
Multifunctional Metallothioneins as a Target for Neuroprotection in Parkinson’s Disease


Abstract
:
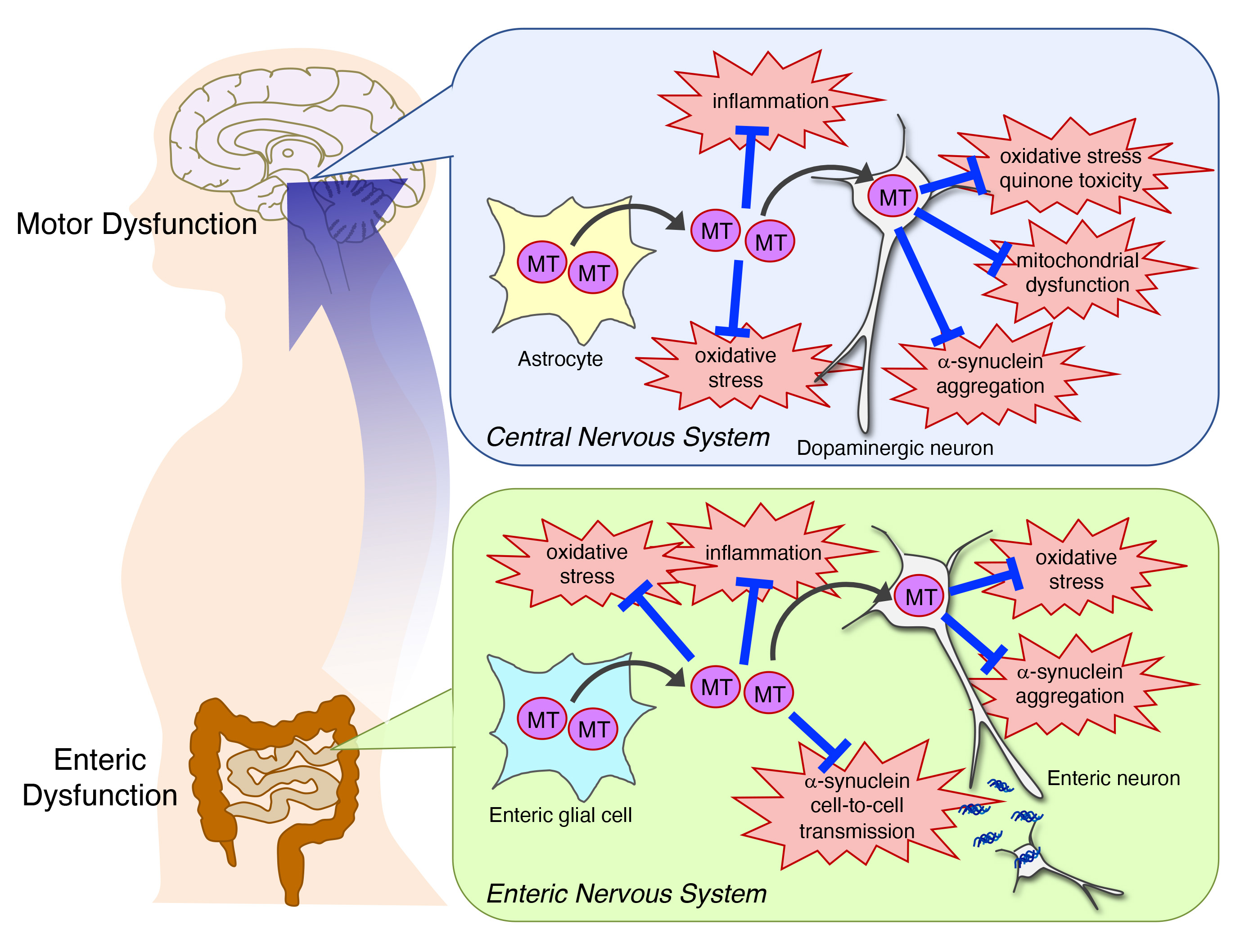{kind=link}
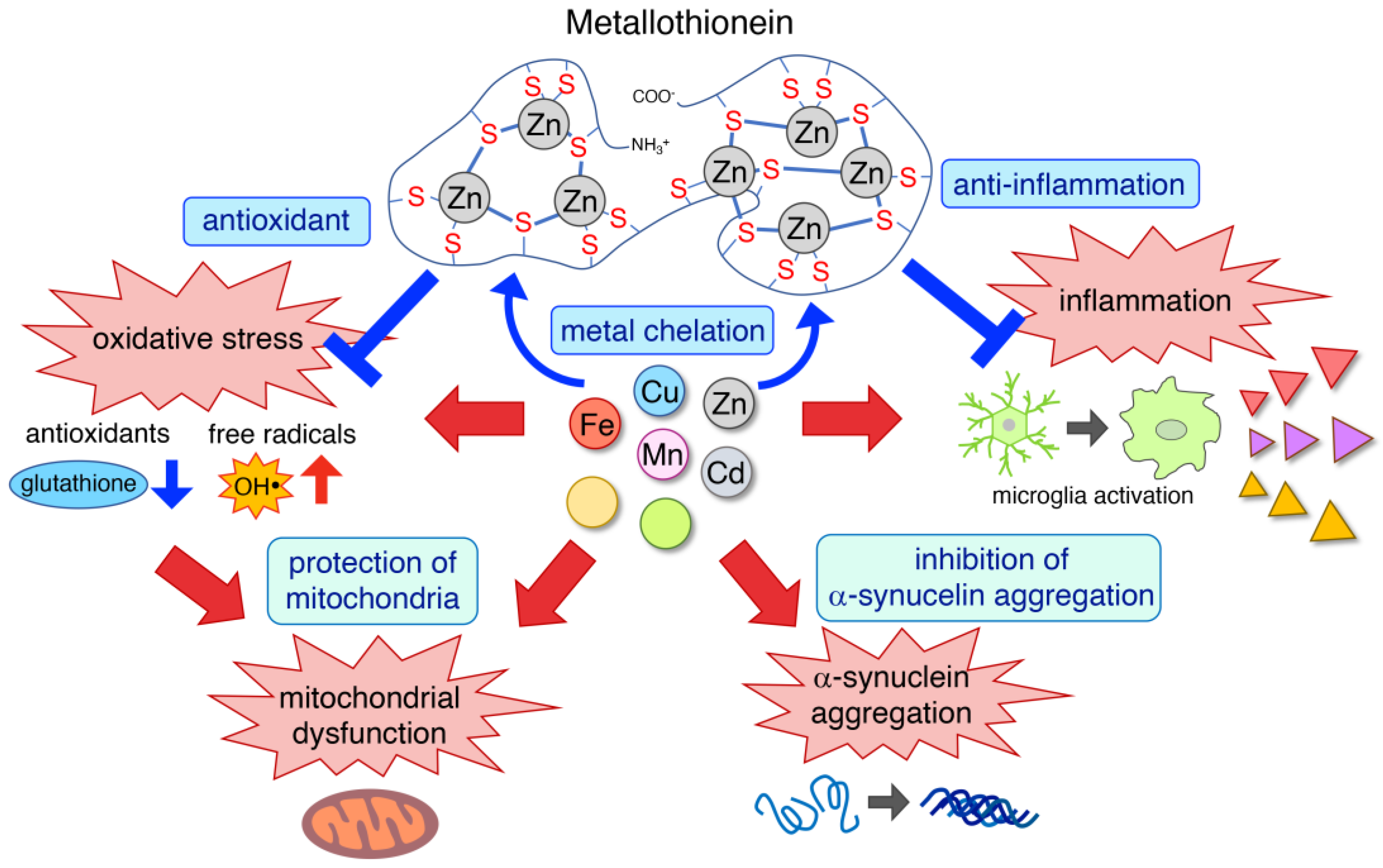{kind=link}
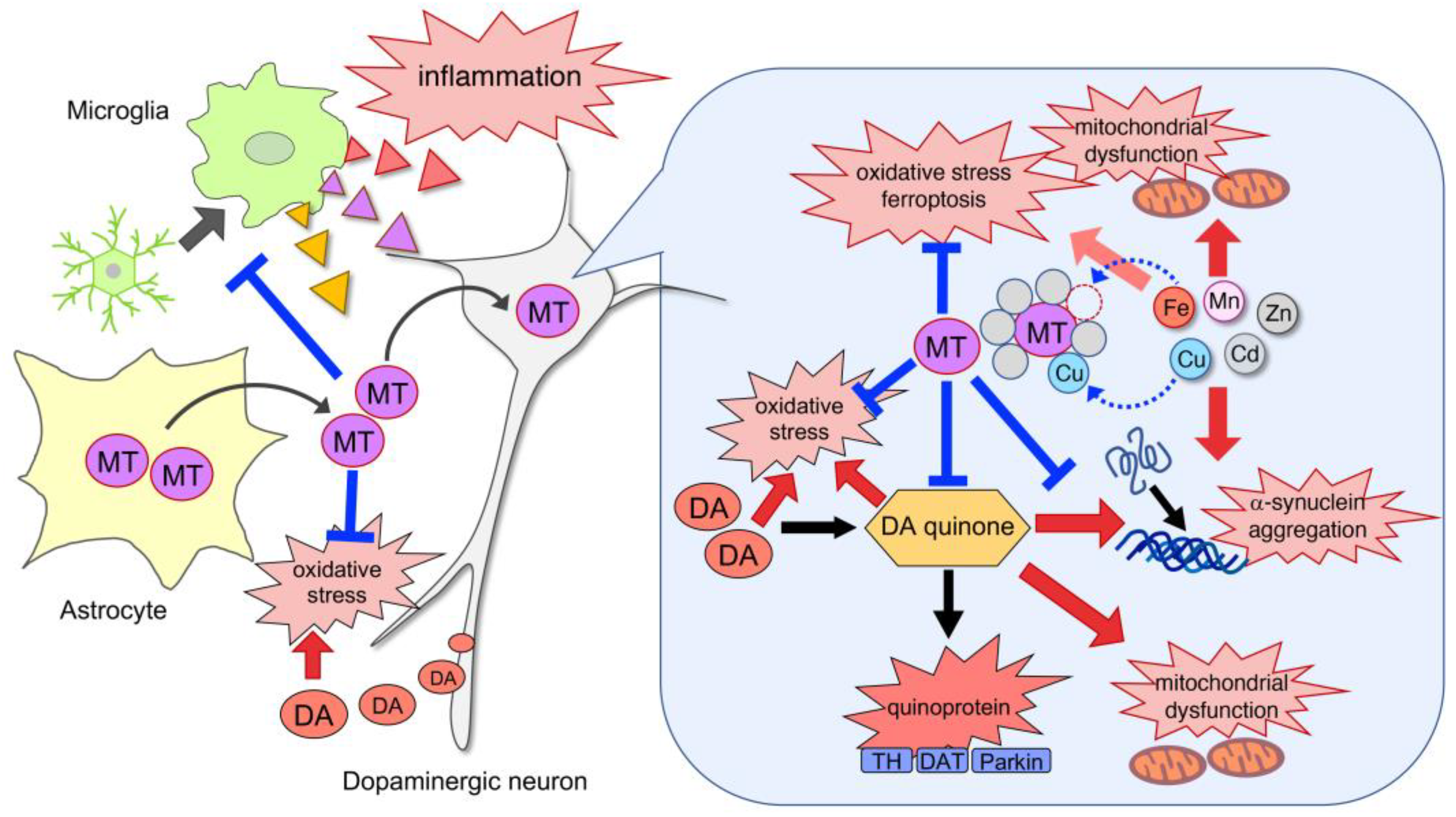{kind=link}
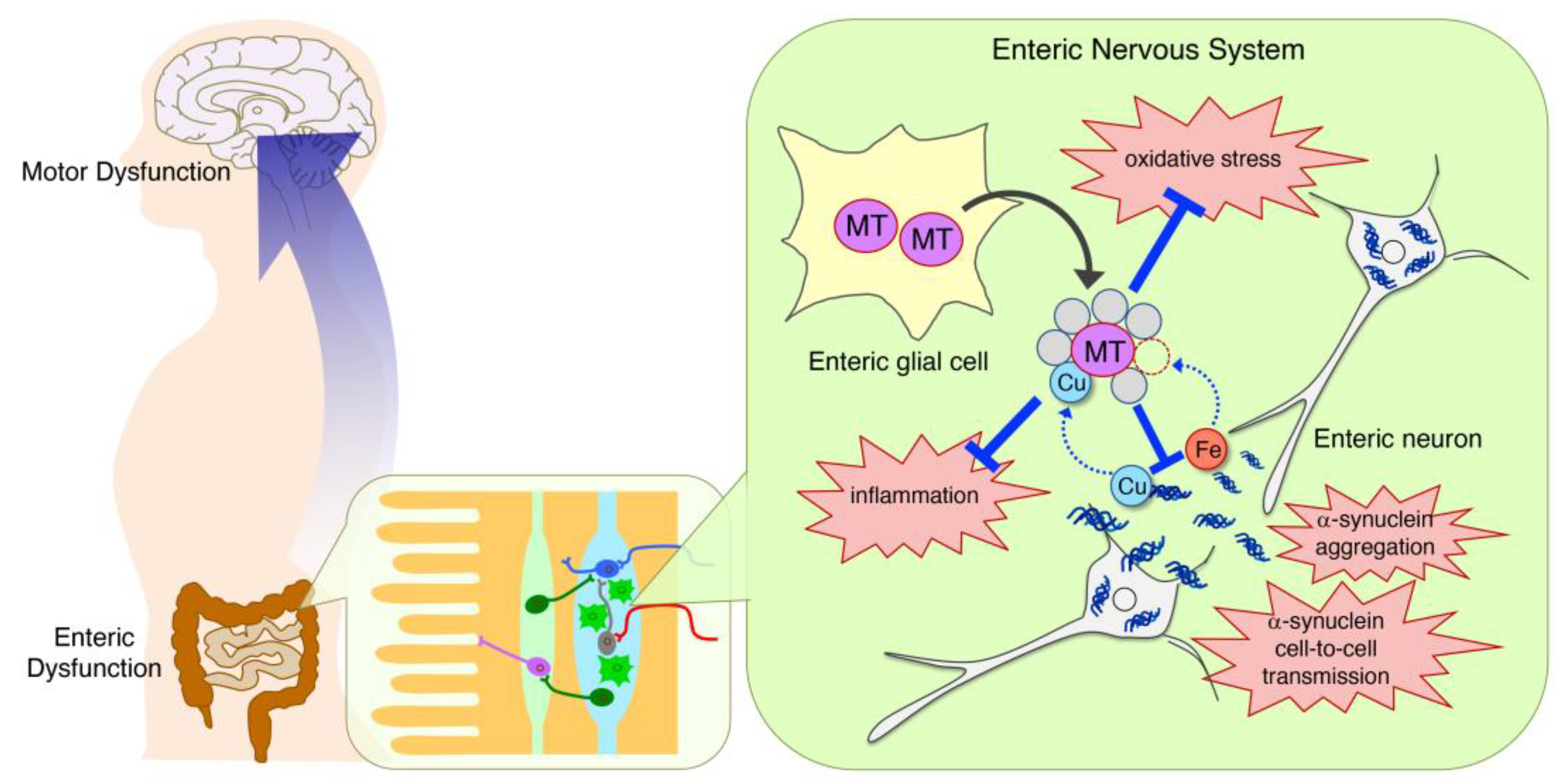{kind=link}
1. Introduction
2. Gene Expression and Induction of MTs by Stimulation
2.1. Expression and Induction of MTs in the CNS
2.2. Expression and Induction of MTs in the ENS
2.3. Expression of MTs in the Brains of PD Patients
3. Protective Functions of MTs against Etiopathogenesis in PD
3.1. Metal Chelation
3.1.1. Iron
3.1.2. Copper
3.1.3. Zinc
3.1.4. Manganese
3.1.5. Cadmium
3.1.6. Other Metals
3.2. Antioxidant
3.3. Anti-Inflammation
3.4. Protection of Mitochondria
3.5. Inhibition of α-Synuclein Aggregation
4. Neuroprotective Approaches Targeting MTs in PD
4.1. Neuroprotection against Nigral Dopaminergic Neurodegeneration
4.2. Neuroprotection in the Intestinal Myenteric Plexus
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogra, N.; Mani, R.J.; Katare, D.P. The Gut-Brain Axis: Two Ways Signaling in Parkinson’s Disease. Cell. Mol. Neurobiol. 