

KDELC2 Upregulates Glioblastoma Angiogenesis via Reactive Oxygen Species Activation and Tumor-Associated Macrophage Proliferation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Silico Study
2.2. Cell Cultures
2.3. Stable Expression Clones of shRNAs
2.4. Overexpression of KDELC2
2.5. RNA Isolation and qRT-PCR
2.6. ROS Detection
2.7. Western Blot Analysis
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Tube Formation
2.10. Orthotopic Xenograft Animal Model
2.11. Statistical Analysis
3. Results
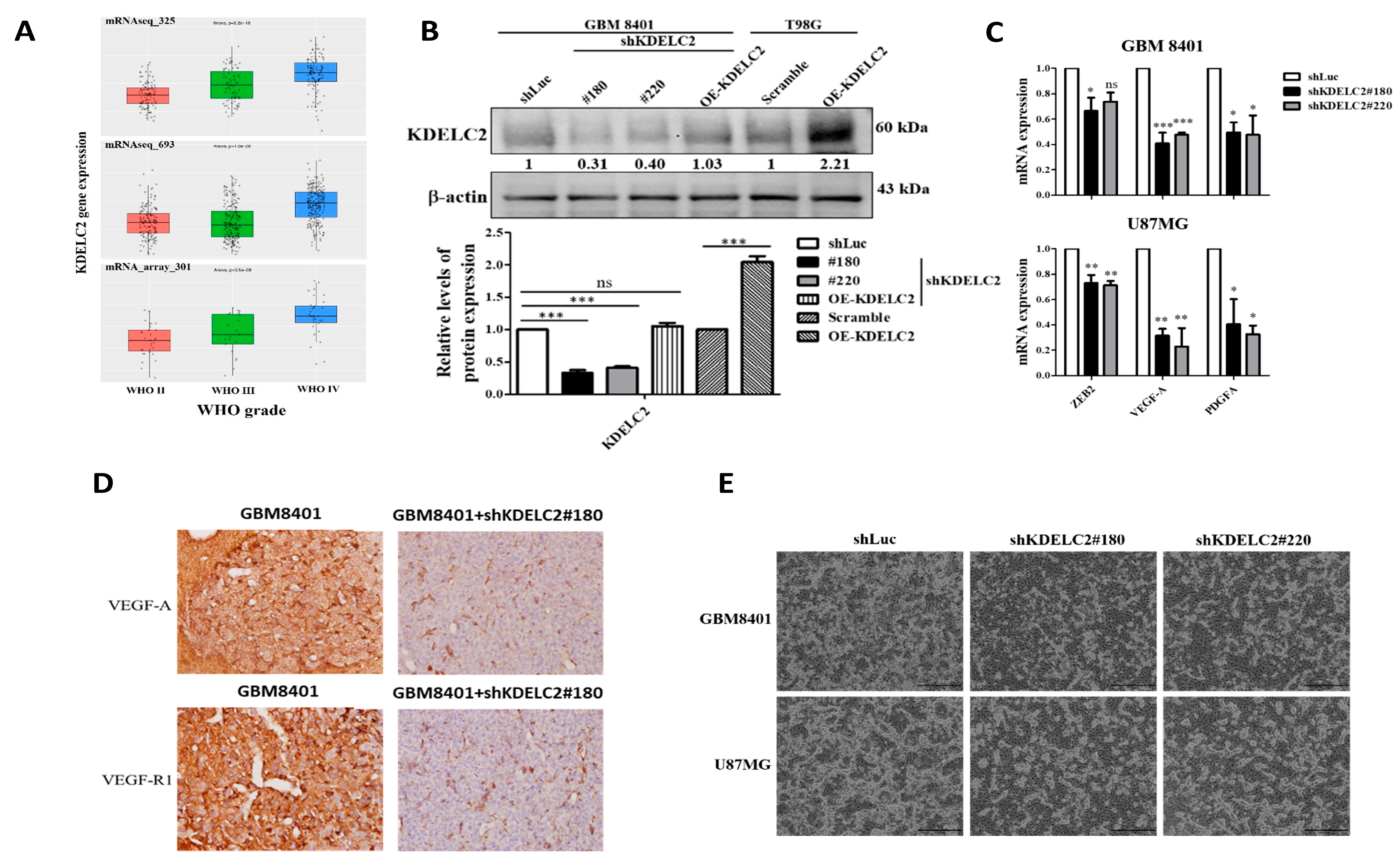
3.1. KDELC2 Expression Correlates with Glioma Tumor Grade and Glioblastoma Neovascularization
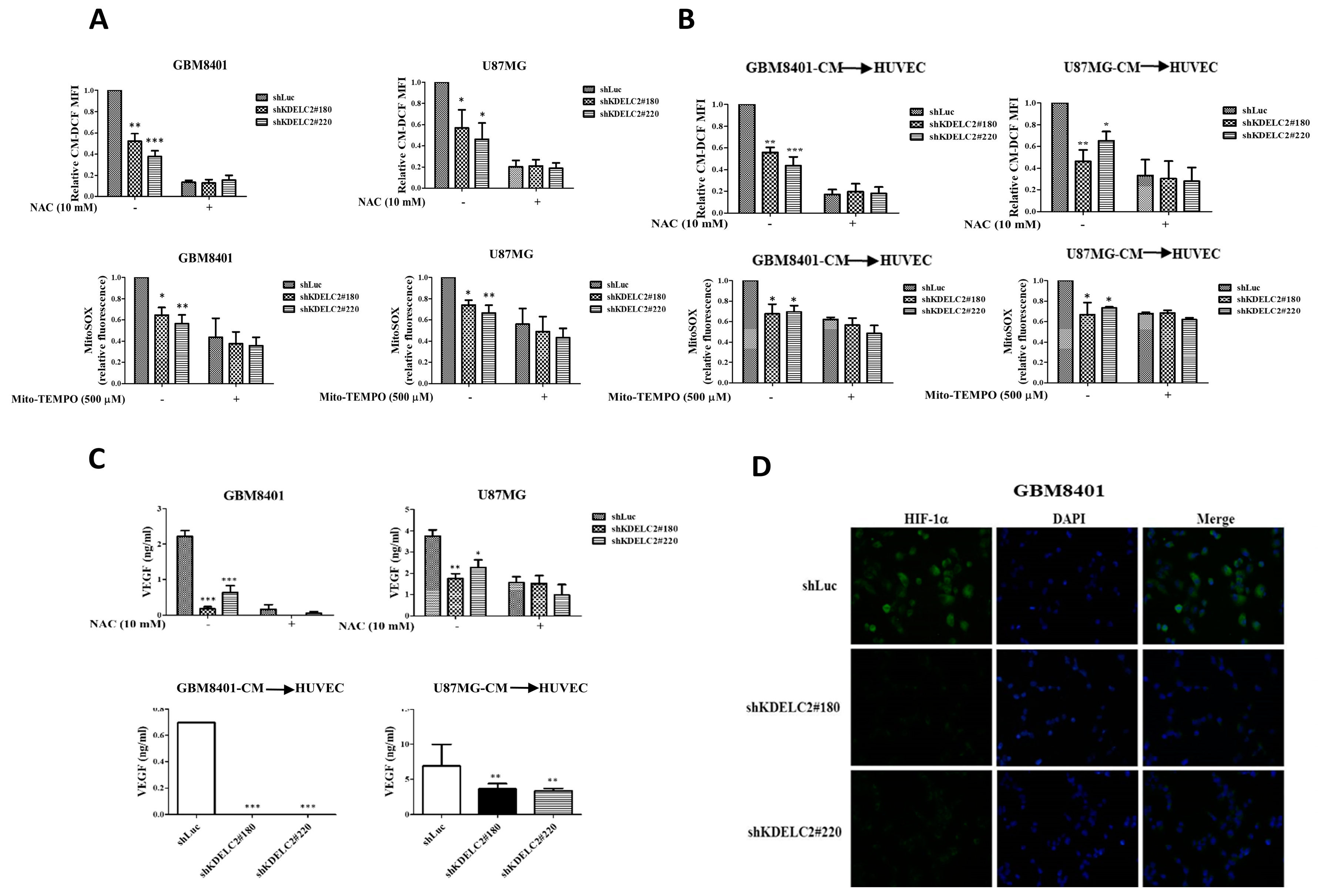
3.2. KDELC2 Induces Angiogenesis by Elevating Mitochondrial ROS in Glioblastoma Cells and HUVECs
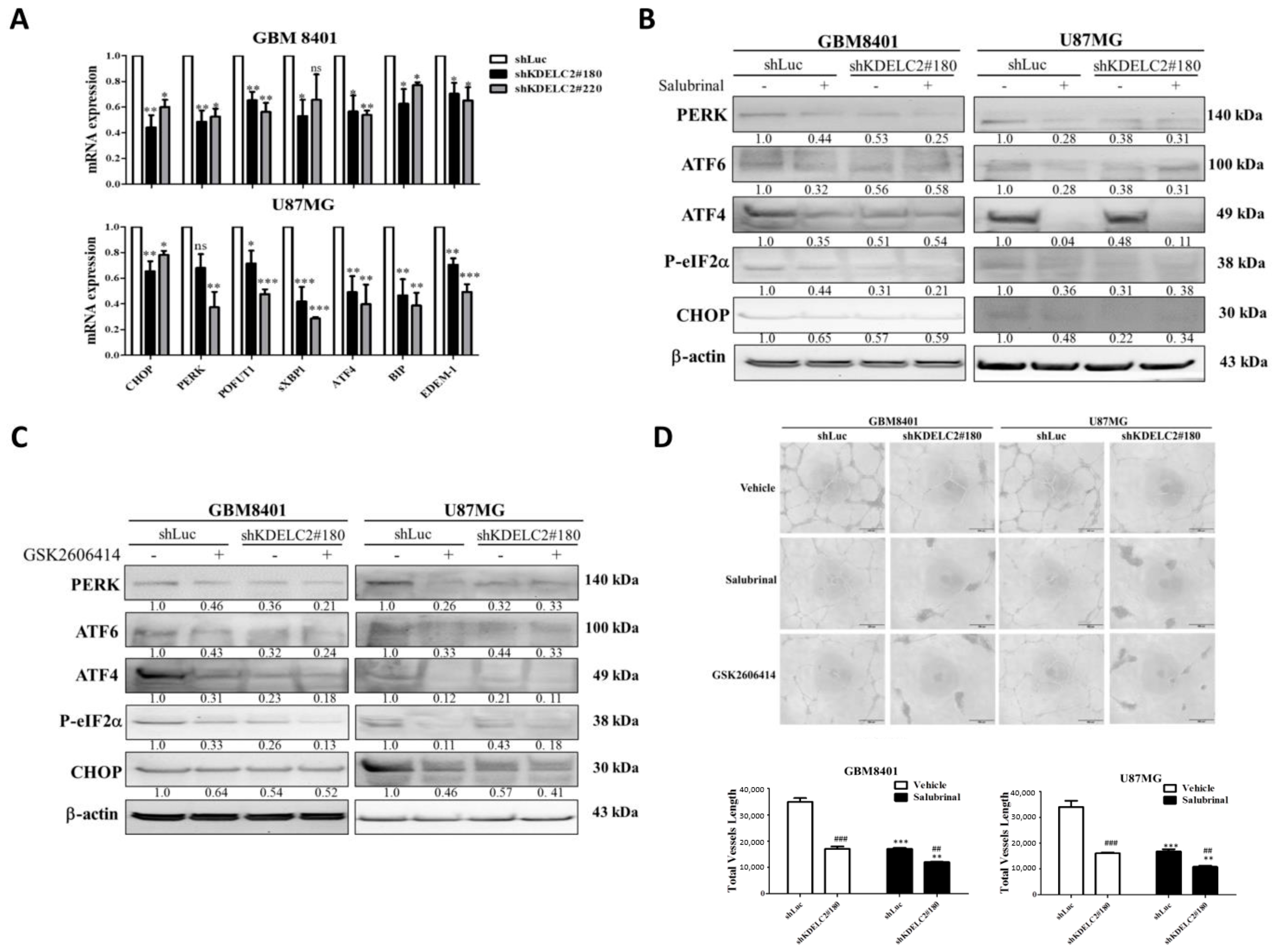
3.3. Upregulation of ER Stress and HIF-1α Expression by KDELC2 Overexpression in Glioblastoma Cells
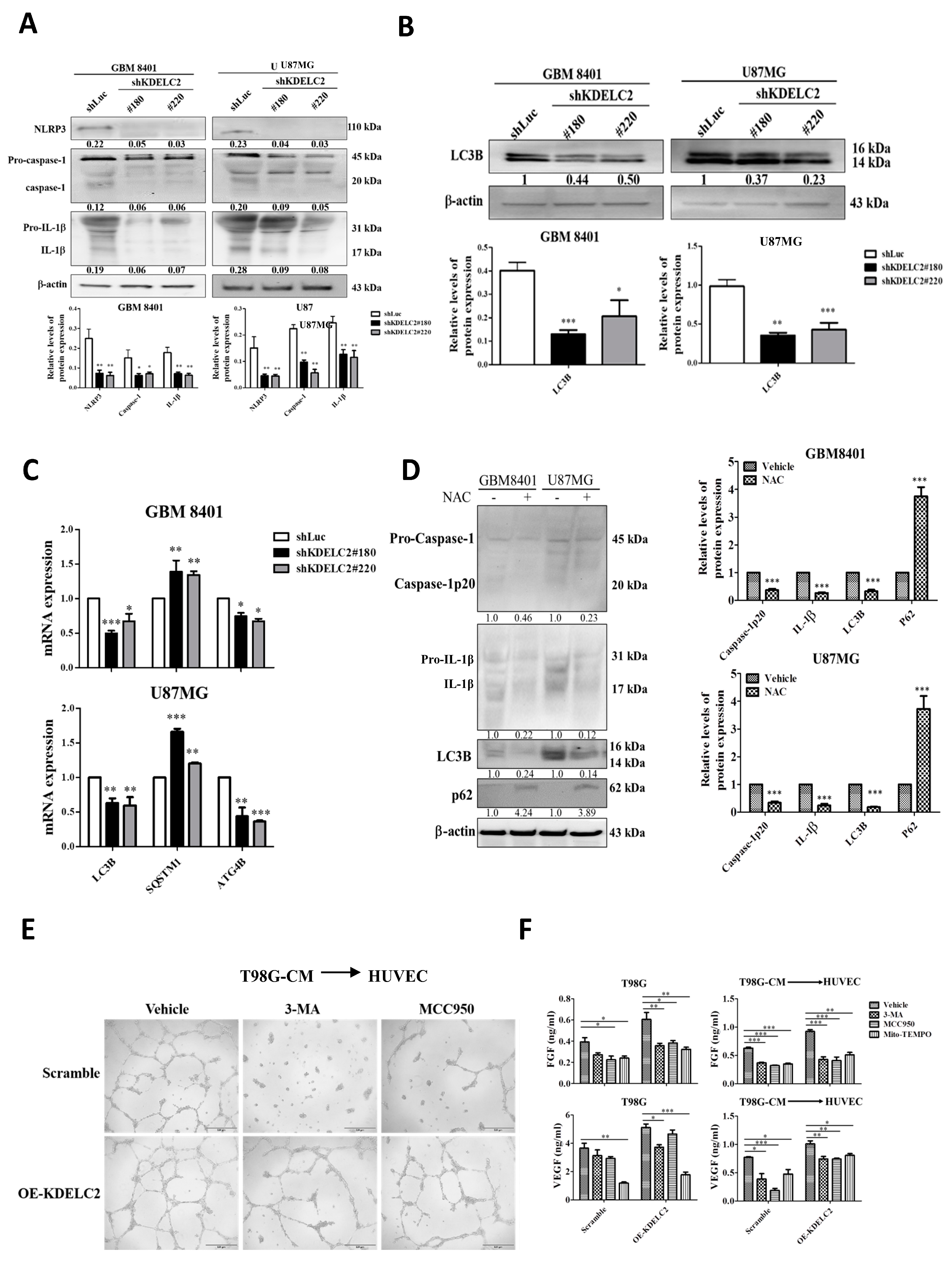
3.4. KDELC2 Overexpression Activates NLRP3 Inflammasome and Upregulates Glioblastoma Autophagy via Increased Mitochondrial ROS Production
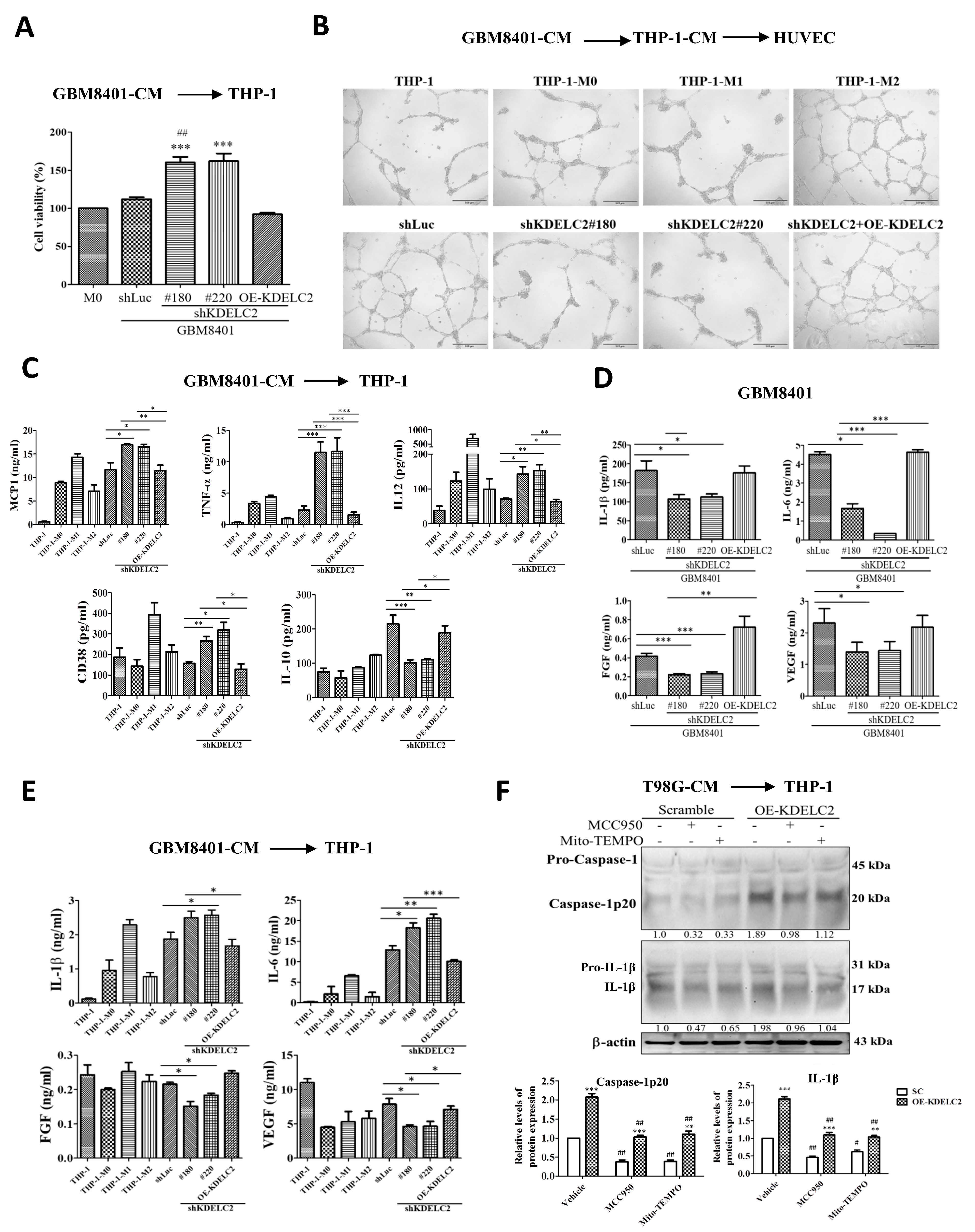
3.5. KDELC2 Stimulates the Transformation of M2 Macrophages and Promotes Tumor Angiogenesis
4. Discussion
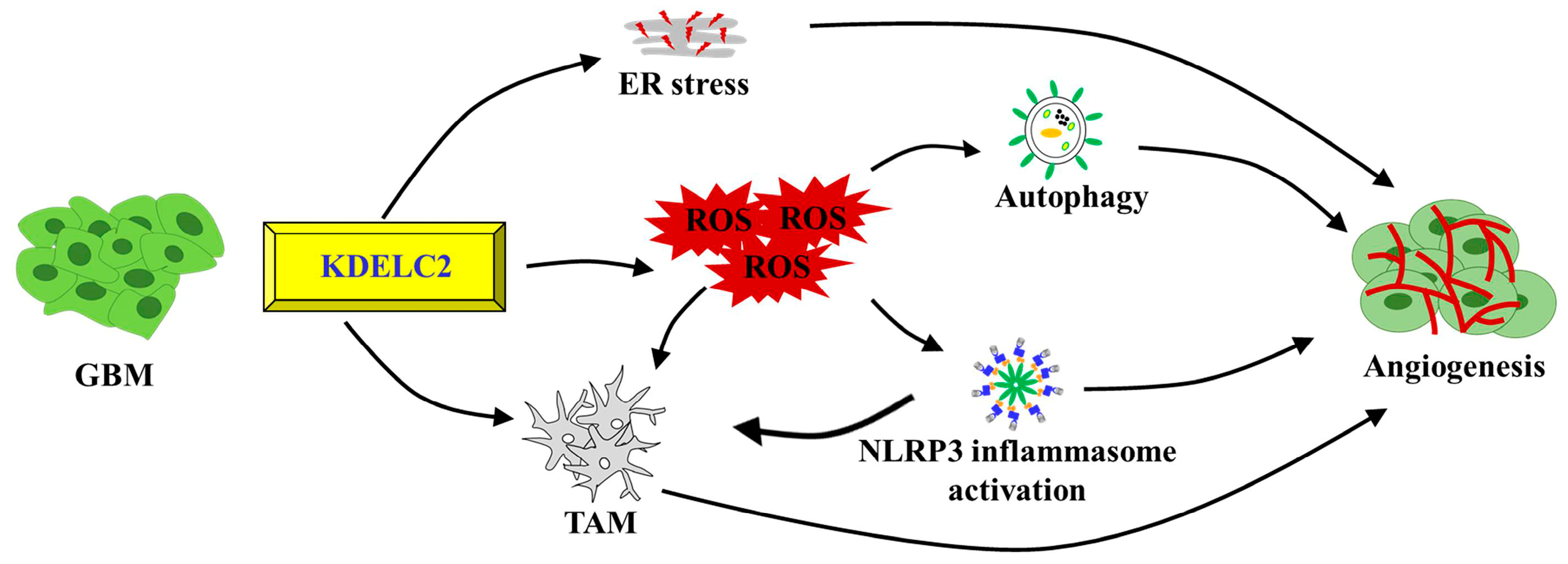
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncology 2005, 7, 134–153. [Google Scholar] [CrossRef] [Green Version]
