [CrossRef] [PubMed]
- Smedby, K.E.; Vajdic, C.M.; Falster, M.; Engels, E.A.; Martinez-Maza, O.; Turner, J.; Hjalgrim, H.; Vineis, P.; Costantini, A.S.; Bracci, P.M.; et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: A pooled analysis within the InterLymph Consortium. Blood 2008, 111, 4029–4038. [Google Scholar] [CrossRef] [Green Version]
- Anderson, L.A.; Gadalla, S.; Morton, L.M.; Landgren, O.; Pfeiffer, R.; Warren, J.L.; Berndt, S.I.; Ricker, W.; Parsons, R.; Engels, E.A. Population-based study of autoimmune conditions and the risk of specific lymphoid malignancies. Int. J. Cancer 2009, 125, 398–405. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Basso, K.; Dalla-Favera, R. Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172. [Google Scholar] [PubMed]
- Stebegg, M.; Kumar, S.D.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the germinal center response. Front. Immunol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janz, S.; Potter, M.; Rabkin, C.S. Lymphoma- and leukemia-associated chromosomal translocations in healthy individuals. Genes Chromosomes Cancer 2003, 36, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2017.
- Marculescu, R.; Le, T.; Simon, P.; Jaeger, U.; Nadel, B. V(D)J-mediated translocations in lymphoid neoplasms: A functional assessment of genomic instability by cryptic sites. J. Exp. Med. 2002, 195, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; von Keudell, G.; López-Guillermo, A.; Fitzgibbon, J. Follicular lymphoma. Nat. Rev. Dis. Prim. 2019, 5, 83. [Google Scholar] [CrossRef]
- Tellier, J.; Menard, C.; Roulland, S.; Martin, N.; Monvoisin, C.; Chasson, L.; Nadel, B.; Gaulard, P.; Schiff, C.; Tarte, K. Human t(14;18)positive germinal center B cells: A new step in follicular lymphoma pathogenesis? Blood 2014, 123, 3462–3465. [Google Scholar] [CrossRef] [Green Version]
- Sungalee, S.; Mamessier, E.; Morgado, E.; Grégoire, E.; Brohawn, P.Z.; Morehouse, C.A.; Jouve, N.; Monvoisin, C.; Menard, C.; Debroas, G.; et al. Germinal center reentries of BCL2-overexpressing B cells drive follicular lymphoma progression. J. Clin. Investig. 2014, 124, 5337–5351. [Google Scholar] [CrossRef] [Green Version]
- Araf, S.; Wang, J.; Korfi, K.; Pangault, C.; Kotsiou, E.; Rio-Machin, A.; Rahim, T.; Heward, J.; Clear, A.; Iqbal, S.; et al. Genomic profiling reveals spatial intra-tumor heterogeneity in follicular lymphoma. Leukemia 2018, 32, 1261–1265. [Google Scholar] [CrossRef] [Green Version]
- Auten, R.L.; Davis, J.M. Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef] [PubMed]
- Leong, X.F. Lipid Oxidation Products on Inflammation-Mediated Hypertension and Atherosclerosis: A Mini Review. Front. Nutr. 2021, 8, 717740. [Google Scholar] [CrossRef]
- Mantena, S.K.; Vaughn, D.P.; Andringa, K.K.; Eccleston, H.B.; King, A.L.; Abrams, G.A.; Doeller, J.E.; Kraus, D.W.; Darley-Usmar, V.M.; Bailey, S.M. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem. J. 2009, 417, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.R.; Kakimoto, P.A.; Kowaltowski, A.J. Diet-sensitive sources of reactive oxygen species in liver mitochondria: Role of very long chain acyl-CoA dehydrogenases. PLoS ONE 2013, 8, e77088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashida Gnanaprakasam, J.N.; Wu, R.; Wang, R. Metabolic Reprogramming in Modulating T Cell Reactive Oxygen Species Generation and Antioxidant Capacity. Front. Immunol. 