
The Effect of Bioactive Aliment Compounds and Micronutrients on Non-Alcoholic Fatty Liver Disease

Abstract
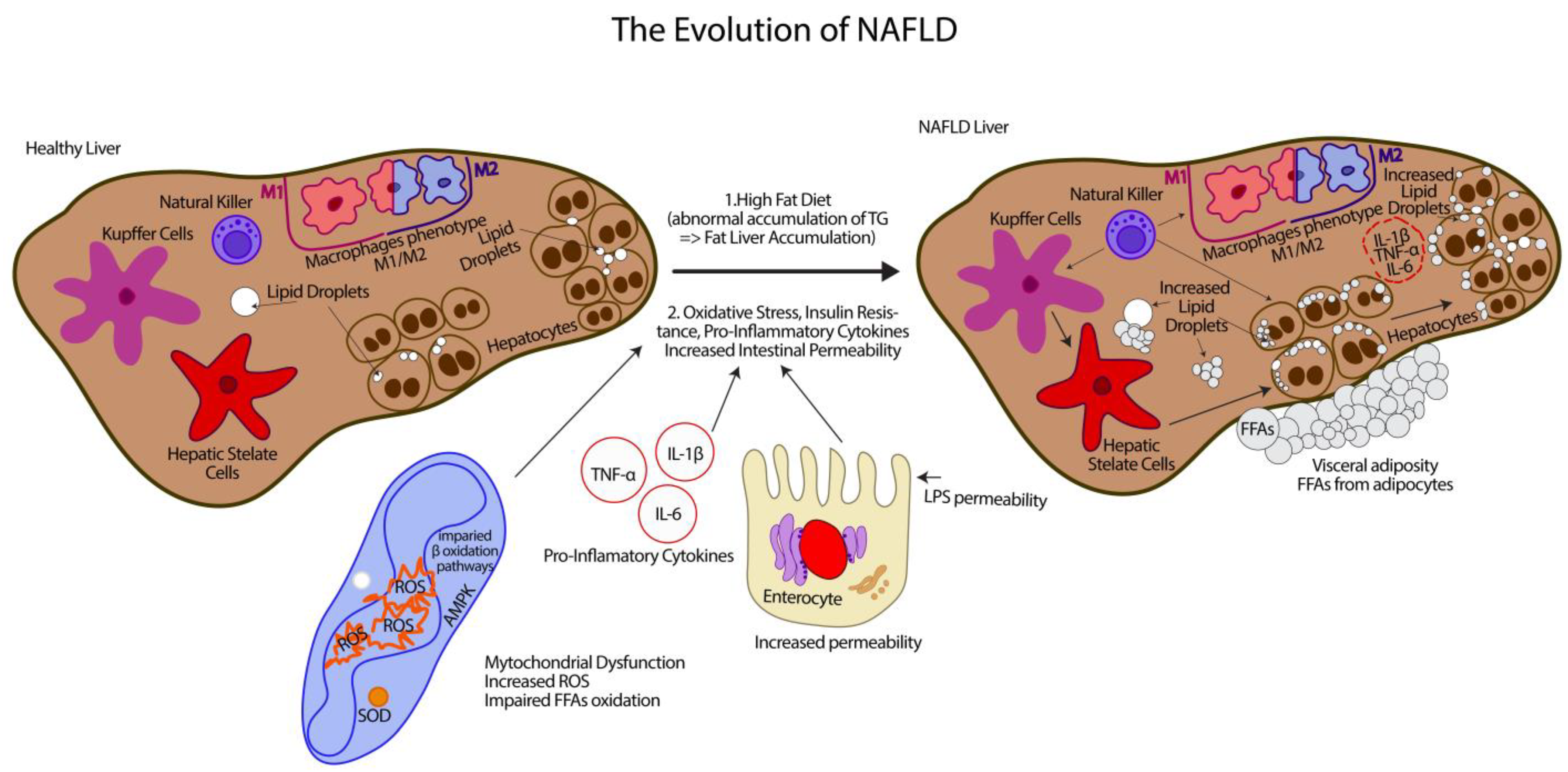
:1. Non-Alcoholic Fatty Liver Disease
2. Potential Bioactive Nutrients That May Interfere with NAFLD
2.1. Dark Chocolate
2.2. Cocoa Butter
2.3. Peanut Butter
2.4. Caffeine
3. Sweeteners
3.1. Stevia
3.2. Sucralose and Saccharin
3.3. Maltitol
3.4. Erythritol
4. Glutathione and NAFLD
5. Whole Milk or Low-Fat Milk for Fatty Liver
6. Soluble Dietary Fiber (FDS)

{kind=link}
{kind=link}
| Nutrients/Category | Effects on Liver | Effects on Intestinal Microbiota | References |
|---|---|---|---|
| Dark chocolate | 1. Positive effects on the lipid profile, reducing total and LDL cholesterol levels and increasing HDL levels | [36] | |
| 2. Improve insulin resistance through reducing oxidative stress, improving endothelial function, and/or altering glucose metabolism | [39] | ||
| 3. Decrease aspartate aminotransferase (AST) levels in the serum of NAFLD patients | [44] | ||
| 4. Increase glucose uptake, increase fatty acid and glucose oxidation, inhibit lipid synthesis | [45] | ||
| 5. Anti-inflammatory properties, which can regulate the TNF-κB gene expression and reduce inflammatory biomarkers and ROS production | [50] | ||
| Cocoa butter | 1. Lowers cholesterol LDL levels and increases cholesterol HDL levels | [56] | |
| Peanut butter | 1. Lowers cholesterol LDL levels and increases protective cholesterol HDL levels. | [67] | |
| 2. Prevents cell damage and induces cell repair, effects associated with reduced risk of chronic diseases such as NAFLD | [59,61] | ||
| Caffeine | 1. Lowers the risk of NAFLD in healthy people | [75,77] | |
| 2. Reduces the risk of developing fibrosis | |||
| Sweeteners | |||
| Stevia | 1. Plays a role in glucose metabolism and has even been reported to improve the postprandial glucose–insulin index | [87] | |
| 2. Significant improvement of liver enzymes blood levels, improvement of liver steatosis and liver fibrosis | 1. Causes changes in the composition of the microbiome. | [88] | |
| 3. Decreased inflammation associated with oxidative stress | 2. An inverse relationship with Akkermansia abundance associated with body weight of mice and humans | [96] | |
| 4. Lower gene expression related to oxidative stress. Improve fasting glucose levels and improve insulin sensitivity | |||
| Sucralose and saccharin | 1. Sucralose may cause dysbiosis by reducing the total number of aerobic and anaerobic species, bifidobacteria, lactobacilli, Bacteriodes, and Clostridiales | [102] | |
| 2. Saccharin may inhibit the growth of six bacterial strains: three species of lactobacilli and three strains of E. coli in animal models | [95] | ||
| 3. Associations between the consumption of sweeteners and a disturbed microbiota | [98] | ||
| Maltitol | 1.Suppress cholesterol synthesis in the liver leading to a decrease in circulating cholesterol levels | [106,107] | |
| 2. Maltitol mimics indigestible fibers, absorbs bile acids in the intestine, and reduces circulating bile acid levels, which leads to the activation of bile receptors in the liver and an increase in circulating bile acid levels | [108] | ||
| 3. Prevents obesity, hyperglycemia, hypercholesterolemia, and fatty liver degeneration in mice fed a high-fat diet | [109] | ||
| Erythritol | 1. Long-term administration of Ery has no effect on body weight and glucose tolerance of young/adolescent mice | [110] | |
| 2. Alleviate metabolic disorders in mice induced by a high-fat diet (HFD), including dyslipidemia, impaired glucose tolerance, and obesity | [111] | ||
| 3. Inhibits hepatic lipid accumulation and alleviate hepatic oxidative damage in HepG2 cells induced by fatty acid treatment and in high-fat diet-induced NAFLD models | [113] | ||
| 4. Exerts an antioxidant function by activating the Nrf2 signaling pathway, thus inhibiting endoplasmic reticulum stress and lipid accumulation and then playing a role in alleviating NAFLD. | |||
| Soluble dietary fiber (FDS) | 1.Improvement caused by the administration of fructo-oligosaccharide (FOS) in improving NASH disease in mice | 1. Improvement of intestinal barrier function | [139] |
| 2. Regulate the accumulation of lipids in the liver | [141] | ||
| 3. Inhibit fat accumulation in adipose tissue as well as promote lipid and glucose metabolism in the liver | [144] | ||
| 4. Provide potential ligands for the peroxisome proliferator-activated receptor γ- (PPARγ), and as a result can result in improved insulin sensitivity | [145] | ||
| 5. Decrease expression of pro-inflammatory markers such as interleukin-6 and nuclear factor-kappa-beta (NF-κB), thereby raising the threshold for inflammatory reactions in the liver of rats fed a high-fat diet | [146] |
7. Soy Lecithin as a Source for Choline and Inositol
8. Turmeric and Curcumin Extracts
9. Silymarin
10. Selenium (Se)
10.1. Evidence Suggesting a Beneficial Effect of Se on NAFLD
10.2. Evidence Suggesting a Negative Effect of Se Administration on NAFLD
11. The Enzyme Stearoyl-CoA Desaturase 1 (SCD1) in NAFLD and the Use of Supplements That Lower Its Activity
Adverse Effects of SCD1 Inhibitor Treatments
12. Sterculic Acid (SA)
13. Aquamin
14. Oleic Acid
| Compounds | Effects on Liver | Effects on Intestinal Microbiota | References | |
|---|---|---|---|---|
| Glutathione | 1. Therapeutic effect of oral glutathione in patients with NAFLD | [121] | ||
| 2. Improvement of ALT blood levels | [122] | |||
| 3. Decrease in triglycerides, NEFAs, and ferritin levels | [127] | |||
| 4. Lowers protein-bound glutathione to normal basal levels | ||||
| 5. Improvement of hyperferritinemia and oxidative stress, and exertion of therapeutic effects in patients with NAFLD | ||||
| Soy lecithin as a source of choline and inositol | 1. Lecithin in the context of NAFLD is considered one of the most important sources of choline and inositol | [151] | ||
| 2. Choline, phosphatidylcholine, and lecithin are associated with the prevention of the development of fatty liver | [152] | |||
| 3. Choline and betaine have been shown in animal and human studies to prevent and even ameliorate NAFLD | [155] | |||
| 4. Lecithin ensures adequate TG export from the liver | [157] | |||
| Turmeric and curcumin extracts | 1. Antioxidant, anti-inflammatory, and anti-fibrotic properties, as well as insulin-sensitizing effects | 1. Probiotic-like effects | [158] | |
| 2. Induces increased energy metabolism of adipocytes and/or induction of apoptosis, increased expression of neseptin levels in serum | [165,167] | |||
| 3. Loss of appetite, reduction in body fat, anti-inflammatory activities, anti-hyperglycemic activity, metabolic and neuroendocrine regulation | [168,169] | |||
| 4. Reduces body fat mass by inhibiting adipocyte differentiation through suppression of peroxisome proliferator-activated receptor-γ and by increasing adenosine monophosphate-activated protein kinase resulting in lipolysis | [174] | |||
| 5. Enhances the activities of detoxifying enzymes such as glutathione-S-transferase, glutathione peroxidase, glutathione reductase, catalase, and he-oxygenase-1 in the liver and thus suppresses oxidative stress in the liver | [179,180,181] | |||
| 6. Blocks the activation of key mediators of cellular inflammation such as NF-κB, 5-lipoxygenase (5-LOX), and cyclooxygenase-2 (COX-2) | [182,183] | |||
| 7. Inhibits the activation and proliferation of stellate cells in the liver, which have a known role in the progression of liver fibrosis | [184] | |||
| Silymarin | 1. Anti-inflammatory, antioxidant, and anti-fibrotic activity | [188] | ||
| 2. Reduces insulin resistance | [193] | |||
| 3. Reduces liver inflammation by inhibiting lipooxygenase activity and reducing the function of leukotrienes and their effect on Kupffer cells in the liver and by reducing oxidative stress by increasing glutathione levels | ||||
| 4. Improvement of serum superoxide dismutase activity and malondaldehyde (MDA) levels in rats in which NAFLD was induced | [192] | |||
| 5. Reduce serum aspartate aminotransferase enzyme levels and levels of triglycerides (TGs) and cholesterol including VLDL in NAFLD-induced rats | ||||
| Sterculic acid (SA) | 1. Improvement in glucose tolerance and blood pressure, reduction in body mass, and benefit in serum levels of triglycerides and adiponectin | [228] | ||
| Aquamin [241] | 1. Prevents and can even help stop the progression of NAFLD | 1. Regulates expression of several proteins related to cell differentiation in the colon mucosa | [235,241] | |
| 2. Reduce the formation of tumors | [236,237] | |||
| 3. Reduces inflammation, damage to hepatocytes, and the appearance of collagen deposits | 2. Change the intestinal bacterial profile | [241] | ||
| Oleic acid | 1. Prevents the accumulation of lipids in the liver | [248] | ||
| 2. De novo synthesized oleic acid contributes to the protection of hepatocytes against insulin resistance | ||||
| 3. Modulates the activity of liver X receptors (LXRs) | [249] | |||
| 4. Protects against the cytotoxic activity caused by treatment with palmitic acid-induced steatosis in primary rat hepatocytes in culture. | [259] | |||
| Bilirel (BIL) | 1. Rapid improvement in liver fat accumulation, improvement in glucose levels and metabolism | [260] | ||
| Cannabinoids | 1. Decreases fibrosis | [261] | ||
| 2. Stimulation of adipocyte metabolism | [262] | |||
| 3. Improvement on the insulin–glucose circuit and inhibition of weight gain | [263] | |||
| 4. Suppresses the development of NAFLD | [264,265,266] | |||
| 5. Decreases hepatic TG synthesis, as does VLDL synthesis, and increases insulin sensitivity |
15. Antioxidants and NAFLD
15.1. Oxidative Stress and NAFLD
15.2. Antioxidants Effects on NAFLD
15.2.1. Bilirel (BIL)
15.2.2. Additional Antioxidants
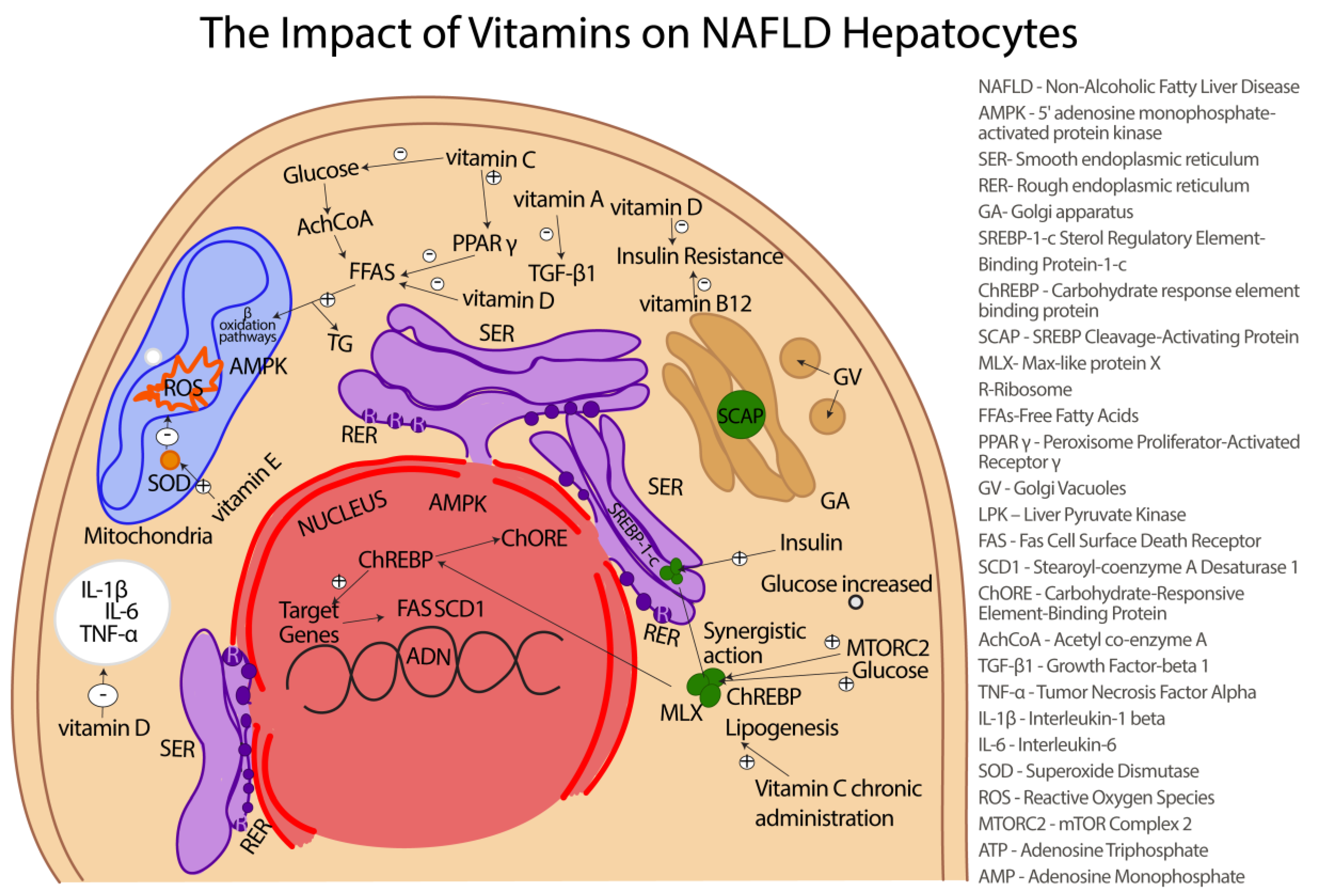
16. Vitamins with Antioxidant Activity in NAFLD
16.1. Vitamin A and NAFLD
16.2. Vitamin C and NAFLD
16.3. Vitamin E and NAFLD
16.4. Vitamin D and NAFLD
16.5. Vitamin B12 and NAFLD
| Type of Micronutrient | Effects on Liver | Effects on Intestinal Microbiota | References |
|---|---|---|---|
| Se | 1. Administration of a combination of Se and zinc improved the lipid profile, liver functions, and liver steatosis in rats | [206] | |
| 2. Se and probiotics reversed the negative effect of feeding in mice on HFD and improved liver functions and steatosis | [207] | ||
| 3. Decreases the number of hepatic stellate cells (HSCs) and liver fibrosis induced by CCl4 treatment in mice | [209] | ||
| Vitamin A | 1. Retinoic acid (RA) administration has been shown to be an effective antioxidant by reducing mitochondrial ROS and by increasing SOD2 in mice | [282] | |
| 2. Protects the liver against hepatic steatosis itself, as well as against liver damage in NAFLD populations | [283] | ||
| 3. Reduces the release of transforming growth factor beta 1 (TGF-β1) | [285] | ||
| 4. Suppresses the activation of hepatic stellate cells and fibrogenesis | |||
| Vitamin C | 1. Reduces hepatic fatty acid load by promoting the gene expression of PPARα-dependent β-fatty acid genes in HFD-induced NAFLD mice | [286] | |
| 2. Attenuates steatosis and NAFLD in mice | |||
| 3. Improves adiponectin levels and reduces liver TG levels and thus prevents NASH progression in NAFLD patients | [288] | ||
| 4. Significant inverse association between ingested Vitamin C and NAFLD | [289] | ||
| Vitamin E | 1. Normalizes cholesterol metabolism and reduces inflammation and fibrosis associated with oxidative stress | [292] | |
| 2. Attenuates fructose diet-induced NAFLD by activating the Nrf2/carboxylesterase 1 pathway involved in lipogenesis | [293] | ||
| 3. Is considered an effective inhibitor of steatohepatitis | [295] | ||
| 4. Inhibits the expression of genes responsible for fibrosis, inflammation, and apoptosis | [296] | ||
| 5. Improves the blood levels of ALT and AST | [299] | ||
| 6. Responsible for regulating the cellular signaling of different enzymes essential in molecular signal translation, such as 5-lipoxygenase, cyclooxygenase-2 (COX-2), protein kinase C (PKC), and protein phosphate 2A (PP2A) | [300] | ||
| 7. Stimulates the expression of adiponectin | [302] | ||
| 8. Induces favorable modifications in intestinal disturbed microbiota by the increases of portal LPS | |||