- Krcek, R.; Matschke, V.; Theis, V.; Adamietz, I.A.; Bühler, H.; Theiss, C. Vascular Endothelial Growth Factor, Irradiation, and Axitinib Have Diverse Effects on Motility and Proliferation of Glioblastoma Multiforme Cells. Front. Oncol. 2017, 7, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardizzone, A.; Scuderi, S.A.; Giuffridam, D.; Colarossi, C.; Puglisi, C.; Campolo, M.; Cuzzocrea, S.; Esposito, E.; Paterniti, I. Role of Fibroblast Growth Factors Receptors (FGFRs) in Brain Tumors, Focus on Astrocytoma and Glioblastoma. Cancers 2020, 12, 3825. [Google Scholar] [CrossRef]
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O’Rourke, D.M.; Gong, Y.; et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 2018, 9, 3439. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Sulman, E.P.; Mehta, M.P. Bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 2048–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Cai, H.; Liu, W.; Liu, X.; Li, Z.; Feng, T.; Xue, Y.; Liu, Y. Advances and Prospects of Vasculogenic Mimicry in Glioma: A Potential New Therapeutic Target? OncoTargets Ther. 2020, 13, 4473–4483. [Google Scholar] [CrossRef]
- Chen, X.; Fang, J.; Wang, S.; Liu, H.; Du, X.; Chen, J.; Li, X.; Yang, Y.; Zhang, B.; Zhang, W. A new mosaic pattern in glioma vascularization: Exogenous endothelial progenitor cells integrating into the vessels containing tumor-derived endothelial cells. Oncotarget 2014, 5, 1955–1968. [Google Scholar] [CrossRef]
- Guarnaccia, L.; Navone, S.E.; Trombetta, E.; Cordiglieri, C.; Cherubini, A.; Crisà, F.M.; Rampini, P.; Miozzo, M.; Fontana, L.; Caroli, M.; et al. Angiogenesis in human brain tumors: Screening of drug response through a patient-specific cell platform for personalized therapy. Sci. Rep. 2018, 8, 8748. [Google Scholar] [CrossRef] [Green Version]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef] [PubMed]