2018, 9, 1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity—From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.-S.; Jang, J.-H.; Kim, D.-H. Perspectives Regarding the Intersections between STAT3 and Oxidative Metabolism in Cancer. Cells 2020, 9, 2202. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Goel, A.; Spitz, D.R.; Weiner, G.J. Manipulation of cellular redox parameters for improving therapeutic responses in B-cell lymphoma and multiple myeloma. J. Cell. Biochem. 2012, 113, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Domka, K.; Goral, A.; Firczuk, M. cROSsing the Line: Between Beneficial and Harmful Effects of Reactive Oxygen Species in B-Cell Malignancies. Front. Immunol. 2020, 11, 1538. [Google Scholar] [CrossRef]
- Wang, S.S.; Davis, S.; Cerhan, J.R.; Hartge, P.; Severson, R.K.; Cozen, W.; Lan, Q.; Welch, R.; Chanock, S.J.; Rothman, N. Polymorphisms in oxidative stress genes and risk for non-Hodgkin lymphoma. Carcinogenesis 2006, 27, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Lan, Q.; Zheng, T.; Shen, M.; Zhang, Y.; Wang, S.S.; Zahm, S.H.; Holford, T.R.; Leaderer, B.; Boyle, P.; Chanock, S. Genetic polymorphisms in the oxidative stress pathway and susceptibility to non-Hodgkin lymphoma. Hum. genet. 2007, 121, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farawela, H.; Khorshied, M.; Shaheen, I.; Gouda, H.; Nasef, A.; Abulata, N.; Mahmoud, H.A.; Zawam, H.M.; Mousa, S.M. The association between hepatitis C virus infection, genetic polymorphisms of oxidative stress genes and B-cell non-Hodgkin’s lymphoma risk in Egypt. Infect. Genet. Evol. 2012, 12, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, T.J.; Skibola, C.F.; Smith, A.G.; Forrest, M.S.; Adamson, P.J.; Morgan, G.J.; Bracci, P.M.; Roman, E.; Smith, M.T.; Holly, E.A. Polymorphisms in the oxidative stress genes, superoxide dismutase, glutathione peroxidase and catalase and risk of non-Hodgkin’s lymphoma. Haematologica 2006, 91, 1222–1227. [Google Scholar]
- Khorshied, M.M.; Shaheen, I.A.; Selim, Y.M.M.; Elshahawy, A.O.; Youssry, I. Impact of Superoxide Dismutase Genetic Polymorphism (SOD2 Val16Ala) and Superoxide Dismutase Level on Disease Severity in a Cohort of Egyptian Sickle Cell Disease Patients. Mediterr. J. Hematol. Infect. Dis. 2022, 14, e2022037. [Google Scholar] [CrossRef] [PubMed]
- Peroja, P.; Haapasaari, K.M.; Mannisto, S.; Miinalainen, I.; Koivunen, P.; Leppä, S.; Karjalainen-Lindsberg, M.L.; Kuusisto, M.E.; Turpeenniemi-Hujanen, T.; Kuittinen, O.; et al. Total peroxiredoxin expression is associated with survival in patients with follicular lymphoma. Virchows Arch. 2016, 468, 623–630. [Google Scholar] [CrossRef]
- Gustafson, H.L.; Yao, S.; Goldman, B.H.; Lee, K.; Spier, C.M.; LeBlanc, M.L.; Rimsza, L.M.; Cerhan, J.R.; Habermann, T.M.; Link, B.K.; et al. Genetic polymorphisms in oxidative stress-related genes are associated with outcomes following treatment for aggressive B-cell non-Hodgkin lymphoma. Am. J. Hematol. 2014, 89, 639–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weniger, M.A.; Rizzatti, E.G.; Pérez-Galán, P.; Liu, D.; Wang, Q.; Munson, P.J.; Raghavachari, N.; White, T.; Tweito, M.M.; Dunleavy, K.; et al. Treatment-induced oxidative stress and cellular antioxidant capacity determine response to bortezomib in mantle cell lymphoma. Clin. Cancer Res. 2011, 17, 5101–5112. [Google Scholar] [CrossRef] [Green Version]