| Vitamin D | 1. Protects the liver against the inflammation induced by different chronic hepatitises | [306] | |
| 2. Increases insulin sensitivity | [307] | ||
| 3. Induces anti-fibrotic, anti-inflammatory, and anti-cirrhotic properties | [308] | ||
| 4. Decreases secretion of the pro-inflammatory cytokines IL-1β and IL-6 | [314] | ||
| 5. Decreases TG levels in NAFLD patients | [315] | ||
| 6. Improves blood lipid profile | [317] | ||
| Vitamin B12 | 1. Affects the disruption of mitochondrial metabolism, involved in the pathology of NAFLD | [318] | |
| 2. Therapeutic effects regarding the pathology of NAFLD | [320] | ||
| 3. Significantly decreases the serum concentration of MDA | [322] | ||
| 4. Reduces fasting blood glucose (FBG) | [323] |
17. Bile Acids and NAFLD
18. Imbalance of the Intestinal Microbiota, Choline Metabolism, and NAFLD
19. The Activity of the Enzyme AMPK (5′ Adenosine Monophosphate-Activated Protein Kinase) in NAFLD
| Type of Enzyme | Positive Effects of Enzymes and/or Supplements on Liver | Adverse Effects of Enzymes Inhibitor Treatments | References |
|---|---|---|---|
| Stearoyl-CoA desaturase 1 (SCD1) inhibitors | 1. SCD1 inhibitors are associated with amelioration of NAFLD, diabetes, dyslipidemic failure, and hepatitis C virus infections | 1. Knockout mice develop multiple skin eczemas accompanied by weight disturbances [223] | [220,223] |
| 2. SCD1 inhibitors reduces the accumulation of triglycerides in the liver of rats suffering from NASH by 80% | 2. Failure of thermoregulation, transepidermal water loss, and metabolic problems [224] | [222] | |
| 3. Attenuate the increase in aspartate aminotransferase (AST) enzyme levels and alanine transaminase (ALT) by 86% and 78%, respectively | 3. Cause atrophy of sebocyte cells (epithelial cells in the skin), effects reflected in hair loss and dry eyes causation | [226] | |
| AMPK (5′ adenosine monophosphate-activated protein kinase) | 1. Inhibits different signaling pathways such as TNF-α and IL-1β | [359] | |
| 2. Suppresses inflammation | [361] | ||
| 3. Inhibits the transcription of lipogenic genes | [364] | ||
| 4. Improves NAFLD by stimulating metabolism at the mitochondrial level and intensifying the oxidation of fatty acids in the liver |
20. Cannabinoids and NAFLD
21. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pouwels, S.; Sakran, N.; Graham, Y.; Leal, A.; Pintar, T.; Yang, W.; Kassir, R.; Singhal, R.; Mahwar, K.; Ramnarain, D. Non-alcoholic fatty liver disease (NAFLD): A review of pathophysiology, clinical management and effects of weight loss. BMC Endocr. Disord. 2022, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Townsend, S.A.; Newsome, P.N. Non-alcoholic fatty liver disease in 2016. Br. Med. Bull. 2016, 119, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Tian, X.; Wu, H.; Huang, J.; Li, M.; Mei, Z.; Zhou, L.; Xie, H.; Zheng, S. Metabolic changes of hepatocytes in NAFLD. Front. Physiol. 2021, 12, 710420. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.S.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Bedossa, P. Diagnosis of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: Why liver biopsy is essential. Liver Int. 2018, 38, 64–66. [Google Scholar] [CrossRef] [Green Version]
- Reccia, I.; Kumar, J.; Akladios, C.; Virdis, F.; Pai, M.; Habib, N.; Spalding, D. Non-alcoholic fatty liver disease: A sign of systemic disease. Metabolism 2017, 72, 94–108. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism 2016, 65, 1109–1123. [Google Scholar] [CrossRef]
- Rinaldi, L.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Morone, M.V.; Silvestri, C.; Giordano, M.; Salvatore, T.; Sasso, F.C. Mechanisms of non-alcoholic fatty liver disease in the metabolic syndrome. A narrative review. Antioxidants 2021, 10, 270. [Google Scholar] [CrossRef]
- Martins, I.J. Caffeine with links to NAFLD and accelerated brain aging. In Non-Alcoholic Fatty Liver Disease-Molecular Bases, Prevention and Treatment; IntechOpen: London, UK, 2017. [Google Scholar]
- Perumpail, B.J.; Khan, M.A.; Yoo, E.R.; Cholankeril, G.; Kim, D.; Ahmed, A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 8263. [Google Scholar] [CrossRef]
- Tiwari, P. Recent trends in therapeutic approaches for diabetes management: A comprehensive update. J. Diabetes Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qui, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef]
- Dai, W.; Ye, L.; Liu, A.; Wen, S.W.; Deng, J.; Wu, X.; Lai, Z. Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus. Medicine 2017, 96, e8179. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of nonalcoholic steatohepatitis: An overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Henry, L. Epidemiology of non-alcoholic fatty liver disease and hepatocellular carcinoma. JHEP Rep. 2021, 3, 100305. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic fatty liver disease and insulin resistance: New insights and potential new treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Lee, G.; Heo, S.Y.; Roh, Y.S. Oxidative stress is a key modulator in the development of nonalcoholic fatty liver disease. Antioxidants 2021, 11, 91. [Google Scholar] [CrossRef]
- Arrese, M.; Arab, J.P.; Barrera, F.; Kaufmann, B.; Valenti, L.; Feldstein, A.E. Insights into nonalcoholic fatty-liver disease heterogeneity. In Seminars in Liver Disease; Thieme Medical Publishers Inc.: Leipzig, Germany, 2021; Volume 41, pp. 421–434. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.Q.-H.; Wang, D.; Sperandio, M.; Di Ciaula, A. Gut microbiota and short chain fatty acids: Implications in glucose homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef]
- Wells, J.M.; Brummer, R.J.; Derrien, M.; MacDonald, T.T.; Troost, F.; Cani, P.D.; Theodorou, V.; Dekker, J.; Meheust, A.; de Vos, W.M.; et al. Homeostasis of the gut barrier and potential biomarkers. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G171–G193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. 2019, 1, 30–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecher, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Gebru, Y.A.; Gupta, H.; Kim, H.S.; Eom, J.A.; Kwon, G.H.; Park, E.; Jeong, J.J.; Won, S.M.; Sharma, S.P.; Ganesan, R.; et al. T Cell Subsets and Natural Killer Cells in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 22, 12190. [Google Scholar] [CrossRef]
- Jalil, A.M.M.; Ismail, A. Polyphenols in cocoa and cocoa products: Is there a link between antioxidant properties and health? Molecules 2008, 13, 2190–2219. [Google Scholar] [CrossRef] [Green Version]
- Petyaev, I.M.; Bashmakov, Y.K. Dark Chocolate: Opportunity for an Alliance between Medical Science and the Food Industry? Front. Nutr. 2017, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, F.M.; Bearden, M.M.; Keen, C.L. Cocoa and chocolate flavonoids: Implications for cardiovascular health. J. Am. Diet. Assoc. 2003, 103, 215–223. [Google Scholar] [CrossRef]
- Barišić, V.; Kopjar, M.; Jozinović, A.; Flanjak, I.; Ačkar, Đ.; Miličević, B.; Šubarić, D.; Jokić, S.; Babić, J. The chemistry behind chocolate production. Molecules 2019, 24, 3163. [Google Scholar] [CrossRef] [Green Version]
- Katz, D.L.; Doughty, K.; Ali, A. Cocoa and chocolate in human health and disease. Antioxid. Redox Signal. 2011, 15, 2779–2811. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.W.; Kim, Y.J.; Lee, H.J.; Lee, C.Y. Cocoa has more phenolic phytochemicals and a higher antioxidant capacity than teas and red wine. J. Agric. Food Chem. 2003, 51, 7292–7295. [Google Scholar] [CrossRef]
- Tan, T.Y.C.; Lim, X.Y.; Yeo, J.H.H.; Lee, S.W.H.; Lai, N.M. The health effects of chocolate and cocoa: A systematic review. Nutrients 2021, 13, 2909. [Google Scholar] [CrossRef]
- de Paula Silva, T.; Silva, A.A.; Toffolo, M.C.F.; de Aguiar, A.S. The action of phytochemicals present in cocoa in the prevention of vascular dysfunction and atherosclerosis. J. Clin. Transl. Res. 2022, 8, 509. [Google Scholar] [CrossRef]
- Magrone, T.; Russo, M.A.; Jirillo, E. Cocoa and dark chocolate polyphenols: From biology to clinical applications. Front. Immunol. 2017, 8, 677. [Google Scholar] [CrossRef] [Green Version]
- Samanta, S.; Sarkar, T.; Chakraborty, R.; Rebezov, M.; Shariati, M.A.; Thiruvengadam, M.; Rengasamy, K.R. Dark chocolate: An overview of its biological activity, processing, and fortificationapproaches. Curr. Res. Food Sci. 2022, 8, 677. [Google Scholar] [CrossRef]
- Gasmi, A.; Mujawdiya, P.K.; Noor, S.; Lysiuk, R.; Darmohray, R.; Piscopo, S.; Lenchyk, L.; Antonyak, H.; Dehtiarova, K.; Shanaida, M.; et al. Polyphenols in metabolic diseases. Molecules 2022, 27, 6280. [Google Scholar] [CrossRef]
- Janevski, M.; Antonas, K.N.; Sullivan-Gunn, M.J.; McGlynn, M.A.; Lewandowski, P.A. The effect of cocoa supplementation on hepatic steatosis, reactive oxygen species and LFABP in a rat model of NASH. Comp. Hepatol. 2011, 10, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Kwok, C.S.; Boekholdt, S.M.; Lentjes, M.A.; Loke, Y.K.; Luben, R.N.; Yeong, J.K.; Wareham, J.N.; Myint, K.P.; Khaw, K.T. Habitual chocolate consumption and risk of cardiovascular disease among healthy men and women. Heart 2015, 101, 1279–1287. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.Á.; Ramos, S. Impact of cocoa flavanols on human health. Food Chem. Toxicol. 2021, 151, 112121. [Google Scholar] [CrossRef]
- Wiese, M.; Bashmakov, Y.; Chalyk, N.; Nielsen, D.S.; Krych, Ł.; Kot, W.; Klochkov, V.; Pristensky, D.; Bandaletova, T.; Chernyshova, M.; et al. Prebiotic effect of lycopene and dark chocolate on gut microbiome with systemic changes in liver metabolism, skeletal muscles and skin in moderately obese persons. BioMed. Res. Int. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alavinejad, P.; Farsi, F.; Rezazadeh, A.; Mahmoodi, M.; Hajiani, E.; Masjedizadeh, A.R.; Mard Ali, S.; Neisi, N.; Hoseini, H.; Haghighizadeh, M.H.; et al. The effects of dark chocolate consumption on lipid profile, fasting blood sugar, liver enzymes, inflammation, and antioxidant status in patients with non-alcoholic fatty liver disease: A randomized, placebo-controlled, pilot study. J. Gastroenterol. Hepatol. Res. 2015, 4, 1858–1864. [Google Scholar] [CrossRef]
- Massolt, E.T.; Van Haard, P.M.; Rehfeld, J.F.; Posthuma, E.F.; van der Veer, E.; Schweitzer, D.H. Appetite suppression through smelling of dark chocolate correlates with changes in ghrelin in young women. Regul. Pept. 2010, 161, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Matsui, N.; Ito, R.; Nishimura, E.; Yoshikawa, M.; Kato, M.; Kamei, M.; Shibata, H.; Matsumoto, I.; Abe, K.; Hashizume, S. Ingested cocoa can prevent high-fat diet-induced obesity by regulating the expression of genes for fatty acid metabolism. Nutrition 2005, 21, 594–601. [Google Scholar] [CrossRef]
- Hamed, M.S.; Gambert, S.; Bliden, K.P.; Bailon, O.; Singla, A.; Antonino, M.J.; Hamed, F.; Tantry, S.U.; Gurbel, P.A. Dark chocolate effect on platelet activity, C-reactive protein and lipid profile: A pilot study. South. Med. J. 2008, 101, 1203–1208. [Google Scholar] [CrossRef] [Green Version]
- Mellor, D.D.; Sathyapalan, T.; Kilpatrick, E.S.; Beckett, S.; Atkin, S. High-cocoa polyphenol-rich chocolate improves HDL cholesterol in Type 2 diabetes patients. Diabet. Med. 2010, 27, 1318–1321. [Google Scholar] [CrossRef]
- Mathur, S.; Devaraj, S.; Grundy, S.M.; Jialal, I. Cocoa products decrease low density lipoprotein oxidative susceptibility but do not affect biomarkers of inflammation in humans. J. Nutr. 2002, 132, 3663–3667. [Google Scholar] [CrossRef] [Green Version]
- Goya, L.; Martín, M.Á.; Sarriá, B.; Ramos, S.; Mateos, R.; Bravo, L. Effect of cocoa and its flavonoids on biomarkers of inflammation: Studies of cell culture, animals and humans. Nutrients 2016, 8, 212. [Google Scholar] [CrossRef]
- Rector, R.S.; Thyfault, J.P.; Wei, Y.; Ibdah, J.A. Non-alcoholic fatty liver disease and the metabolic syndrome: An update. World J. Gastroenterol. WJG 2008, 14, 185. [Google Scholar] [CrossRef]
- McKim, S.E.; Konno, A.; Gäbele, E.; Uesugi, T.; Froh, M.; Sies, H.; Thurman, G.R.; Arteel, G.E. Cocoa extract protects against early alcohol-induced liver injury in the rat. Arch. Biochem. Biophys. 2002, 406, 40–46. [Google Scholar] [CrossRef]