- Alhayaza, R.; Haque, E.; Karbasiafshar, C.; Sellke, F.W.; Abid, M.R. The Relationship Between Reactive Oxygen Species and Endothelial Cell Metabolism. Front. Chem. 2020, 8, 592688. [Google Scholar] [CrossRef]
- Takeuchi, H.; Schneider, M.; Williamson, D.B.; Ito, A.; Takeuchi, M.; Handford, P.A.; Haltiwanger, R.S. Two novel protein O-glucosyltransferases that modify sites distinct from POGLUT1 and affect Notch trafficking and signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E8395–E8402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.L.; Chang, H.H.; Chen, Y.C.; Chang, Y.C.; Chen, Y.; Tsai, W.C. Molecular Mechanisms of KDELC2 on Glioblastoma Tumorigenesis and Temozolomide Resistance. Biomedicines 2020, 8, 339. [Google Scholar] [CrossRef] [PubMed]
- Kofler, N.M.; Shawber, C.J.; Kangsamaksin, T.; Reed, H.O.; Galatioto, J.; Kitajewski, J. Notch signaling in developmental and tumor angiogenesis. Genes Cancer 2011, 2, 1106–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.B.; Mozaffari, K.; Aguirre, B.; Li, V.; Kubba, R.; Desai, N.C.; Wei, D.; Yang, I.; Wadehra, M. Exploring the Past, Present, and Future of Anti-Angiogenic Therapy in Glioblastoma. Cancers 2023, 15, 830. [Google Scholar] [CrossRef]