2022, 42, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Ball, N.; Teo, W.P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Pyatha, S.; Kim, H.; Lee, D.; Kim, K. Association between Heavy Metal Exposure and Parkinson’s Disease: A Review of the Mechanisms Related to Oxidative Stress. Antioxidants 2022, 11, 2467. [Google Scholar] [CrossRef] [PubMed]
- Ghassaban, K.; He, N.; Sethi, S.K.; Huang, P.; Chen, S.; Yan, F.; Haacke, E.M. Regional High Iron in the Substantia Nigra Differentiates Parkinson’s Disease Patients From Healthy Controls. Front. Aging Neurosci. 2019, 11, 106. [Google Scholar] [CrossRef] [Green Version]
- Sikora, J.; Ouagazzal, A.M. Synaptic Zinc: An Emerging Player in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4724. [Google Scholar] [CrossRef]
- Ziller, A.; Fraissinet-Tachet, L. Metallothionein diversity and distribution in the tree of life: A multifunctional protein. Metallomics 2018, 10, 1549–1559. [Google Scholar] [CrossRef]
- Penkowa, M. Metallothioneins are multipurpose neuroprotectants during brain pathology. FEBS J. 2006, 273, 1857–1870. [Google Scholar] [CrossRef]
- Okita, Y.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Chung, R.S.; Faller, P.; Pountney, D.L. Metallothionein, Copper and Alpha-Synuclein in Alpha-Synucleinopathies. Front. Neurosci. 2017, 11, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imafuku, F.; Miyazaki, I.; Sun, J.; Kamimai, S.; Shimizu, T.; Toyota, T.; Okamoto, Y.; Isooka, N.; Kikuoka, R.; Kitamura, Y.; et al. Central and Enteric Neuroprotective Effects by Eucommia ulmoides Extracts on Neurodegeneration in Rotenone-induced Parkinsonian Mouse. Acta Med. Okayama 2022, 76, 373–383. [Google Scholar] [CrossRef]
- Isooka, N.; Miyazaki, I.; Kikuoka, R.; Wada, K.; Nakayama, E.; Shin, K.; Yamamoto, D.; Kitamura, Y.; Asanuma, M. Dopaminergic neuroprotective effects of rotigotine via 5-HT1A receptors: Possibly involvement of metallothionein expression in astrocytes. Neurochem. Int. 2020, 132, 104608. [Google Scholar] [CrossRef] [PubMed]
- Kikuoka, R.; Miyazaki, I.; Kubota, N.; Maeda, M.; Kagawa, D.; Moriyama, M.; Sato, A.; Murakami, S.; Kitamura, Y.; Sendo, T.; et al. Mirtazapine exerts astrocyte-mediated dopaminergic neuroprotection. Sci. Rep. 2020, 10, 20698. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Murakami, S.; Takeshima, M.; Torigoe, N.; Kitamura, Y.; Miyoshi, K. Targeting 5-HT(1A) receptors in astrocytes to protect dopaminergic neurons in Parkinsonian models. Neurobiol. Dis. 2013, 59, 244–256. [Google Scholar] [CrossRef]