- Cooper, K.A.; Donovan, J.L.; Waterhouse, A.L.; Williamson, G. Cocoa and health: A decade of research. Br. J. Nutr. 2008, 99, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Quek, R.Y.C.; Peh, E.W.Y.; Henry, C.J. Effects of cocoa butter and cocoa butter equivalent in a chocolate confectionery on human blood triglycerides, glucose and insulin. Foods 2020, 9, 455. [Google Scholar] [CrossRef] [Green Version]
- Ding, E.L.; Hutfless, S.M.; Ding, X.; Girotra, S. Chocolate and prevention of cardiovascular disease: A systematic review. Nutr. Metab. 2006, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Loganathan, R.; Nagapan, G.; Teng, K.T.; Voon, P.T.; Yap, S.Y.; Ng, Y.T.; Choo, M.Y.; Ong, H.S.A.; Ong, H.S.; Selvaduray, K.R. Diets enriched with palm olein, cocoa butter, and extra virgin olive oil exhibited similar lipid response: A randomized controlled study in young healthy adults. Nutr. Res. 2022, 105, 113–125. [Google Scholar] [CrossRef]
- Chang, H.Y.; Chen, J.R.; Chen, Y.H.; Xiao, Q.; Chen, Y.L.; Yang, S.C. The Preliminary Results for Evaluating Cocoa Butter’s Hepatoprotective Effects against Lipid Accumulation and Inflammation in Adult Male Rats Chronically Fed Ethanol. Bioengineering 2022, 9, 526. [Google Scholar] [CrossRef]
- Torres-Moreno, M.; Torrescasana, E.; Salas-Salvadó, J.; Blanch, C. Nutritional composition and fatty acids profile in cocoa beans and chocolates with different geographical origin and processing conditions. Food Chem. 2015, 166, 125–132. [Google Scholar] [CrossRef]
- Arya, S.S.; Salve, A.R.; Chauhan, S. Peanuts as functional food: A review. J. Food Sci. Technol. 2016, 53, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, S.; Khalid, N.; Ahmed, I.; Shahzad, A.; Suleria, H.A.R. Physicochemical characteristics, functional properties, and nutritional benefits of peanut oil: A review. Crit. Rev. Food Sci. Nutr. 2014, 54, 1562–1575. [Google Scholar] [CrossRef]
- Toomer, O.T. Nutritional chemistry of the peanut (Arachis hypogaea). Crit. Rev. Food Sci. Nutr. 2018, 58, 3042–3053. [Google Scholar] [CrossRef]
- Hou, Y.Y.; Ojo, O.; Wang, L.L.; Wang, Q.; Jiang, Q.; Shao, X.Y.; Wang, X.H. A randomized controlled trial to compare the effect of peanuts and almonds on the cardio-metabolic and inflammatory parameters in patients with type 2 diabetes mellitus. Nutrients 2018, 10, 1565. [Google Scholar] [CrossRef] [Green Version]
- Parilli-Moser, I.; Hurtado-Barroso, S.; Guasch-Ferre, M.; Lamuela-Raventos, R.M. Effect of Peanut Consumption on Cardiovascular Risk Factors: A Randomized Clinical Trial and Meta-Analysis. Front. Nutr. 2022, 9, 853378. [Google Scholar] [CrossRef] [PubMed]
- Coates, A.M.; Hill, A.M.; Tan, S.Y. Nuts and cardiovascular disease prevention. Curr. Atheroscler. Rep. 2018, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.S.; Murphy, J.; Whitbread, J.; Clifton, P.M.; Keogh, J.B. The Effect of a Peanut-Enriched Weight Loss Diet Compared to a Low-Fat Weight Loss Diet on Body Weight, Blood Pressure, and Glycemic Control: A Randomized Controlled Trial. Nutrients 2022, 14, 2986. [Google Scholar] [CrossRef] [PubMed]
- Shramko, V.S.; Polonskaya, Y.V.; Kashtanova, E.V.; Stakhneva, E.M.; Ragino, Y.I. The short overview on the relevance of fatty acids for human cardiovascular disorders. Biomolecules 2020, 10, 1127. [Google Scholar] [CrossRef]
- Vassiliou, E.K.; Gonzalez, A.; Garcia, C.; Tadros, J.H.; Chakraborty, G.; Toney, J.H. Oleic acid and peanut oil high in oleic acid reverse the inhibitory effect of insulin production of the inflammatory cytokine TNF-α both in vitro and in vivo systems. Lipids Health Dis. 2009, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Parilli-Moser, I.; Domínguez-López, I.; Arancibia-Riveros, C.; Marhuenda-Muñoz, M.; Vallverdú-Queralt, A.; Hurtado-Barroso, S.; Lamuela-Raventós, R.M. Effect of crushing peanuts on fatty acid and phenolic bioaccessibility: A long-term study. Antioxidants 2022, 11, 423. [Google Scholar] [CrossRef]
- Reis, C.E.; Ribeiro, D.N.; Costa, N.M.; Bressan, J.; Alfenas, R.C.; Mattes, R.D. Acute and second-meal effects of peanuts on glycaemic response and appetite in obese women with high type 2 diabetes risk: A randomised cross-over clinical trial. Br. J. Nutr. 2013, 109, 2015–2023. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.J. Appetite Control with Relevance to Mitochondrial Biogenesis and Activation of Post-Prandial Lipid Metabolism in Obesity Linked Diabetes. Ann. Obes. Disord 2016, 1, 1012. [Google Scholar]
- Martins, I.J. Diabetes and cholesterol dyshomeostasis involve abnormal α-synuclein and amyloid beta transport in neurodegenerative diseases. Austin Alzheimer’s J. Park. Dis. 2015, 2, 1020. [Google Scholar]
- Jessen, A.; Buemann, B.; Toubro, S.; Skovgaard, I.M.; Astrup, A. The appetite-suppressant effect of nicotine is enhanced by caffeine. Diabetes Obes. Metab. 2005, 7, 327–333. [Google Scholar] [CrossRef]
- Martins, I.J. Nutritional and genotoxic stress contributes to diabetes and neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases. In Frontiers in Clinical Drug Research—CNS and Neurological Disorders; Rahman, A., Ed.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2015; Volume 3, pp. 158–192. [Google Scholar]
- Shen, H.; Rodriguez, A.C.; Shiani, A.; Lipka, S.; Shahzad, G.; Kumar, A.; Mustacchia, P. Association between caffeine consumption and nonalcoholic fatty liver disease: A systemic review and meta-analysis. Ther. Adv. Gastroenterol. 2016, 9, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Hayat, U.; Siddiqui, A.A.; Okut, H.; Afroz, S.; Tasleem, S.; Haris, A. The effect of coffee consumption on the non-alcoholic fatty liver disease and liver fibrosis: A meta-analysis of 11 epidemiological studies. Ann. Hepatol. 2021, 20, 100254. [Google Scholar] [CrossRef]
- Shim, S.G.; Jun, D.W.; Kim, E.K.; Saeed, W.K.; Lee, K.N.; Lee, H.L.; Lee, O.Y.; Choi, H.S.; Yoon, B.C. Caffeine attenuates liver fibrosis via defective adhesion of hepatic stellate cells in cirrhotic model. J. Gastroenterol. Hepatol. 2013, 28, 1877–1884. [Google Scholar] [CrossRef]
- Molloy, J.W.; Calcagno, C.J.; Williams, C.D.; Jones, F.J.; Torres, D.M.; Harrison, S.A. Association of coffee and caffeine consumption with fatty liver disease, nonalcoholic steatohepatitis, and degree of hepatic fibrosis. Hepatology 2012, 55, 429–436. [Google Scholar] [CrossRef]
- Mansour, A.; Mohajeri-Tehrani, M.R.; Samadi, M.; Qorbani, M.; Merat, S.; Adibi, H.; Poustchi, H.; Hekmatdoost, A. Effects of supplementation with main coffee components including caffeine and/or chlorogenic acid on hepatic, metabolic, and inflammatory indices in patients with non-alcoholic fatty liver disease and type 2 diabetes: A randomized, double-blind, placebo-controlled, clinical trial. Nutr. J. 2021, 20, 35. [Google Scholar] [CrossRef]
- MacKenzie, T.; Comi, R.; Sluss, P.; Keisari, R.; Manwar, S.; Kim, J.; Larson, R.; Baron, J.A. Metabolic and hormonal effects of caffeine: Randomized, double-blind, placebo-controlled crossover trial. Metab. Clin. Exp. 2007, 56, 1694–1698. [Google Scholar] [CrossRef]
- Thong, F.S.; Derave, W.; Kiens, B.; Graham, T.E.; Ursø, B.; Wojtaszewski, J.F.; Hansen, B.F.; Richter, E.A. Caffeine-induced impairment of insulin action but not insulin signaling in human skeletal muscle is reduced by exercise. Diabetes 2002, 51, 583–590. [Google Scholar] [CrossRef]
- Arnlöv, J.; Vessby, B.; Risérus, U. Coffee consumption and insulin sensitivity. JAMA 2004, 291, 1199–1201. [Google Scholar] [CrossRef]
- van Dam, R.M.; Hu, F.B. Coffee consumption and risk of type 2 diabetes: A systematic review. JAMA 2005, 294, 97–104. [Google Scholar] [CrossRef]
- Peteliuk, V.; Rybchuk, L.; Bayliak, M.; Storey, K.B.; Lushchak, O. Natural sweetener Stevia rebaudiana: Functionalities, health benefits and potential risks. EXCLI J. 2021, 20, 1412. [Google Scholar] [CrossRef]
- Magnuson, B.A.; Carakostas, M.C.; Moore, N.H.; Poulos, S.P.; Renwick, A.G. Biological fate of low-calorie sweeteners. Nutr. Rev. 2016, 74, 670–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwell, M. Stevia, nature’s zero-calorie sustainable sweetener: A new player in the fight against obesity. Nutr. Today 2015, 50, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandel, K.R. Sugar substitutes: Health controversy over perceived benefits. J. Pharmacol. Pharmacother. 2011, 2, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anker, C.C.B.; Rafiq, S.; Jeppesen, P.B. Effect of steviol glycosides on human health with emphasis on type 2 diabetic biomarkers: A systematic review and meta-analysis of randomized controlled trials. Nutrients 2019, 11, 1965. [Google Scholar] [CrossRef] [Green Version]
- Xi, D.; Bhattacharjee, J.; Salazar-Gonzalez, R.M.; Park, S.; Jang, A.; Warren, M.; Merritt, R.; Michail, S.; Bouret, S.; Kohli, R. Rebaudioside affords hepatoprotection ameliorating sugar sweetened beverage-induced nonalcoholic steatohepatitis. Sci. Rep. 2020, 10, 6689. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef]
- Zhang, P. Influence of Foods and Nutrition on the Gut Microbiome and Implications for Intestinal Health. Int. J. Mol. Sci. 2022, 23, 9588. [Google Scholar] [CrossRef]
- Yan, J.; Sheng, L.; Li, H. Akkermansia muciniphila: Is it the Holy Grail for ameliorating metabolic diseases? Gut Microbes 2021, 13, 1984104. [Google Scholar] [CrossRef]
- Richardson, I.L.; Frese, S.A. Non-nutritive sweeteners and their impacts on the gut microbiome and host physiology. Front. Nutr. 2022, 9, 988144. [Google Scholar] [CrossRef]
- Renwick, A.G.; Tarka, S.M. Microbial hydrolysis of steviol glycosides. Food Chem. Toxicol. 2008, 46, S70–S74. [Google Scholar] [CrossRef]
- Casas-Grajales, S.; Reyes-Gordillo, K.; Cerda-García-Rojas, C.M.; Tsutsumi, V.; Lakshman, M.R.; Muriel, P. Rebaudioside A administration prevents experimental liver fibrosis: An in vivo and in vitro study of the mechanisms of action involved. J. Appl. Toxicol. 2019, 39, 1118–1131. [Google Scholar] [CrossRef]
- Ruiz-Ojeda, F.J.; Plaza-Díaz, J.; Sáez-Lara, M.J.; Gil, A. Effects of sweeteners on the gut microbiota: A review of experimental studies and clinical trials. Adv. Nutr. 2019, 10 (Suppl. 1), S31–S48. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, N.; Tan, H.Y.; Li, S.; Zhang, C.; Feng, Y. Function of Akkermansia muciniphila in obesity: Interactions with lipid metabolism, immune response and gut systems. Front. Microbiol. 2020, 11, 219. [Google Scholar] [CrossRef] [Green Version]
- Nettleton, J.E.; Reimer, R.A.; Shearer, J. Reshaping the gut microbiota: Impact of low calorie sweeteners and the link to insulin resistance? Physiol. Behav. 2016, 164, 488–493. [Google Scholar] [CrossRef]
- Suez, J.; Cohen, Y.; Valdés-Mas, R.; Mor, U.; Dori-Bachash, M.; Federici, S.; Zmora, N.; Leshem, A.; Heinemann, M.; Linevsky, R.; et al. Personalized microbiome-driven effects of non-nutritive sweeteners on human glucose tolerance. Cell 2022, 185, 3307–3328. [Google Scholar] [CrossRef]
- Pang, M.D.; Goossens, G.H.; Blaak, E.E. The impact of artificial sweeteners on body weight control and glucose homeostasis. Front. Nutr. 2021, 7, 598340. [Google Scholar] [CrossRef]
- Olofsson, L.E.; Bäckhed, F. The metabolic role and therapeutic potential of the microbiome. Endocr. Rev. 2022, 43, 907–926. [Google Scholar] [CrossRef]
- Hrncir, T. Gut Microbiota Dysbiosis: Triggers, Consequences, Diagnostic and Therapeutic Options. Microorganisms 2022, 10, 578. [Google Scholar] [CrossRef]
- Del Pozo, S.; Gómez-Martínez, S.; Díaz, L.E.; Nova, E.; Urrialde, R.; Marcos, A. Potential effects of Sucralose and saccharin on gut microbiota: A review. Nutrients 2022, 14, 1682. [Google Scholar] [CrossRef]
- Uebanso, T.; Ohnishi, A.; Kitayama, R.; Yoshimoto, A.; Nakahashi, M.; Shimohata, T.; Mawatari, K.; Takahashi, A. Effects of low-dose non-caloric sweetener consumption on gut microbiota in mice. Nutrients 2017, 9, 560. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, A.; Carrascosa, C.; Raheem, D.; Ramos, F.; Raposo, A. Maltitol: Analytical determination methods, applications in the food industry, metabolism and health impacts. Int. J. Environ. Res. Public Health 2020, 17, 5227. [Google Scholar] [CrossRef] [PubMed]