- Yang, Y.P.; Chien, C.S.; Yarmishyn, A.A.; Chan, M.S.; Lee, A.C.L.; Chen, Y.W.; Huang, P.I.; Ma, H.I.; Lo, W.L.; Chien, Y.; et al. Musashi-1 Regulates MIF1-Mediated M2 Macrophage Polarization in Promoting Glioblastoma Progression. Cancers 2021, 13, 1799. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Unfolded protein response. Curr. Biol. 2012, 22, R622–R626. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Yin, X.F.; Zhang, Q.; Chen, Z.Y.; Wang, H.F.; Li, X.; Wang, H.X.; Li, H.X.; Kang, C.M.; Chu, S.; Li, K.F.; et al. NLRP3 in human glioma is correlated with increased WHO grade, and regulates cellular proliferation, apoptosis and metastasis via epithelial-mesenchymal transition and the PTEN/AKT signaling pathway. Int. J. Oncol. 2018, 53, 973–986. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, X.; Chun, J.; Vilaysane, A.; Clark, S.; French, G.; Bracey, N.A.; Trpkov, K.; Bonni, S.; Duff, H.J.; et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J. Immunol. 2013, 190, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Selvakumaran, M.; Amaravadi, R.K.; Vasilevskaya, I.A.; O’Dwyer, P.J. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin. Cancer Res. 2013, 19, 2995–3007. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2021, 12, 775758. [Google Scholar] [CrossRef]
- Caffo, M.; Caruso, G.; Fata, G.L.; Barresi, V.; Visalli, M.; Venza, M.; Venza, I. Heavy metals and epigenetic alterations in brain tumors. Curr. Genomics 2014, 15, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.T.; Shen, S.C.; Chow, J.M.; Lin, C.W.; Shia, L.T.; Chen, Y.C. Contribution of reactive oxygen species to migration/invasion of human glioblastoma cells U87 via ERK-dependent COX-2/PGE(2) activation. Neurobiol. Dis. 2010, 37, 118–122. [Google Scholar] [CrossRef]