- Sallustio, B.C.; Boddy, A.V. Is there scope for better individualisation of anthracycline cancer chemotherapy? Br. J. Clin. Pharmacol. 2021, 87, 295–305. [Google Scholar] [CrossRef]
- Kiebala, M.; Skalska, J.; Casulo, C.; Brookes, P.S.; Peterson, D.R.; Hilchey, S.P.; Dai, Y.; Grant, S.; Maggirwar, S.B.; Bernstein, S.H. Dual targeting of the thioredoxin and glutathione antioxidant systems in malignant B cells: A novel synergistic therapeutic approach. Exp. Hematol. 2015, 43, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Barr, P.M.; Miller, T.P.; Friedberg, J.W.; Peterson, D.R.; Baran, A.M.; Herr, M.; Spier, C.M.; Cui, H.; Roe, D.J.; Persky, D.O.; et al. Phase 2 study of imexon, a prooxidant molecule, in relapsed and refractory B-cell non-Hodgkin lymphoma. Blood 2014, 124, 1259–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Pittman, E.F.; Romaguera, J.; Fayad, L.; Wang, M.; Neelapu, S.S.; McLaughlin, P.; Kwak, L.; McCarty, N. Nuclear translocation of B-cell-specific transcription factor, BACH2, modulates ROS mediated cytotoxic responses in mantle cell lymphoma. PLoS ONE 2013, 8, e69126. [Google Scholar] [CrossRef]
- Graczyk-Jarzynka, A.; Goral, A.; Muchowicz, A.; Zagozdzon, R.; Winiarska, M.; Bajor, M.; Trzeciecka, A.; Fidyt, K.; Krupka, J.A.; Cyran, J.; et al. Inhibition of thioredoxin-dependent H(2)O(2) removal sensitizes malignant B-cells to pharmacological ascorbate. Redox Biol. 2019, 21, 101062. [Google Scholar] [CrossRef]
- Fritsche, K.L. The science of fatty acids and inflammation. Adv. Nutr. 2015, 6, 293s–301s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calder, P.C. Fatty acids and inflammation: The cutting edge between food and pharma. Eur. J. Pharmacol. 2011, 668 (Suppl. S1), S50–S58. [Google Scholar] [CrossRef]
- Bock, M.; Karber, M.; Kuhn, H. Ketogenic diets attenuate cyclooxygenase and lipoxygenase gene expression in multiple sclerosis. EBioMedicine 2018, 36, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, A.J.; Lim, U. The role of dietary factors in the epidemiology of non-Hodgkin’s lymphoma. Leuk. Lymphoma 2006, 47, 2477–2487. [Google Scholar] [CrossRef]
- Bernard, M.P.; Bancos, S.; Sime, P.J.; Phipps, R.P. Targeting cyclooxygenase-2 in hematological malignancies: Rationale and promise. Curr. Pharm. Des. 2008, 14, 2051–2060. [Google Scholar] [CrossRef] [Green Version]
- Hazar, B.; Ergin, M.; Seyrek, E.; Erdoğan, S.; Tuncer, I.; Hakverdi, S. Cyclooxygenase-2 (Cox-2) expression in lymphomas. Leuk. Lymphoma 2004, 45, 1395–1399. [Google Scholar] [CrossRef]
- Smyth, L.; Blunt, D.N.; Gatov, E.; Nagamuthu, C.; Croxford, R.; Mozessohn, L.; Cheung, M.C. Statin and cyclooxygenase-2 inhibitors improve survival in newly diagnosed diffuse large B-cell lymphoma: A large population-based study of 4913 subjects. Br. J. Haematol. 2020, 191, 396–404. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, X.; Wang, X. Targeting the tumor microenvironment in B-cell lymphoma: Challenges and opportunities. J. Hematol. Oncol. 