- Sima, P.; Vannucci, L.; Vetvicka, V. β-glucans and cholesterol. Int. J. Mol. Med. 2018, 41, 1799–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, R.S.; Anderson, J.W.; Bridges, S.R. Propionate inhibits hepatocyte lipid synthesis. Proc. Soc. Exp. Biol. Med. 1990, 195, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Chen, X.; Yu, C.; Deng, Y.; Zhang, Y.; Chen, S.; Chen, X.; Chen, K.; Yang, Y.; Ling, W. Gut microbially produced indole-3-propionic acid inhibits atherosclerosis by promoting reverse cholesterol transport and its deficiency is causally related to atherosclerotic cardiovascular disease. Circ. Res. 2022, 131, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, M.J.; Do, H.V.; Mun, S.; Jun, H.J.; Lee, J.H.; Kim, Y.R.; Lee, S.J. Hypocholesterolemic and hypoglycemic effects of enzymatically modified carbohydrates from rice in high-fat-fed C57BL/6J mice. Mol. Nutr. Food Res. 2011, 55, S214–S226. [Google Scholar] [CrossRef]
- Urushima, H.; Sanada, Y.; Sakaue, M.; Matsuzawa, Y.; Ito, T.; Maeda, K. Maltitol prevents the progression of fatty liver degeneration in mice fed high-fat diets. J. Med. Food 2015, 18, 1081–1087. [Google Scholar] [CrossRef]
- Ortiz, S.R.; Field, M.S. Chronic dietary erythritol exposure elevates plasma erythritol concentration in mice but does not cause weight gain or modify glucose homeostasis. J. Nutr. 2021, 151, 2114–2124. [Google Scholar] [CrossRef]
- Kawano, R.; Okamura, T.; Hashimoto, Y.; Majima, S.; Senmaru, T.; Ushigome, E.; Asano, M.; Yamazaki, M.; Takakuwa, M.; Sasano, R.; et al. Erythritol ameliorates small intestinal inflammation induced by high-fat diets and improves glucose tolerance. Int. J. Mol. Sci. 2021, 22, 5558. [Google Scholar] [CrossRef]
- Yokozawa, T.; Kim, H.Y.; Cho, E.J. Erythritol attenuates the diabetic oxidative stress through modulating glucose metabolism and lipid peroxidation in streptozotocin-induced diabetic rats. J. Agric. Food Chem. 2002, 50, 5485–5489. [Google Scholar] [CrossRef]
- Jin, M.; Wei, Y.; Yu, H.; Ma, X.; Yan, S.; Zhao, L.; Ding, L.; Cheng, J.; Feng, H. Erythritol improves nonalcoholic fatty liver disease by activating Nrf2 antioxidant capacity. J. Agric. Food Chem. 2021, 69, 13080–13092. [Google Scholar] [CrossRef]
- Oku, T.; Nakamura, S. Threshold for transitory diarrhea induced by ingestion of xylitol and lactitol in young male and female adults. J. Nutr. Sci. Vitaminol. 2007, 53, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Storey, D.; Lee, A.; Bornet, F.; Brouns, F.J.P.H. Gastrointestinal tolerance of erythritol and xylitol ingested in a liquid. Eur. J. Clin. Nutr. 2007, 61, 349–354. [Google Scholar] [CrossRef]
- Anderson, M.E. Glutathione: An overview of biosynthesis and modulation. Chem. Biol. Interact. 1998, 111, 1–14. [Google Scholar] [CrossRef]
- Honda, Y.; Kessoku, T.; Sumida, Y.; Kobayashi, T.; Kato, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M. Efficacy of glutathione for the treatment of nonalcoholic fatty liver disease: An open-label, single-arm, multicenter, pilot study. BMC Gastroenterol. 2017, 17, 1–8. [Google Scholar] [CrossRef]
- Allen, J.; Bradley, R.D. Effects of oral glutathione supplementation on systemic oxidative stress biomarkers in human volunteers. J. Altern. Complement. Med. 2011, 17, 827–833. [Google Scholar] [CrossRef] [Green Version]
- Kovacs-Nolan, J.; Rupa, P.; Matsui, T.; Tanaka, M.; Konishi, T.; Sauchi, Y.; Sato, K.; Ono, S.; Mine, Y. In vitro and ex vivo uptake of glutathione (GSH) across the intestinal epithelium and fate of oral GSH after in vivo supplementation. J. Agric. Food Chem. 2014, 62, 9499–9506. [Google Scholar] [CrossRef]
- Park, E.Y.; Shimura, N.; Konishi, T.; Sauchi, Y.; Wada, S.; Aoi, W.; Nakamura, Y.; Sato, K. Increase in the protein-bound form of glutathione in human blood after the oral administration of glutathione. J. Agric. Food Chem. 2014, 62, 6183–6189. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Söderlund, S.; Ståhlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kiricarslan, M.; et al. Personal model-assisted identification of NAD+ and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef]
- Park, S.H.; Han, A.L.; Kim, N.H.; Shin, S.R. Liver histological improvement after administration of high-dose vitamin C in guinea pig with nonalcoholic steatohepatitis. Int. J. Vitam. Nutr. Res. 2019, 88, 263–269. [Google Scholar] [CrossRef]
- Souza, M.R.D.A.; Diniz, M.D.F.F.D.M.; Medeiros-Filho, J.E.M.D.; Araújo, M.S.T.D. Metabolic syndrome and risk factors for non-alcoholic fatty liver disease. Arq. De Gastroenterol. 2012, 49, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Aoi, W.; Ogaya, Y.; Takami, M.; Konishi, T.; Sauchi, Y.; Park, E.Y.; Wada, S.; Sato, K.; Higashi, A. Glutathione supplementation suppresses muscle fatigue induced by prolonged exercise via improved aerobic metabolism. J. Int. Soc. Sport. Nutr. 2015, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapenna, D.; Pierdomenico, S.D.; Ciofani, G.; Ucchino, S.; Neri, M.; Giamberardino, M.A.; Cuccurullo, F. Association of body iron stores with low molecular weight iron and oxidant damage of human atherosclerotic plaques. Free. Radic. Biol. Med. 2007, 42, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L.; Fracanzani, A.L.; Gatti, S.; Cairo, G.; Fargion, S. Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. Am. J. Pathol. 2008, 172, 738–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. D J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziani, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009, 301, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Lee, H.S.; Ahn, S.B.; Kwon, Y.J. Dairy protein intake is inversely related to development of non-alcoholic fatty liver disease. Clin. Nutr. 2021, 40, 5252–5260. [Google Scholar] [CrossRef]
- Léveillé, M.; Estall, J.L. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019, 9, 233. [Google Scholar] [CrossRef] [Green Version]
- Irie, M.; Sohda, T.; Anan, A.; Fukunaga, A.; Takata, K.; Tanaka, T.; Yokoyama, K.; Morihara, D.; Takeyama, Y.; Shakado, S.; et al. Reduced glutathione suppresses oxidative stress in nonalcoholic fatty liver disease. Euroasian J. Hepato-Gastroenterol. 2016, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Hamad, E.M.; Taha, S.H.; Abou Dawood, A.G.I.; Sitohy, M.Z.; Abdel-Hamid, M. Protective effect of whey proteins against nonalcoholic fatty liver in rats. Lipids Health Dis. 2011, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Morton, R.W.; Murphy, K.T.; McKellar, S.R.; Schoenfeld, B.J.; Henselmans, M.; Helms, E.; Helms, E.; Aragon, A.A.; Devries, M.C.; Banfield, L.; et al. A systematic review, meta-analysis and meta-regression of the effect of protein supplementation on resistance training-induced gains in muscle mass and strength in healthy adults. Br. J. Sport. Med. 2018, 52, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.A.; Jacobs, D.R., Jr.; Van Horn, L.; Slattery, M.L.; Kartashov, A.I.; Ludwig, D.S. Dairy consumption, obesity, and the insulin resistance syndrome in young adults: The CARDIA Study. JAMA 2002, 287, 2081–2089. [Google Scholar] [CrossRef]
- Duarte, S.M.; Stefano, J.T.; Oliveira, C.P. Microbiota and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis (NAFLD/NASH). Ann. Hepatol. 2019, 18, 416–421. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, X. The Role of Gut–Liver Axis in Gut Microbiome Dysbiosis Associated NAFLD and NAFLD-HCC. Biomedicines 2022, 10, 524. [Google Scholar] [CrossRef]
- Khan, A.; Ding, Z.; Ishaq, M.; Bacha, A.S.; Khan, I.; Hanif, A.; Li, W.; Guo, X. Understanding the effects of gut microbiota dysbiosis on nonalcoholic fatty liver disease and the possible probiotics role: Recent updates. Int. J. Biol. Sci. 2021, 17, 818. [Google Scholar] [CrossRef]
- Wang, H.; Mehal, W.; Nagy, L.E.; Rotman, Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell. Mol. Immunol. 2021, 18, 73–91. [Google Scholar] [CrossRef]
- Takai, A.; Kikuchi, K.; Ichimura, M.; Tsuneyama, K.; Moritoki, Y.; Matsumoto, K.; Tsunashima, H.; Onda, T.; Kuniyoshi, N.; Nariyama, T.; et al. Fructo-oligosaccharides ameliorate steatohepatitis, visceral adiposity, and associated chronic inflammation via increased production of short-chain fatty acids in a mouse model of non-alcoholic steatohepatitis. BMC Gastroenterol. 2020, 20, 1–10. [Google Scholar] [CrossRef]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein–coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Yabut, J.M.; Drucker, D.J. Glucagon-like peptide-1 receptor-based therapeutics for metabolic liver disease. Endocr. Rev. 2023, 44, 14–32. [Google Scholar] [CrossRef]
- Seghieri, M.; Christensen, A.S.; Andersen, A.; Solini, A.; Knop, F.K.; Vilsbøll, T. Future perspectives on GLP-1 receptor agonists and GLP-1/glucagon receptor co-agonists in the treatment of NAFLD. Front. Endocrinol. 2018, 9, 649. [Google Scholar] [CrossRef]
- Kimura, I.; Inoue, D.; Hirano, K.; Tsujimoto, G. The SCFA receptor GPR43 and energy metabolism. Front. Endocrinol. 2014, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngound, D.J.; et al. Short-chain fatty acids protect against high-fat diet–induced obesity via a PPARγ-dependent switch from lipogenesis to fat oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattace Raso, G.; Simeoli, R.; Russo, R.; Iacono, A.; Santoro, A.; Paciello, O.; Ferrante, M.C.; Canani, R.B.; Calignano, A.; Meli, R. Effects of sodium butyrate and its synthetic amide derivative on liver inflammation and glucose tolerance in an animal model of steatosis induced by high fat diet. PLoS ONE 2013, 8, e68626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Loh, K.; Song, Z.Y.; Yang, H.Q.; Zhang, Y.; Lin, S. Update on glycerol-3-phosphate acyltransferases: The roles in the development of insulin resistance. Nutr. Diabetes 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Weitkunat, K.; Schumann, S.; Nickel, D.; Kappo, K.A.; Petzke, K.J.; Kipp, A.P.; Blaut, M.; Klaus, S. Importance of propionate for the repression of hepatic lipogenesis and improvement of insulin sensitivity in high-fat diet-induced obesity. Mol. Nutr. Food Res. 2016, 60, 2611–2621. [Google Scholar] [CrossRef] [Green Version]
- Al-Lahham, S.; Roelofsen, H.; Rezaee, F.; Weening, D.; Hoek, A.; Vonk, R.; Venema, K. Propionic acid affects immune status and metabolism in adipose tissue from overweight. Eur. J. Clin. Investig. 2012, 42, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826. [Google Scholar] [CrossRef]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Castiglione, A.; Croce, L.S.; Tiribelli, C.; Bellentani, S. Incidence and natural course of fatty liver in the general population: The Dionysos study. Hepatology 2007, 46, 1387–1391. [Google Scholar] [CrossRef]
- Singh, S.; Osna, N.A.; Kharbanda, K.K. Treatment options for alcoholic and non-alcoholic fatty liver disease: A review. World J. Gastroenterol. 2017, 23, 6549. [Google Scholar] [CrossRef]
- Sherriff, J.L.; O’Sullivan, T.A.; Properzi, C.; Oddo, J.L.; Adams, L.A. Choline, its potential role in nonalcoholic fatty liver disease, and the case for human and bacterial genes. Adv. Nutr. 2016, 7, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Craig, S.A. Betaine in human nutrition. Am. J. Clin. Nutr. 2004, 80, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.M.; Vance, D.E. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J. Biol. Chem. 1988, 263, 2998–3004. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Wiedeman, A.M.; Barr, S.I.; Green, T.J.; Xu, Z.; Innis, S.M.; Kitts, D.D. Dietary choline intake: Current state of knowledge across the life cycle. Nutrients 2018, 10, 1513. [Google Scholar] [CrossRef] [Green Version]
- Lobo, R.; Prabhu, K.S.; Shirwaikar, A.; Shirwaikar, A. Curcuma zedoaria Rosc.(white turmeric): A review of its chemical, pharmacological and ethnomedicinal properties. J. Pharm. Pharmacol. 2009, 61, 13–21. [Google Scholar] [CrossRef]
- Baziar, N.; Parohan, M. The effects of curcumin supplementation on body mass index, body weight, and waist circumference in patients with nonalcoholic fatty liver disease: A systematic review and dose–response meta-analysis of randomized controlled trials. Phytother. Res. 2020, 34, 464–474. [Google Scholar] [CrossRef]