- Benhar, M. Oxidants, Antioxidants and Thiol Redox Switches in the Control of Regulated Cell Death Pathways. Antioxidants 2020, 9, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, M.; Caffo, M.; Minutoli, L.; Marini, H.; Abbritti, R.V.; Squadrito, F.; Trichilo, V.; Valenti, A.; Barresi, V.; Altavilla, D.; et al. ROS and Brain Gliomas: An Overview of Potential and Innovative Therapeutic Strategies. Int. J. Mol. Sci. 2016, 17, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tudek, B.; Winczura, A.; Janik, J.; Siomek, A.; Foksinski, M.; Oliński, R. Involvement of oxidatively damaged DNA and repair in cancer development and aging. Am. J. Transl. Res. 2010, 2, 254–284. [Google Scholar] [PubMed]
- Shi, T.; Dansen, T.B. Reactive Oxygen Species Induced p53 Activation: DNA Damage, Redox Signaling, or Both? Antioxid. Redox Signal 2020, 33, 839–859. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Neill, D.R.; McKenzie, A.N. Nuocytes and beyond: New insights into helminth expulsion. Trends Parasitol. 2011, 27, 214–221. [Google Scholar] [CrossRef]
- Alexandre, Y.O.; Devi, S.; Park, S.L.; Mackay, L.K.; Heath, W.R.; Mueller, S.N. Systemic Inflammation Suppresses Lymphoid Tissue Remodeling and B Cell Immunity during Concomitant Local Infection. Cell Rep. 2020, 33, 108567. [Google Scholar] [CrossRef]
- Shim, D.W.; Lee, K.H. Posttranslational Regulation of the NLR Family Pyrin Domain-Containing 3 Inflammasome. Front. Immunol. 2018, 9, 1054. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.; Seo, W.; Silwal, P.; Jo, E.K. Crosstalks between inflammasome and autophagy in cancer. J. Hematol. Oncol. 2020, 13, 100. [Google Scholar] [CrossRef]