2021, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Chagas, T.R.; Borges, D.S.; de Oliveira, P.F.; Mocellin, M.C.; Barbosa, A.M.; Camargo, C.Q.; Del Moral JÂ, G.; Poli, A.; Calder, P.C.; Trindade, E.; et al. Oral fish oil positively influences nutritional-inflammatory risk in patients with haematological malignancies during chemotherapy with an impact on long-term survival: A randomised clinical trial. J. Hum. Nutr. Diet 2017, 30, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Moloudizargari, M.; Mortaz, E.; Asghari, M.H.; Adcock, I.M.; Redegeld, F.A.; Garssen, J. Effects of the polyunsaturated fatty acids, EPA and DHA, on hematological malignancies: A systematic review. Oncotarget 2018, 9, 11858–11875. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Wang, X.; Li, Y.; Bai, O. Function and regulation of lipid signaling in lymphomagenesis: A novel target in cancer research and therapy. Crit. Rev. Oncol. Hematol. 2020, 154, 103071. [Google Scholar] [CrossRef]
- Uddin, S.; Hussain, A.; Bavi, P.; Al-Kuraya, K. Fatty Acid Synthase and Diffuse Large B Cell Lymphoma. Blood 2008, 112, 4979. [Google Scholar] [CrossRef]
- Gelebart, P.; Zak, Z.; Anand, M.; Belch, A.; Lai, R. Blockade of Fatty Acid Synthase Triggers Significant Apoptosis in Mantle Cell Lymphoma. PLoS ONE 2012, 7, e33738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhayaza, R.; Haque, E.; Karbasiafshar, C.; Sellke, F.W.; Abid, M.R. The Relationship Between Reactive Oxygen Species and Endothelial Cell Metabolism. Front. Chem. 2020, 8, 592688. [Google Scholar] [CrossRef]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Effects of glutamine deprivation on oxidative stress and cell survival in breast cell lines. Biol. Res. 2019, 52, 15. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Ackerman, D.; Quinn, Z.L.; Mancuso, A.; Gruber, M.; Liu, L.; Giannoukos, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; et al. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 2013, 27, 1115–1131. [Google Scholar] [CrossRef] [Green Version]
- Mikalayeva, V.; Ceslevičienė, I.; Sarapinienė, I.; Žvikas, V.; Skeberdis, V.A.; Jakštas, V.; Bordel, S. Fatty Acid Synthesis and Degradation Interplay to Regulate the Oxidative Stress in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 1348. [Google Scholar] [CrossRef] [Green Version]
- Torres-Mendoza, B.M.G.; Ortiz, G.G.; Sánchez-Romero, L.; Delgado-Lara, D.L.C.; García Martínez, M.T.; Mireles-Ramírez, M.A.; Cruz Serrano, J.A.; Pacheco Moisés, F.P. Dietary fish oil increases catalase activity in patients with probable Alzheimer’s disease. Nutr. Hosp. 2022, 39, 1364–1368. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa-Pimenta, M.; Estevinho, M.M.; Sousa Dias, M.; Martins, Â.; Estevinho, L.M. Oxidative Stress and Inflammation in B-Cell Lymphomas. Antioxidants 2023, 12, 936. https://doi.org/10.3390/antiox12040936
Sousa-Pimenta M, Estevinho MM, Sousa Dias M, Martins Â, Estevinho LM. Oxidative Stress and Inflammation in B-Cell Lymphomas. Antioxidants. 2023; 12(4):936. https://doi.org/10.3390/antiox12040936
Chicago/Turabian StyleSousa-Pimenta, Mário, Maria Manuela Estevinho, Miguel Sousa Dias, Ângelo Martins, and Letícia M. Estevinho. 2023. "Oxidative Stress and Inflammation in B-Cell Lymphomas" Antioxidants 12, no. 4: 936. https://doi.org/10.3390/antiox12040936