- White, C.M.; Lee, J.Y. The impact of turmeric or its curcumin extract on nonalcoholic fatty liver disease: A systematic review of clinical trials. Pharm. Pract. 2019, 17, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Cicero, A.F.; Sahebkar, A.; Fogacci, F.; Bove, M.; Giovannini, M.; Borghi, C. Effects of phytosomal curcumin on anthropometric parameters, insulin resistance, cortisolemia and non-alcoholic fatty liver disease indices: A double-blind, placebo-controlled clinical trial. Eur. J. Nutr. 2020, 59, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Saadati, S.; Hatami, B.; Yari, Z.; Shahrbaf, M.A.; Eghtesad, S.; Mansour, A.; Poustchi, H.; Hedayati, M.; Aghajanpoor-Pasha, M.; Sadeghi, A.; et al. The effects of curcumin supplementation on liver enzymes, lipid profile, glucose homeostasis, and hepatic steatosis and fibrosis in patients with non-alcoholic fatty liver disease. Eur. J. Clin. Nutr. 2019, 73, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhai, Y.; Heng, X.; Che, F.Y.; Chen, W.; Sun, D.; Zhai, G. Oral bioavailability of curcumin: Problems and advancements. J. Drug Target. 2016, 24, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Song, Z.; Weng, J.; Fantus, I.G. Curcumin and other dietary polyphenols: Potential mechanisms of metabolic actions and therapy for diabetes and obesity. Am. J. Physiol. -Endocrinol. Metab. 2018, 314, E201–E205. [Google Scholar] [CrossRef] [PubMed]
- Marton, L.T.; Pescinini-e-Salzedas, L.M.; Camargo, M.E.C.; Barbalho, S.M.; Haber, J.F.D.S.; Sinatora, R.V.; Detregiachi, C.R.P.; Girio, R.J.S.; Buchaim, D.V.; Cincotto dos Santos Bueno, P. The effects of curcumin on diabetes mellitus: A systematic review. Front. Endocrinol. 2021, 12, 669448. [Google Scholar] [CrossRef] [PubMed]
- Navekar, R.; Rafraf, M.; Ghaffari, A.; Asghari-Jafarabadi, M.; Khoshbaten, M. Turmeric supplementation improves serum glucose indices and leptin levels in patients with nonalcoholic fatty liver diseases. J. Am. Coll. Nutr. 2017, 36, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Xu, H.; Xu, H.; Wang, P.F.; Cai, G.J.; Song, H.F.; Wang, C.; Dong, Z.; Ju, Y.; Jiang, Z.Y. Nesfatin-1 stimulates fatty-acid oxidation by activating AMP-activated protein kinase in STZ-induced type 2 diabetic mice. PLoS ONE 2013, 8, e83397. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.; Han, Y.; Cai, B.; Wang, Y. Decreased levels of serum nesfatin-1 in patients with obstructive sleep apnea syndrome. Sleep Breath. 2015, 19, 515–522. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X.; Ye, Z.; Xu, C.; Zhang, M.; Ruan, B.; Wei, M.; Jiang, J.; Zhang, Y.; Wang, L.; et al. Curcumin promotes browning of white adipose tissue in a norepinephrine-dependent way. Biochem. Biophys. Res. Commun. 2015, 466, 247–253. [Google Scholar] [CrossRef]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar] [CrossRef] [Green Version]
- Kumari, M.; Chandola, T.; Brunner, E.; Kivimaki, M. A nonlinear relationship of generalized and central obesity with diurnal cortisol secretion in the Whitehall II study. J. Clin. Endocrinol. Metab. 2010, 95, 4415–4423. [Google Scholar] [CrossRef]
- Hu, G.X.; Lin, H.; Lian, Q.Q.; Zhou, S.H.; Guo, J.; Zhou, H.Y.; Chu, Y.; Ge, R.S. Curcumin as a potent and selective inhibitor of 11β-hydroxysteroid dehydrogenase 1: Improving lipid profiles in high-fat-diet-treated rats. PLoS ONE 2013, 8, e49976. [Google Scholar] [CrossRef] [Green Version]
- Bradford, P.G. Curcumin and obesity. Biofactors 2013, 39, 78–87. [Google Scholar] [CrossRef]
- Qin, S.; Huang, L.; Gong, J.; Shen, S.; Huang, J.; Tang, Y.; Ren, H.; Hu, H. Meta-analysis of randomized controlled trials of 4 weeks or longer suggest that curcumin may afford some protection against oxidative stress. Nutr. Res. 2018, 60, 1–12. [Google Scholar] [CrossRef]
- Alavinejad, P.; Farsi, F.; Husain, D.; Rezazadeh, A. The effect of turmeric on lipid profile, malondialdehyde, liver echogenicity and enzymes among patients with nonalcoholic fatty liver disease: A randomized double blind clinical trial. Diabetol. Metab. Syndr. 2021, 13, 1–9. [Google Scholar] [CrossRef]
- Rao, M.N.A. Curcuminoids as potent inhibitors of lipid peroxidation. J. Pharm. Pharmacol. 1994, 46, 1013–1016. [Google Scholar] [CrossRef]
- Reddy, A.C.P.; Lokesh, B.R. Studies on the inhibitory effects of curcumin and eugenol on the formation of reactive oxygen species and the oxidation of ferrous iron. Mol. Cell. Biochem. 1994, 137, 1–8. [Google Scholar] [CrossRef]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Eccleston, H.B.; Bailey, S.M. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol-and obesity-induced fatty liver diseases. Free. Radic. Biol. Med. 2008, 44, 1259–1272. [Google Scholar] [CrossRef] [Green Version]
- Motterlini, R.; Foresti, R.; Bassi, R.; Green, C.J. Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free. Radic. Biol. Med. 2000, 28, 1303–1312. [Google Scholar] [CrossRef]
- Piper, J.T.; Singhal, S.S.; Salameh, M.S.; Torman, R.T.; Awasthi, Y.C.; Awasthi, S. Mechanisms of anticarcinogenic properties of curcumin: The effect of curcumin on glutathione linked detoxification enzymes in rat liver. Int. J. Biochem. Cell Biol. 1998, 30, 445–456. [Google Scholar] [CrossRef]
- Barnes, P.J.; Karin, M. Nuclear factor-κB—A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Nanji, A.A.; Jokelainen, K.; Rahemtulla, A.; Miao, L.; Fogt, F.; Matsumoto, H.; Tahan, S.R.; Su, G.L. Activation of nuclear factor kappa B and cytokine imbalance in experimental alcoholic liver disease in the rat. Hepatology 1999, 30, 934–943. [Google Scholar] [CrossRef]
- Kang, H.C.; Nan, J.X.; Park, P.H.; Kim, J.Y.; Lee, S.H.; Woo, S.W.; Zhao, Y.Z.; Park, E.J.; Sohn, D.H. Curcumin inhibits collagen synthesis and hepatic stellate cell activation in-vivo and in-vitro. J. Pharm. Pharmacol. 2002, 54, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Zarei, M.; Acharya, P.; Talahalli, R.R. Ginger and turmeric lipid-solubles attenuate heated oil-induced hepatic inflammation via the downregulation of NF-kB in rats. Life Sci. 2021, 265, 118856. [Google Scholar] [CrossRef] [PubMed]
- Graf, T.N.; Wani, M.C.; Agarwal, R.; Kroll, D.J.; Oberlies, N.H. Gram-scale purification of flavonolignan diastereoisomers from Silybum marianum (Milk Thistle) extract in support of preclinical in vivo studies for prostate cancer chemoprevention. Planta Med. 2007, 73, 1495–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehér, J.; Lengyel, G. Silymarin in the prevention and treatment of liver diseases and primary liver cancer. Curr. Pharm. Biotechnol. 2012, 13, 210–217. [Google Scholar] [CrossRef]
- Fried, M.W.; Navarro, V.J.; Afdhal, N.; Belle, S.H.; Wahed, A.S.; Hawke, R.L.; Doo, E.; Meyers, C.M.; Reddy, K.R. Effect of silymarin (milk thistle) on liver disease in patients with chronic hepatitis C unsuccessfully treated with interferon therapy: A randomized controlled trial. JAMA 2012, 308, 274–282. [Google Scholar] [CrossRef]
- Polyak, S.J.; Ferenci, P.; Pawlotsky, J.M. Hepatoprotective and antiviral functions of silymarin components in HCV infection. Hepatology 2013, 57, 1262. [Google Scholar] [CrossRef] [Green Version]
- Abenavoli, L.; Izzo, A.A.; Milić, N.; Cicala, C.; Santini, A.; Capasso, R. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother. Res. 2018, 32, 2202–2213. [Google Scholar] [CrossRef]
- Loguercio, C.; Andreone, P.; Brisc, C.; Brisc, M.C.; Bugianesi, E.; Chiaramonte, M.; Cursaro, C.; Danila, M.; de Sio, I.; Floreani, A.; et al. Silybin combined with phosphatidylcholine and vitamin E in patients with nonalcoholic fatty liver disease: A randomized controlled trial. Free. Radic. Biol. Med. 2012, 52, 1658–1665. [Google Scholar] [CrossRef] [Green Version]
- Mengesha, T.; Gnanasekaran, N.; Mehare, T. Hepatoprotective effect of silymarin on fructose induced nonalcoholic fatty liver disease in male albino wistar rats. BMC Complement. Med. Ther. 2021, 21, 1–13. [Google Scholar] [CrossRef]
- Ross, S.M. Milk thistle (Silybum marianum): An ancient botanical medicine for modern times. Holist. Nurs. Pract. 2008, 22, 299–300. [Google Scholar] [CrossRef]
- Navarro, V.J.; Belle, S.H.; D’Amato, M.; Adfhal, N.; Brunt, E.M.; Fried, M.W.; Reddy, K.R.; Wahed, A.S.; Harrison, S. Silymarin in non-cirrhotics with non-alcoholic steatohepatitis: A randomized, double-blind, placebo controlled trial. PLoS ONE 2019, 14, e0221683. [Google Scholar] [CrossRef] [Green Version]
- Kheong, C.W.; Mustapha, N.R.N.; Mahadeva, S. A randomized trial of silymarin for the treatment of nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2017, 15, 1940–1949. [Google Scholar] [CrossRef] [Green Version]
- Solhi, H.; Ghahremani, R.; Kazemifar, A.M.; Yazdi, Z.H. Silymarin in treatment of non-alcoholic steatohepatitis: A randomized clinical trial. Casp. J. Intern. Med. 2014, 5, 9. [Google Scholar]
- Abenavoli, L.; Greco, M.; Nazionale, I.; Peta, V.; Milic, N.; Accattato, F.; Foti, D.; Gulletta, E.; Luzza, F. Effects of Mediterranean diet supplemented with silybin–vitamin E–phospholipid complex in overweight patients with non-alcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 519–527. [Google Scholar] [CrossRef]
- Loguercio, C.; Federico, A.; Trappoliere, M.; Tuccillo, C.; Sio, I.D.; Leva, A.D.; Niosi, M.; D’auria, V.M.; Capasso, R.; Blanci, C.D.V.; et al. The effect of a silybin-vitamin e-phospholipid complex on nonalcoholic fatty liver disease: A pilot study. Dig. Dis. Sci. 2007, 52, 2387–2395. [Google Scholar] [CrossRef]
- Geillinger, K.E.; Rathmann, D.; Köhrle, J.; Fiamoncini, J.; Daniel, H.; Kipp, A.P. Hepatic metabolite profiles in mice with a suboptimal selenium status. J. Nutr. Biochem. 2014, 25, 914–922. [Google Scholar] [CrossRef]
- Schäfer, K.; Kyriakopoulos, A.; Gessner, H.; Grune, T.; Behne, D. Effects of selenium deficiency on fatty acid metabolism in rats fed fish oil-enriched diets. J. Trace Elem. Med. Biol. 2004, 18, 89–97. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Nonalcoholic fatty liver disease: The pathogenetic roles of insulin resistance and adipocytokines. Curr. Mol. Med. 2009, 9, 299–314. [Google Scholar] [CrossRef]
- Han, J.; Liang, H.; Yi, J.; Tan, W.; He, S.; Wang, S.; Li, F.; Wu, X.; Ma, J.; Shi, X.; et al. Long-term selenium-deficient diet induces liver damage by altering hepatocyte ultrastructure and MMP1/3 and TIMP1/3 expression in growing rats. Biol. Trace Elem. Res. 2017, 175, 396–404. [Google Scholar] [CrossRef]
- George, J. Determination of selenium during pathogenesis of hepatic fibrosis employing hydride generation and inductively coupled plasma mass spectrometry. Biol. Chem. 2018, 399, 499–509. [Google Scholar] [CrossRef]
- Bitiren, M.; Karakılçık, A.Z.; Zerin, M.; Aksoy, N.; Musa, D. Effects of selenium on histopathological and enzymatic changes in experimental liver injury of rats. Exp. Toxicol. Pathol. 2004, 56, 59–64. [Google Scholar] [CrossRef]
- Zhang, Q.; Qian, Z.Y.; Zhou, P.H.; Zhou, X.L.; Zhang, D.L.; He, N.; Zhang, J.; Liu, Y.H.; Gu, Q. Effects of oral selenium and magnesium co-supplementation on lipid metabolism, antioxidative status, histopathological lesions, and related gene expression in rats fed a high-fat diet. Lipids Health Dis. 2018, 17, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shidfar, F.; Faghihi, A.; Amiri, H.L.; Mousavi, S.N. Regression of nonalcoholic fatty liver disease with zinc and selenium co-supplementation after disease progression in rats. Iran. J. Med. Sci. 2018, 43, 26. [Google Scholar] [PubMed]
- Nido, S.A.; Shituleni, S.A.; Mengistu, B.M.; Liu, Y.; Khan, A.Z.; Gan, F.; Kumbhar, S.; Huang, K. Effects of selenium-enriched probiotics on lipid metabolism, antioxidative status, histopathological lesions, and related gene expression in mice fed a high-fat diet. Biol. Trace Elem. Res. 2016, 171, 399–409. [Google Scholar] [CrossRef]
- González-Reimers, E.; Monedero-Prieto, M.J.; González-Pérez, J.M.; Duran-Castellon, M.C.; Galindo-Martín, L.; Abreu-González, P.; Sanchez-Perez, M.J.; Santolaria-Fernández, F. Relative and combined effects of selenium, protein deficiency and ethanol on hepatocyte ballooning and liver steatosis. Biol. Trace Elem. Res. 2013, 154, 281–287. [Google Scholar] [CrossRef]
- Ding, M.; Potter, J.J.; Liu, X.; Torbenson, M.S.; Mezey, E. Selenium supplementation decreases hepatic fibrosis in mice after chronic carbon tetrachloride administration. Biol. Trace Elem. Res. 2010, 133, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipokines in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Bioulac-Sage, P.; Dubuisson, L.; Bedin, C.; Gonzalez, P.; de Tinguy-Moreaud, E.; Garcin, H.; Balabaud, C. Nodular regenerative hyperplasia in the rat induced by a selenium-enriched diet: Study of a model. Hepatology 1992, 16, 418–425. [Google Scholar] [CrossRef]