- Zhao, S.; Chen, F.; Yin, Q.; Wang, D.; Han, W.; Zhang, Y. Reactive Oxygen Species Interact With NLRP3 Inflammasomes and Are Involved in the Inflammation of Sepsis: From Mechanism to Treatment of Progression. Front. Physiol. 2020, 11, 571810. [Google Scholar] [CrossRef]
- Peker, N.; Gozuacik, D. Autophagy as a Cellular Stress Response Mechanism in the Nervous System. J. Mol. Biol. 2020, 432, 2560–2588. [Google Scholar] [CrossRef]
- Li, X.; Lu, Q.; Xie, W.; Wang, Y.; Wang, G. Anti-tumor effects of triptolide on angiogenesis and cell apoptosis in osteosarcoma cells by inducing autophagy via repressing Wnt/β-Catenin signaling. Biochem. Biophys. Res. Commun. 2018, 496, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Du, J.H.; Li, X.; Li, R.; Cheng, B.X.; Kuerbanjiang, M.; Ma, L. Role of Autophagy in Angiogenesis Induced by a High-Glucose Condition in RF/6A Cells. Ophthalmologica 2017, 237, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Rizvi, S.F.; Parveen, S.; Pathak, N.; Nazir, A.; Mir, S.S. Crosstalk Between ROS and Autophagy in Tumorigenesis: Understanding the Multifaceted Paradox. Front. Oncol. 2022, 12, 852424. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Gao, L.; Loveless, J.; Shay, C.; Teng, Y. Targeting ROS-Mediated Crosstalk between Autophagy and Apoptosis in Cancer. Adv. Exp. Med. Biol. 2020, 1260, 1–12. [Google Scholar] [PubMed]