- Dubuisson, L.; Boussarie, L.; Bedin, C.A.; Balabaud, C.; Bioulac-Sage, P. Transformation of sinusoids into capillaries in a rat model of selenium-induced nodular regenerative hyperplasia: An immunolight and immunoelectron microscopic study. Hepatology 1995, 21, 805–814. [Google Scholar]
- Koeberle, A.; Löser, K.; Thürmer, M. Stearoyl-CoA desaturase-1 and adaptive stress signaling. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 1719–1726. [Google Scholar] [CrossRef]
- Song, Z.; Xiaoli, A.M.; Yang, F. Regulation and metabolic significance of de novo lipogenesis in adipose tissues. Nutrients 2018, 10, 1383. [Google Scholar] [CrossRef] [Green Version]
- Poudyal, H.; Brown, L. Stearoyl-CoA desaturase: A vital checkpoint in the development and progression of obesity. Endocr. Metab. Immune Disord. -Drug Targets (Former. Curr. Drug Targets-Immune Endocr. Metab. Disord.) 2011, 11, 217–231. [Google Scholar] [CrossRef]
- Paton, C.M.; Ntambi, J.M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol. -Endocrinol. Metab. 2009, 297, E28–E37. [Google Scholar] [CrossRef] [Green Version]
- Enoch, H.G.; Catalá, A.; Strittmatter, P. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. J. Biol. Chem. 1976, 251, 5095–5103. [Google Scholar] [CrossRef]
- Mosley, E.E.; McGuire, M.A. Methodology for the In Vivo Measurement of the Δ9-Desaturation of Myristic, Palmitic, and Stearic Acids in Lactating Dairy Cattle. Lipids 2007, 42, 939–945. [Google Scholar] [CrossRef]
- Miyazaki, M.A.; Dobrzyn, W.C.; Man, K.; Chu, H.; Sampath, H.J.; Kim, J.M. Ntambi. Stearoyl-CoA desaturase 1 gene expression is necessary for fructose-mediated induction of lipogenic gene expression by sterol regulatory element-binding protein-1c-dependent and -independent mechanisms. J. Biol. Chem. 2004, 279, 25164–25171. [Google Scholar] [CrossRef] [Green Version]
- Ntambi, J.M.; Miyazaki, M.; Stoehr, J.P.; Lan, H.; Kendziorski, C.M.; Yandell, B.S.; Song, Y.; Cohen, P.; Friedman, J.M.; Attie, A.D. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA 2002, 99, 11482–11486. [Google Scholar] [CrossRef] [Green Version]
- Oballa, R.M.; Belair, L.; Black, W.C.; Bleasby, K.; Chan, C.C.; Desroches, C.; Du, X.; Gordon, R.; Guay, J.; Guiral, S.; et al. Development of a liver-targeted stearoyl-CoA desaturase (SCD) inhibitor (MK-8245) to establish a therapeutic window for the treatment of diabetes and dyslipidemia. J. Med. Chem. 2011, 54, 5082–5096. [Google Scholar] [CrossRef]
- Uto, Y.; Ogata, T.; Kiyotsuka, Y.; Ueno, Y.; Miyazawa, Y.; Kurata, H.; Deguchi, T.; Watanabe, N.; Konishi, M.; Okuyama, R.; et al. Novel benzoylpiperidine-based stearoyl-CoA desaturase-1 inhibitors: Identification of 6-[4-(2-methylbenzoyl) piperidin-1-yl]pyridazine-3-carboxylic acid (2-hydroxy-2-pyridin-3-ylethyl)amide and its plasma triglyceride-lowering effects in Zucker fatty rats. Bioorg. Med. Chem. Lett. 2010, 20, 341–345. [Google Scholar] [CrossRef]
- Flowers, M.T.; Paton, C.M.; O’Byrne, S.M.; Schiesser, K.; Dawson, J.A.; Blaner, W.S.; Kendziorski, C.; Ntambi, J.M. Metabolic changes in skin caused by Scd1 deficiency: A focus on retinol metabolism. PLoS ONE 2011, 9, e19734. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Rudel, L.L. Stearoyl-coenzyme A desaturase 1 inhibition and the metabolic syndrome: Considerations for future drug discovery. Curr. Opin. Lipidol. 2010, 21, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Chung, S.; Sawyer, J.K.; Degirolamo, C.; Alger, H.M.; Nguyen, T.; Zhu, X.; Duong, M.N.; Wibley, A.L.; Shah, R.; et al. Inhibition of stearoyl-coenzyme A desaturase 1 dissociates insulin resistance and obesity from atherosclerosis. Circulation 2008, 118, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Dales, N.A.; Winther, M.D. Opportunities and challenges in developing stearoyl-coenzyme A desaturase-1 inhibitors as novel therapeutics for human disease. J. Med. Chem. 2014, 57, 5039–5056. [Google Scholar] [CrossRef] [PubMed]
- Major, C.A.; Ryan, K.; Bennett, A.J.; Lock, A.L.; Bauman, D.E.; Salter, A.M. Inhibition of stearoyl CoA desaturase activity induces hypercholesterolemia in the cholesterol-fed hamster. J. Lipid Res. 2008, 49, 1456–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortinau, L.C.; Nickelson, K.J.; Stromsdorfer, K.L.; Naik, C.Y.; Pickering, R.T.; Haynes, R.A.; Fritsche, K.L.; Perfield, J.W., II. Sterculic Oil, a natural inhibitor of SCD1, improves the metabolic state of obese OLETF rats. Obesity 2013, 21, 344–352. [Google Scholar] [CrossRef]
- Gomez, F.E.; Bauman, D.E.; Ntambi, J.M.; Fox, B.G. Effects of sterculic acid on stearoyl-CoA desaturase in differentiating 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2003, 300, 316–326. [Google Scholar] [CrossRef]
- Raju, P.K.; Reiser, R. Inhibition of fatty acyl desaturase by cyclopropene fatty acids. J. Biol. Chem. 1967, 242, 379–384. [Google Scholar] [CrossRef]
- Jeffcoat, R.; Pollard, M.R. Studies on the inhibition of the desaturases by cyclopropenoid fatty acids. Lipids 1977, 12, 480–485. [Google Scholar] [CrossRef]
- Ortinau, L.C.; Pickering, R.T.; Nickelson, K.J.; Stromsdorfer, K.L.; Naik, C.Y.; Haynes, R.A.; Bauman, D.E.; Rector, R.S.; Fritsche, K.L. Sterculic Oil, a Natural SCD1 Inhibitor, Improves Glucose Tolerance in Obese ob/ob Mice. Perfield JW 2nd. ISRN Endocrinol. 2012, 2012, 947323. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.J.; Blond, P.; Bézard, J. Inhibition of fatty acid Δ6-and Δ5-desaturation by cyclopropene fatty acids in rat liver microsomes. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1993, 1210, 27–34. [Google Scholar] [CrossRef]
- Lock, A.L.; Corl, B.A.; Barbano, D.M.; Bauman, D.E.; Ip, C. The anticarcinogenic effect of trans-11 18:1 is dependent on its conversion to cis-9, trans-11 CLA by delta9-desaturase in rats. J. Nutr. 2004, 134, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Varani, J.; McClintock, S.D.; Knibbs, R.N.; Harber, I.; Zeidan, D.; Jawad-Makki, M.A.H.; Aslam, M.N. Liver Protein Expression in NASH Mice on a High-Fat Diet: Response to Multi-Mineral Intervention. Front. Nutr. 2022, 11, 859292. [Google Scholar] [CrossRef]
- Aslam, M.N.; Bergin, I.; Naik, M.; Hampton, A.; Allen, R.; Kunkel, S.L.; Rush, H.; Varani, J. A multi-mineral natural product inhibits liver tumor formation in C57BL/6 mice. Biol. Trace Elem. Res. 2012, 147, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.N.; Bassis, C.M.; Zhang, L.; Zaidi, S.; Varani, J.; Bergin, I.L. Correction: Calcium Reduces Liver Injury in Mice on a High-Fat Diet: Alterations in Microbial and Bile Acid Profiles. PLoS ONE 2017, 12, e0170136. [Google Scholar] [CrossRef]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [Green Version]
- Canaff, L.; Petit, J.L.; Kisiel, M.; Watson, P.H.; Gascon-Barré, M.; Hendy, G.N. Extracellular calcium-sensing receptor is expressed in rat hepatocytes. coupling to intracellular calcium mobilization and stimulation of bile flow. J. Biol. Chem. 2001, 276, 4070–4079. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Herforth, A.; Masters, W.A. Global variation in the cost of a nutrient-adequate diet by population group: An observational study. Lancet Planet Health 2022, 6, e19–e28. [Google Scholar] [CrossRef]
- Aslam, M.N.; Bassis, C.M.; Bergin, I.L.; Knuver, K.; Zick, S.M.; Sen, A.; Turgeon, D.K.; Varani, J. A Calcium-Rich Multimineral Intervention to Modulate Colonic Microbial Communities and Metabolomic Profiles in Humans: Results from a 90-Day Trial. Cancer Prev. Res. 2020, 13, 101–116. [Google Scholar] [CrossRef]
- Ducheix, S.; Montagner, A.; Polizzi, A.; Lasserre, F.; Régnier, M.; Marmugi, A.; Benhamed, F.; Bertrand-Michel, J.; Mselli-Lakhal, L.; Loiseau, N.; et al. Dietary oleic acid regulates hepatic lipogenesis through a liver X receptor-dependent signaling. PLoS ONE 2017, 12, e0181393. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Shah, Y.M.; Morimura, K.; Krausz, K.W.; Miyazaki, M.; Richardson, T.A.; Morgan, E.T.; Ntambi, J.M.; Idle, J.R.; Gonzalez, F.J. Metabolomics reveals that hepatic stearoyl-CoA desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab. 2008, 7, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Flowers, M.T.; Ade, L.; Strable, M.S.; Ntambi, J.M. Combined deletion of SCD1 from adipose tissue and liver does not protect mice from obesity. J. Lipid Res. 2012, 53, 1646–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burhans, M.S.; Flowers, M.T.; Harrington, K.R.; Bond, L.M.; Guo, C.A.; Anderson, R.M.; Ntambi, J.M. Hepatic oleate regulates adipose tissue lipogenesis and fatty acid oxidation. J. Lipid Res. 2015, 56, 304–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Kono, H.; Ishii, K.; Hosomura, N.; Fujii, H. Dietary olive oil prevents carbon tetrachloride-induced hepatic fibrosis in mice. J. Gastroenterol. 2009, 44, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Moon, J.H.; Park, J.S.; Lee, B.W.; Kang, E.S.; Ahn, C.W.; Lee, H.C.; Cha, B.S. Dietary oleate has beneficial effects on every step of non-alcoholic Fatty liver disease progression in a methionine- and choline-deficient diet-fed animal model. Diabetes Metab. J. 2011, 35, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Quiroz, M.E.; Alba, G.; Saenz, J.; Santa-María, C.; Geniz, I.; Jiménez, J.; Ramírez, R.; Martín-Nieto, J.; Pintado, E.; Sobrino, F. Oleic acid modulates mRNA expression of liver X receptor (LXR) and its target genes ABCA1 and SREBP1c in human neutrophils. Eur. J. Nutr. 2014, 53, 1707–1717. [Google Scholar] [CrossRef]
- Ou, J.; Tu, H.; Shan, B.; Luk, A.; DeBose-Boyd, R.A.; Bashmakov, Y.; Goldstein, J.L.; Brown, M.S. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc. Natl. Acad. Sci. USA 2001, 98, 6027–6032. [Google Scholar] [CrossRef] [Green Version]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.B.; Laffitte, B.A.; Patel, P.H.; Watson, M.A.; Matsukuma, K.E.; Walczak, R.; Collins, J.L.; Osborne, T.F.; Tontonoz, P. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef] [Green Version]
- Chu, K.; Miyazaki, M.; Man, W.C.; Ntambi, J.M. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol. Cell. Biol. 2006, 26, 6786–6798. [Google Scholar] [CrossRef] [Green Version]
- Grefhorst, A.; Elzinga, B.M.; Voshol, P.J.; Plösch, T.; Kok, T.; Bloks, V.W.; van der Sluijs, F.H.; Havekes, L.M.; Romijn, J.A.; Verkade, H.J.; et al. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J. Biol. Chem. 2002, 277, 34182–34190. [Google Scholar] [CrossRef] [Green Version]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Zelcer, N.; Tontonoz, P. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Investig. 2006, 116, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Beaven, S.W.; Wroblewski, K.; Wang, J.; Hong, C.; Bensinger, S.; Tsukamoto, H.; Tontonoz, P. Liver X receptor signaling is a determinant of stellate cell activation and susceptibility to fibrotic liver disease. Gastroenterology 2011, 140, 1052–1062. [Google Scholar] [CrossRef] [Green Version]
- Moravcová, A.; Červinková, Z.; Kučera, O.; Mezera, V.; Rychtrmoc, D.; Lotková, H. The effect of oleic and palmitic acid on induction of steatosis and cytotoxicity on rat hepatocytes in primary culture. Physiol. Res. 2015, 64 (Suppl. 5), S627–S636. [Google Scholar] [CrossRef]
- Abenavoli, L.; Peta, V.; Milic, N. Lifestyle changes associated with a new antioxidant formulation in non-alcoholic fatty liver disease: A case series. Ann. Hepatol. 2015, 14, 121–126. [Google Scholar] [CrossRef]
- Wasmuth, H.E.; Trautwein, C. CB1 cannabinoid receptor antagonism: A new strategy for the treatment of liver fibrosis. Hepatology 2007, 45, 543–544. [Google Scholar] [CrossRef]
- Palomares, B.; Ruiz-Pino, F.; Garrido-Rodriguez, M.; Eugenia Prados, M.; Sánchez-Garrido, M.A.; Velasco, I.; Vazquez, M.J.; Nadal, X.; Ferreiro-Vera, C.; Morrugares, R.; et al. Tetrahydrocannabinolic acid A (THCA-A) reduces adiposity and prevents metabolic disease caused by diet-induced obesity. Biochem. Pharmacol. 2020, 171, 113693. [Google Scholar] [CrossRef]