- Kim, D.; Kim, K.A.; Kim, J.H.; Kim, E.H.; Bae, O.N. Methylglyoxal-Induced Dysfunction in Brain Endothelial Cells via the Suppression of Akt/HIF-1α Pathway and Activation of Mitophagy Associated with Increased Reactive Oxygen Species. Antioxidants 2020, 9, 820. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Tiainen, S.; Masarwah, A.; Oikari, S.; Rilla, K.; Hämäläinen, K.; Sudah, M.; Sutela, A.; Vanninen, R.; Ikonen, J.; Tammi, R.; et al. Tumor microenvironment and breast cancer survival: Combined effects of breast fat, M2 macrophages and hyaluronan create a dismal prognosis. Breast Cancer Res. Treat 2020, 179, 565–575. [Google Scholar] [CrossRef]
- Kashfi, K.; Kannikal, J.; Nath, N. Macrophage Reprogramming and Cancer Therapeutics: Role of iNOS-Derived NO. Cells 2021, 10, 3194. [Google Scholar] [CrossRef]
- Geindreau, M.; Bruchard, M.; Vegran, F. Role of Cytokines and Chemokines in Angiogenesis in a Tumor Context. Cancers 2022, 14, 2446. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; He, F.; Feng, F.; Liu, X.W.; Dong, G.Y.; Qin, H.Y.; Hu, X.B.; Zheng, M.H.; Liang, L.; Feng, L.; et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010, 70, 4840–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.; Lewis, C.E. Macrophage migration and gene expression in response to tumor hypoxia. Int. J. Cancer 2005, 117, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020, 5, 108. [Google Scholar] [CrossRef]
- Huang, R.; Dong, R.; Wang, N.; He, Y.; Zhu, P.; Wang, C.; Lan, B.; Gao, Y.; Sun, L. Adaptive Changes Allow Targeting of Ferroptosis for Glioma Treatment. Cell Mol. Neurobiol. 2022, 42, 2055–2074. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kalimuthu, K.; Seok Park, Y.; Makala, H.; Watkins, S.C.; Choudry, M.H.A.; Bartlett, D.L.; Tae Kwon, Y.; Lee, Y.J. Ferroptotic agent-induced endoplasmic reticulum stress response plays a pivotal role in the autophagic process outcome. J. Cell Physiol. 2020, 235, 6767–6778. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, Y.; Chen, C.; Sui, X.; Yang, J.; Wang, L.; Zhou, J. The crosstalk between autophagy and ferroptosis: What can we learn to target drug resistance in cancer? Cancer Biol. Med. 2019, 16, 630–646. [Google Scholar] [CrossRef]
- Liu, Y.; Duan, C.; Dai, R.; Zeng, Y. Ferroptosis-mediated Crosstalk in the Tumor Microenvironment Implicated in Cancer Progression and Therapy. Front. Cell Dev. Biol. 2021, 9, 739392. [Google Scholar] [CrossRef]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, Y.-L.; Chen, Y.; Chen, Y.-C.; Tsai, W.-C. KDELC2 Upregulates Glioblastoma Angiogenesis via Reactive Oxygen Species Activation and Tumor-Associated Macrophage Proliferation. Antioxidants 2023, 12, 923. https://doi.org/10.3390/antiox12040923
Tsai Y-L, Chen Y, Chen Y-C, Tsai W-C. KDELC2 Upregulates Glioblastoma Angiogenesis via Reactive Oxygen Species Activation and Tumor-Associated Macrophage Proliferation. Antioxidants. 2023; 12(4):923. https://doi.org/10.3390/antiox12040923
Chicago/Turabian StyleTsai, Yu-Ling, Ying Chen, Ying-Chuan Chen, and Wen-Chiuan Tsai. 2023. "KDELC2 Upregulates Glioblastoma Angiogenesis via Reactive Oxygen Species Activation and Tumor-Associated Macrophage Proliferation" Antioxidants 12, no. 4: 923. https://doi.org/10.3390/antiox12040923