- Rajavashisth, T.B.; Shaheen, M.; Norris, K.C.; Pan, D.; Sinha, S.K.; Ortega, J.; Friedman, T.C. Decreased prevalence of diabetes in marijuana users: Cross-sectional data from the National Health and Nutrition Examination Survey (NHANES) III. BMJ Open 2012, 2, e000494. [Google Scholar] [CrossRef] [Green Version]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Bátkai, S.; Har-vey-White, J.; Mackie, K.; Offertáler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osei-Hyiaman, D.; Liu, J.; Zhou, L.; Godlewski, G.; Harvey-White, J.; Jeong, W.I.; Bátkai, S.; Marsicano, G.; Lutz, B.; Buettner, C.; et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J. Clin. Investig. 2008, 118, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Liu, J.; Mukhopadhyay, B.; Cinar, R.; Godlewski, G.; Kunos, G. Endocannabinoids in liver disease. Hepatology 2011, 53, 346–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free. Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef] [PubMed]
- Gabbia, D.; Cannella, L.; De Martin, S. The Role of Oxidative Stress in NAFLD-NASH-HCC Transition-Focus on NADPH Oxidases. Biomedicines 2021, 9, 687. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Li Volti, G.; Rosso, C.; Grosso, G.; Bugianesi, E. Unconjugated bilirubin, a potent endogenous antioxidant, is decreased in patients with non-alcoholic steatohepatitis and advanced fibrosis. J. Gastroenterol. Hepatol. 2013, 28, 1202–1208. [Google Scholar] [CrossRef]
- Świderska, M.; Maciejczyk, M.; Zalewska, A.; Pogorzelska, J.; Flisiak, R.; Chabowski, A. Oxidative stress biomarkers in the serum and plasma of patients with non-alcoholic fatty liver disease (NAFLD). Can plasma AGE be a marker of NAFLD? Oxidative stress biomarkers in NAFLD patients. Free. Radic. Res. 2019, 53, 841–850. [Google Scholar] [CrossRef]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nature reviews. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Li, Z.; Yu, P.; Wu, J.; Tao, F.; Zhou, J. Transcriptional Regulation of Early Growth Response Gene-1 (EGR1) is Associated with Progression of Nonalcoholic Fatty Liver Disease (NAFLD) in Patients with Insulin Resistance. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 2293–3004. [Google Scholar] [CrossRef]
- Abenavoli, L.; Larussa, T.; Corea, A.; Procopio, A.C.; Boccuto, L.; Dallio, M.; Federico, A.; Luzza, F. Dietary Polyphenols and Non-Alcoholic Fatty Liver Disease. Nutrients 2021, 13, 494. [Google Scholar] [CrossRef]
- Aljomah, G.; Baker, S.S.; Liu, W.; Kozielski, R.; Oluwole, J.; Lupu, B.; Baker, R.D.; Zhu, L. Induction of CYP2E1 in non-alcoholic fatty liver diseases. Exp. Mol. Pathol. 2015, 99, 677–681. [Google Scholar] [CrossRef] [Green Version]
- Fu, A.; Shi, X.; Zhang, H.; Fu, B. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front. Pharmacol. 2017, 8, 241. [Google Scholar] [CrossRef] [Green Version]
- Koo, J.H.; Han, C.Y. Signaling Nodes Associated with Endoplasmic Reticulum Stress during NAFLD Progression. Biomolecules 2021, 11, 242. [Google Scholar] [CrossRef]
- Madan, K.; Bhardwaj, P.; Thareja, S.; Gupta, S.D.; Saraya, A. Oxidant stress and antioxidant status among patients with nonalcoholic fatty liver disease (NAFLD). J. Clin. Gastroenterol. 2006, 40, 930–935. [Google Scholar] [CrossRef]
- Berenstein, N. Making a global sensation: Vanilla flavor, synthetic chemistry, and the meanings of purity. Hist. Sci. 2016, 54, 399–424. [Google Scholar] [CrossRef]
- Raza, S.; Tewari, A.; Rajak, S.; Sinha, R.A. Vitamins and non-alcoholic fatty liver disease: A Molecular Insight⋆. Liver Res. 2021, 5, 62–71. [Google Scholar] [CrossRef]
- Ezhilarasan, D. Hepatic stellate cells in the injured liver: Perspectives beyond hepatic fibrosis. J. Cell. Physiol. 2022, 237, 436–449. [Google Scholar] [CrossRef]
- Saeed, A.; Bartuzi, P.; Heegsma, J.; Dekker, D.; Kloosterhuis, N.; de Bruin, A.; Jonker, J.W.; van de Sluis, B.; Faber, K.N. Impaired Hepatic Vitamin A Metabolism in NAFLD Mice Leading to Vitamin A Accumulation in Hepatocytes. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 309–325.e3. [Google Scholar] [CrossRef]
- Geng, C.; Xu, H.; Zhang, Y.; Gao, Y.; Li, M.; Liu, X.; Gao, M.; Wang, X.; Liu, X.; Fang, F.; et al. Retinoic acid ameliorates high-fat diet-induced liver steatosis through sirt1. Sci. China Life Sci. 2017, 60, 1234–1241. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, H.; Wang, J.; Zhou, W.; Sun, R.; Xia, M. Association of serum retinoic acid with hepatic steatosis and liver injury in nonalcoholic fatty liver disease. Am. J. Clin. Nutr. 2015, 102, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016, 25, 5212–5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 406–421.e7. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ahn, J.; Shin, S.S.; Yoon, M. Ascorbic acid inhibits visceral obesity and nonalcoholic fatty liver disease by activating peroxisome proliferator-activated receptor α in high-fat-diet-fed C57BL/6J mice. Int. J. Obes. 2019, 43, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Zhao, L.; Meng, C.; Zhao, X.; Liu, Y.; Shi, R.; Han, X.; Wang, T.; Li, J. Prophylactic and therapeutic effects of different doses of vitamin C on high-fat-diet-induced non-alcoholic fatty liver disease in mice. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 131, 110792. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Baek, S.M.; Kang, K.K.; Lee, A.R.; Kim, T.U.; Choi, S.K.; Roh, Y.S.; Hong, I.H.; Park, S.J.; Kim, T.H.; et al. Vitamin C Deficiency Inhibits Nonalcoholic Fatty Liver Disease Progression through Impaired de Novo Lipogenesis. Am. J. Pathol. 2021, 191, 1550–1563. [Google Scholar] [CrossRef]
- Wei, J.; Lei, G.H.; Fu, L.; Zeng, C.; Yang, T.; Peng, S.F. Association between Dietary Vitamin C Intake and Non-Alcoholic Fatty Liver Disease: A Cross-Sectional Study among Middle-Aged and Older Adults. PLoS ONE 2016, 11, e0147985. [Google Scholar] [CrossRef]
- Zhang, Z.C.; Liu, Y.; Xiao, L.L.; Li, S.F.; Jiang, J.H.; Zhao, Y.; Qian, S.W.; Tang, Q.Q.; Li, X. Upregulation of miR-125b by estrogen protects against non-alcoholic fatty liver in female mice. J. Hepatol. 2015, 63, 1466–1475. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency promote fatty liver disease development? Nutrients 2014, 6, 5473–5499. [Google Scholar] [CrossRef]
- Riederer, P.; Konradi, C.; Schay, V.; Kienzl, E.; Birkmayer, G.; Danielczyk, W.; Sofic, E.; Youdim, M.B. Localization of MAO-A and MAO-B in human brain: A step in understanding the therapeutic action of L-deprenyl. Adv. Neurol. 1987, 45, 111–118. [Google Scholar]
- He, W.; Xu, Y.; Ren, X.; Xiang, D.; Lei, K.; Zhang, C.; Liu, D. Vitamin E Ameliorates Lipid Metabolism in Mice with Nonalcoholic Fatty Liver Disease via Nrf2/CES1 Signaling Pathway. Dig. Dis. Sci. 2019, 64, 3182–3191. [Google Scholar] [CrossRef]
- Nan, Y.M.; Wu, W.J.; Fu, N.; Liang, B.L.; Wang, R.Q.; Li, L.X.; Zhao, S.X.; Zhao, J.M.; Yu, J. Antioxidants vitamin E and 1-aminobenzotriazole prevent experimental non-alcoholic steatohepatitis in mice. Scand. J. Gastroenterol. 2009, 44, 1121–1131. [Google Scholar] [CrossRef]
- Phung, N.; Pera, N.; Farrell, G.; Leclercq, I.; Hou, J.Y.; George, J. Pro-oxidant-mediated hepatic fibrosis and effects of antioxidant intervention in murine dietary steatohepatitis. Int. J. Mol. Med. 2009, 24, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Maboud, M.; Menshawy, A.; Menshawy, E.; Emara, A.; Alshandidy, M.; Eid, M. The efficacy of vitamin E in reducing non-alcoholic fatty liver disease: A systematic review, meta-analysis, and meta-regression. Ther. Adv. Gastroenterol. 2020, 13, 1756284820974917. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Peh, H.Y.; Tan, W.S.; Liao, W.; Wong, W.S. Vitamin E therapy beyond cancer: Tocopherol versus tocotrienol. Pharmacol. Ther. 2016, 162, 152–169. [Google Scholar] [CrossRef]
- Rimbach, G.; Minihane, A.M.; Majewicz, J.; Fischer, A.; Pallauf, J.; Virgli, F.; Weinberg, P.D. Regulation of cell signalling by vitamin E. Proc. Nutr. Soc. 2002, 61, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Landrier, J.F.; Gouranton, E.; El Yazidi, C.; Malezet, C.; Balaguer, P.; Borel, P.; Amiot, M.J. Adiponectin expression is induced by vitamin E via a peroxisome proliferator-activated receptor gamma-dependent mechanism. Endocrinology 2009, 150, 5318–5325. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, S.; Kim, S.; Lee, J.; Ha, J.; Oh, H.; Lee, Y.; Kim, Y.; Yoon, Y. Vitamin E (α-tocopherol) consumption influences gut microbiota composition. Int. J. Food Sci. Nutr. 2020, 71, 221–225. [Google Scholar] [CrossRef]
- Ritze, Y.; Bárdos, G.; Claus, A.; Ehrmann, V.; Bergheim, I.; Schwiertz, A.; Bischoff, S.C. Lactobacillus rhamnosus GG protects against non-alcoholic fatty liver disease in mice. PLoS ONE 2014, 9, e80169. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cordero, P.; Nguyen, V.; Oben, J.A. The Role of Vitamins in the Pathogenesis of Non-alcoholic Fatty Liver Disease. Integr. Med. Insights 2016, 11, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Nagashimada, M.; Ota, T. Role of vitamin E in nonalcoholic fatty liver disease. IUBMB life 2019, 71, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ni, Y.; Nagata, N.; Xu, L.; Ota, T. Micronutrient Antioxidants and Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchetta, I.; Carotti, S.; Labbadia, G.; Gentilucci, U.V.; Muda, A.O.; Angelico, F.; Silecchia, G.; Leonetti, F.; Fraioli, A.; Picardi, A.; et al. Liver vitamin D receptor, CYP2R1, and CYP27A1 expression: Relationship with liver histology and vitamin D3 levels in patients with nonalcoholic steatohepatitis or hepatitis C virus. Hepatology 2012, 56, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.G.; Hou, F.F.; Guo, Z.J.; Liang, M.; Wang, G.B.; Zhang, X. 1,25-Dihydroxyvitamin D improved the free fatty-acid-induced insulin resistance in cultured C2C12 cells. Diabetes/Metab. Res. Rev. 2008, 24, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Abramovitch, S.; Dahan-Bachar, L.; Sharvit, E.; Weisman, Y.; Ben Tov, A.; Brazowski, E.; Reif, S. Vitamin D inhibits proliferation and profibrotic marker expression in hepatic stellate cells and decreases thioacetamide-induced liver fibrosis in rats. Gut 2011, 60, 1728–1737. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Wieckowska, A.; Lopez, A.R.; Liu, Y.C.; Zein, N.N.; McCullough, A.J. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: A multicenter validation study. Hepatology 2009, 50, 1072–1078. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, N.; Amani, R.; Hajiani, E.; Cheraghian, B. Does vitamin D improve liver enzymes, oxidative stress, and inflammatory biomarkers in adults with non-alcoholic fatty liver disease? A randomized clinical trial. Endocrine 2014, 47, 70–80. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [Green Version]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Crespo, J.; Cayón, A.; Fernández-Gil, P.; Hernández-Guerra, M.; Mayorga, M.; Domínguez-Díez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef]
- Neyestani, T.R.; Nikooyeh, B.; Alavi-Majd, H.; Shariatzadeh, N.; Kalayi, A.; Tayebinejad, N.; Heravifard, S.; Salekzamani, S.; Zahedirad, M. Improvement of vitamin D status via daily intake of fortified yogurt drink either with or without extra calcium ameliorates systemic inflammatory biomarkers, including adipokines, in the subjects with type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 2005–2011. [Google Scholar] [CrossRef] [Green Version]
- Foroughi, M.; Maghsoudi, Z.; Ghiasvand, R.; Iraj, B.; Askari, G. Effect of Vitamin D Supplementation on C-reactive Protein in Patients with Nonalcoholic Fatty Liver. Int. J. Prev. Med. 2014, 5, 969–975. [Google Scholar]
- Lorvand Amiri, H.; Agah, S.; Tolouei Azar, J.; Hosseini, S.; Shidfar, F.; Mousavi, S.N. Effect of daily calcitriol supplementation with and without calcium on disease regression in non-alcoholic fatty liver patients following an energy-restricted diet: Randomized, controlled, double-blind trial. Clin. Nutr. 2017, 36, 1490–1497. [Google Scholar] [CrossRef]
- Shidfar, F.; Mousavi, S.N.; Lorvand Amiri, H.; Agah, S.; Hoseini, S.; Hajimiresmail, S.J. Reduction of Some Atherogenic Indices in Patients with Non-Alcoholic Fatty Liver by Vitamin D and Calcium Co-Supplementation: A Double Blind Randomized Controlled Clinical Trial. Iranian journal of pharmaceutical research. IJPR 2019, 18, 496–505. [Google Scholar]
- Pickett-Blakely, O.; Young, K.; Carr, R.M. Micronutrients in Nonalcoholic Fatty Liver Disease Pathogenesis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Ai, Y.; Sun, Z.; Peng, C.; Liu, L.; Xiao, X.; Li, J. Homocysteine Induces Hepatic Steatosis Involving ER Stress Response in High Methionine Diet-Fed Mice. Nutrients 2017, 9, 346. [Google Scholar] [CrossRef] [Green Version]
- Talari, H.R.; Molaqanbari, M.R.; Mokfi, M.; Taghizadeh, M.; Bahmani, F.; Tabatabaei, S.M.H.; Sharifi, N. The effects of vitamin B12 supplementation on metabolic profile of patients with non-alcoholic fatty liver disease: A randomized controlled trial. Sci. Rep. 2022, 12, 14047. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Ivancovsky-Wajcman, D.; Fliss-Isakov, N.; Hahn, M.; Webb, M.; Shibolet, O.; Kariv, R.; Tirosh, O. Serum Malondialdehyde is Associated with Non-Alcoholic Fatty Liver and Related Liver Damage Differentially in Men and Women. Antioxidants 2020, 9, 578. [Google Scholar] [CrossRef]
- Racek, J.; Rusnáková, H.; Trefil, L.; Siala, K.K. The influence of folate and antioxidants on homocysteine levels and oxidative stress in patients with hyperlipidemia and hyperhomocysteinemia. Physiol. Res. 2005, 54, 87–95. [Google Scholar] [CrossRef]
- Al-Daghri, N.M.; Rahman, S.; Sabico, S.; Yakout, S.; Wani, K.; Al-Attas, O.S.; Saravanan, P.; Tripathi, G.; McTernan, P.G.; Alokail, M.S. Association of Vitamin B12 with Pro-Inflammatory Cytokines and Biochemical Markers Related to Cardiometabolic Risk in Saudi Subjects. Nutrients 2016, 8, 460. [Google Scholar] [CrossRef] [Green Version]
- Kurt, R.; Yilmaz, Y.; Ermis, F.; Kalayoglu Besisik, S.; Polat, N.; Elitok, A.; Oflaz, H.; Karan, M.A. Folic Acid and vitamin B12 supplementation improves coronary flow reserve in elderly subjects with vitamin B12 deficiency. Arch. Med. Res. 2010, 41, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Setola, E.; Monti, L.D.; Galluccio, E.; Palloshi, A.; Fragasso, G.; Paroni, R.; Magni, F.; Sandoli, E.P.; Lucotti, P.; Costa, S.; et al. Insulin resistance and endothelial function are improved after folate and vitamin B12 therapy in patients with metabolic syndrome: Relationship between homocysteine levels and hyperinsulinemia. Eur. J. Endocrinol. 2004, 151, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, A. The function of bile salts in fat absorption. The solvent properties of dilute micellar solutions of conjugated bile salts. Biochem. J. 1963, 1963. 89, 57. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.D.; Lee, Y.H.; Guo, G.L. The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mol. Asp. Med. 2017, 56, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 50,1955–1966. [Google Scholar] [CrossRef] [Green Version]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef]
- Pierre, J.F.; Martinez, K.B.; Ye, H.; Nadimpalli, A.; Morton, T.C.; Yang, J.; Wang, Q.; Patno, N.; Chang, E.B.; Yin, D.P. Activation of bile acid signaling improves metabolic phenotypes in high-fat diet-induced obese mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G286–G304. [Google Scholar] [CrossRef] [Green Version]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Sannasiddappa, T.H.; Lund, P.A.; Clarke, S.R. In Vitro Antibacterial Activity of Unconjugated and Conjugated Bile Salts on Staphylococcus aureus. Front. Microbiol. 2017, 8, 1581. [Google Scholar] [CrossRef] [Green Version]
- Pineda Torra, I.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.C.; Staels, B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef] [Green Version]
- Nishitsuji, K.; Xiao, J.; Nagatomo, R.; Umemoto, H.; Morimoto, Y.; Akatsu, H.; Tsuneyama, K. Analysis of the gut microbiome and plasma short-chain fatty acid profiles in a spontaneous mouse model of metabolic syndrome. Sci. Rep. 2017, 7, 15876. [Google Scholar] [CrossRef] [Green Version]
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of tight junction permeability by intestinal bacteria and dietary components. J. Nutr. 2011, 141, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Pierantonelli, I.; Rychlicki, C.; Agostinelli, L.; Giordano, D.M.; Gaggini, M.; Fraumene, C.; Saponaro, C.; Manghina, V.; Sartini, L.; Mingarelli, E.; et al. Lack of NLRP3-inflammasome leads to gut-liver axis derangement, gut dysbiosis and a worsened phenotype in a mouse model of NAFLD. Sci. Rep. 2017, 7, 12200. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noga, A.A.; Vance, D.E. A gender-specific role for phosphatidylethanolamine N-methyltransferase-derived phosphatidylcholine in the regulation of plasma high density and very low density lipoproteins in mice. J. Biol. Chem. 2003, 278, 21851–21859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Gammon, M.D.; Zeisel, S.H.; Bradshaw, P.T.; Wetmur, J.G.; Teitelbaum, S.L.; Neugut, A.I.; Santella, R.M.; Chen, J. High intakes of choline and betaine reduce breast cancer mortality in a population-based study. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 4022–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimbeni, F.; Pais, R.; Bellentani, S.; Day, C.P.; Ratziu, V.; Loria, P.; Lonardo, A. From NAFLD in clinical practice to answers from guidelines. J. Hepatol. 2013, 59, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.L.; Guo, H.; Zhang, C.S.; Lin, S.Y.; Yin, Z.; Peng, Y.; Luo, H.; Shi, Y.; Lian, G.; Zhang, C.; et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013, 18, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Scott, J.W.; Galic, S.; Graham, K.L.; Foitzik, R.; Ling, N.X.; Dite, T.A.; Issa, S.M.; Langendorf, C.G.; Weng, Q.P.; Thomas, H.E.; et al. Inhibition of AMP-Activated Protein Kinase at the Allosteric Drug-Binding Site Promotes Islet Insulin Release. Chem. Biol. 2015, 22, 705–711. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Görgün, C.Z.; Carling, D.; et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig. 2004, 113, 1582–1588. [Google Scholar] [CrossRef] [Green Version]
- Vatner, D.F.; Majumdar, S.K.; Kumashiro, N.; Petersen, M.C.; Rahimi, Y.; Gattu, A.K.; Bears, M.; Camporez, J.P.; Cline, G.W.; Jurczak, M.J.; et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc. Natl. Acad. Sci. USA 2015, 112, 1143–1148. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.J.; Gauthier, M.S.; Hess, D.T.; Apovian, C.M.; Cacicedo, J.M.; Gokce, N.; Farb, M.; Valentine, R.J.; Ruderman, N.B. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J. Lipid Res. 2012, 53, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Mottillo, E.P.; Desjardins, E.M.; Crane, J.D.; Smith, B.K.; Green, A.E.; Ducommun, S.; Henriksen, T.I.; Rebalka, I.A.; Razi, A.; Sakamoto, K.; et al. Lack of Adipocyte AMPK Exacerbates Insulin Resistance and Hepatic Steatosis through Brown and Beige Adipose Tissue Function. Cell Metab. 2016, 24, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Mancini, S.J.; White, A.D.; Bijland, S.; Rutherford, C.; Graham, D.; Richter, E.A.; Viollet, B.; Touyz, R.M.; Palmer, T.M.; Salt, I.P. Activation of AMP-activated protein kinase rapidly suppresses multiple pro-inflammatory pathways in adipocytes including IL-1 receptor-associated kinase-4 phosphorylation. Mol. Cell. Endocrinol. 2017, 440, 44–56. [Google Scholar] [CrossRef]
- Caligiuri, A.; Bertolani, C.; Guerra, C.T.; Aleffi, S.; Galastri, S.; Trappoliere, M.; Vizzutti, F.; Gelmini, S.; Laffi, G.; Pinzani, M.; et al. Adenosine monophosphate-activated protein kinase modulates the activated phenotype of hepatic stellate cells. Hepatology 2008, 47, 668–676. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Gleason, C.E.; Lu, D.; Witters, L.A.; Newgard, C.B.; Birnbaum, M.J. The role of AMPK and mTOR in nutrient sensing in pancreatic beta-cells. J. Biol. Chem. 2007, 282, 10341–10351. [Google Scholar] [CrossRef] [Green Version]
- Chotechuang, N.; Azzout-Marniche, D.; Bos, C.; Chaumontet, C.; Gausserès, N.; Steiler, T.; Gaudichon, C.; Tomé, D. mTOR, AMPK, and GCN2 coordinate the adaptation of hepatic energy metabolic pathways in response to protein intake in the rat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1313–E1323. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. American journal of physiology. Endocrinol. Metab. 2016, 311, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Silvestri, C.; Di Marzo, V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 2013, 17, 475–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazier, W.; Saucisse, N.; Gatta-Cherifi, B.; Cota, D. The Endocannabinoid System: Pivotal Orchestrator of Obesity and Metabolic Disease. Trends Endocrinol. Metab. TEM 2015, 26, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Assa-Glazer, T.; Gorelick, J.; Sela, N.; Nyska, A.; Bernstein, N.; Madar, Z. Cannabis Extracts Affected Metabolic Syndrome Parameters in Mice Fed High-Fat/Cholesterol Diet. Cannabis Cannabinoid Res. 2020, 5, 202–214. [Google Scholar] [CrossRef]
- Di Marzo, V.; Piscitelli, F.; Mechoulam, R. Cannabinoids and endocannabinoids in metabolic disorders with focus on diabetes. Handb. Exp. Pharmacol. 2011, 203, 75–104. [Google Scholar] [CrossRef]
- Dibba, P.; Li, A.; Cholankeril, G.; Iqbal, U.; Gadiparthi, C.; Khan, M.A.; Kim, D.; Ahmed, A. Mechanistic Potential and Therapeutic Implications of Cannabinoids in Nonalcoholic Fatty Liver Disease. Medicines 2018, 5, 47. [Google Scholar] [CrossRef] [Green Version]
- Mallat, A.; Lotersztajn, S. Endocannabinoids and their role in fatty liver disease. Dig. Dis. 2010, 28, 261–266. [Google Scholar] [CrossRef]
- Jadoon, K.A.; Ratcliffe, S.H.; Barrett, D.A.; Thomas, E.L.; Stott, C.; Bell, J.D.; O’Sullivan, S.E.; Tan, G.D. Efficacy and Safety of Cannabidiol and Tetrahydrocannabivarin on Glycemic and Lipid Parameters in Patients with Type 2 Diabetes: A Randomized, Double-Blind, Placebo-Controlled, Parallel Group Pilot Study. Diabetes Care 2016, 39, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- McPartland, J.M.; Duncan, M.; Di Marzo, V.; Pertwee, R.G. Are cannabidiol and Δ(9) -tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br. J. Pharmacol. 2015, 172, 737–753. [Google Scholar] [CrossRef] [Green Version]
- Wu, J. Cannabis, cannabinoid receptors, and endocannabinoid system: Yesterday, today, and tomorrow. Acta Pharmacol. Sin. 2019, 40, 297–299. [Google Scholar] [CrossRef]
- Kirkham, T.C. Cannabinoids and appetite: Food craving and food pleasure. Int. Rev. Psychiatry 2009, 21, 163–171. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sable, S.; Ouwerkerk, R.; Mari, A.; Gharib, A.M.; Walter, M.; Courville, A.; Hall, G.; Chen, K.Y.; Volkow, N.D.; et al. Metabolic effects of chronic cannabis smoking. Diabetes Care 2013, 36, 2415–2422. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Mu, W.; Cheng, X.F.; Liu, Y.; Lv, Q.Z.; Liu, G.L.; Zhang, J.G.; Li, X.Y. Potential Nexus of Non-alcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus: Insulin Resistance Between Hepatic and Peripheral Tissues. Front. Pharmacol. 2019, 9, 1566. [Google Scholar] [CrossRef] [Green Version]
- Jorgačević, B.; Vučević, D.; Vesković, M.; Mladenović, D.; Vukićević, D.; Vukićević, R.J.; Todorović, V.; Radosavljević, T. The effect of cannabinoid receptor 1 blockade on adipokine and proinflammatory cytokine concentration in adipose and hepatic tissue in mice with nonalcoholic fatty liver disease. Can. J. Physiol. Pharmacol. 2019, 97, 120–129. [Google Scholar] [CrossRef]
- Hirvonen, J.; Goodwin, R.S.; Li, C.T.; Terry, G.E.; Zoghbi, S.S.; Morse, C.; Pike, V.W.; Volkow, N.D.; Huestis, M.A.; Innis, R.B. Reversible and regionally selective downregulation of brain cannabinoid CB1 receptors in chronic daily cannabis smokers. Mol. Psychiatry 2012, 17, 642–649. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munteanu, C.; Schwartz, B. The Effect of Bioactive Aliment Compounds and Micronutrients on Non-Alcoholic Fatty Liver Disease. Antioxidants 2023, 12, 903. https://doi.org/10.3390/antiox12040903
Munteanu C, Schwartz B. The Effect of Bioactive Aliment Compounds and Micronutrients on Non-Alcoholic Fatty Liver Disease. Antioxidants. 2023; 12(4):903. https://doi.org/10.3390/antiox12040903
Chicago/Turabian StyleMunteanu, Camelia, and Betty Schwartz. 2023. "The Effect of Bioactive Aliment Compounds and Micronutrients on Non-Alcoholic Fatty Liver Disease" Antioxidants 12, no. 4: 903. https://doi.org/10.3390/antiox12040903