- Miyazaki, I.; Isooka, N.; Wada, K.; Kikuoka, R.; Kitamura, Y.; Asanuma, M. Effects of Enteric Environmental Modification by Coffee Components on Neurodegeneration in Rotenone-Treated Mice. Cells 2019, 8, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitriu, A.; Golji, J.; Labadorf, A.T.; Gao, B.; Beach, T.G.; Myers, R.H.; Longo, K.A.; Latourelle, J.C. Integrative analyses of proteomics and RNA transcriptomics implicate mitochondrial processes, protein folding pathways and GWAS loci in Parkinson disease. BMC Med. Genom. 2016, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaab, E.; Schneider, R. Comparative pathway and network analysis of brain transcriptome changes during adult aging and in Parkinson’s disease. Neurobiol. Dis. 2015, 74, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Michael, G.J.; Esmailzadeh, S.; Moran, L.B.; Christian, L.; Pearce, R.K.; Graeber, M.B. Up-regulation of metallothionein gene expression in parkinsonian astrocytes. Neurogenetics 2011, 12, 295–305. [Google Scholar] [CrossRef]
- Chung, R.S.; Adlard, P.A.; Dittmann, J.; Vickers, J.C.; Chuah, M.I.; West, A.K. Neuron-glia communication: Metallothionein expression is specifically up-regulated by astrocytes in response to neuronal injury. J. Neurochem. 2004, 88, 454–461. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Kikkawa, Y.; Takeshima, M.; Murakami, S.; Miyoshi, K.; Sogawa, N.; Kita, T. Astrocyte-derived metallothionein protects dopaminergic neurons from dopamine quinone toxicity. Glia 2011, 59, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y.; Lee, S.J. Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol. Brain 2020, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, M. Metallothioneins: Historical review and state of knowledge. Talanta 1998, 46, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, S.; Kramer, K.K.; Berman, N.E.; Dalton, T.P.; Andrews, G.K.; Klaassen, C.D. Constitutive expression of metallothionein genes in mouse brain. Toxicol. Appl. Pharmacol. 1995, 131, 144–154. [Google Scholar] [CrossRef]
- McLeary, F.A.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Radford, R.A.W.; Okita, Y.; Faller, P.; Chung, R.S.; Pountney, D.L. Switching on Endogenous Metal Binding Proteins in Parkinson’s Disease. Cells 2019, 8, 179. [Google Scholar] [CrossRef] [Green Version]
- Andrews, G.K. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem. Pharmacol. 2000, 59, 95–104. [Google Scholar] [CrossRef]
- Laity, J.H.; Andrews, G.K. Understanding the mechanisms of zinc-sensing by metal-response element binding transcription factor-1 (MTF-1). Arch. Biochem. Biophys. 2007, 463, 201–210. [Google Scholar] [CrossRef]
- Palmiter, R.D. Regulation of metallothionein genes by heavy metals appears to be mediated by a zinc-sensitive inhibitor that interacts with a constitutively active transcription factor, MTF-1. Proc. Natl. Acad. Sci. USA 1994, 91, 1219–1223. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [Green Version]
- Chung, R.S.; Penkowa, M.; Dittmann, J.; King, C.E.; Bartlett, C.; Asmussen, J.W.; Hidalgo, J.; Carrasco, J.; Leung, Y.K.; Walker, A.K.; et al. Redefining the role of metallothionein within the injured brain: Extracellular metallothioneins play an important role in the astrocyte-neuron response to injury. J. Biol. Chem. 2008, 283, 15349–15358. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, J.; Hidalgo, J. Endotoxin and intracerebroventricular injection of IL-1 and IL-6 induce rat brain metallothionein-I and -II. Neurochem. Int. 1998, 32, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wang, L.; Li, L.; Huang, Z.; Ye, L. Metallothionein 1: A New Spotlight on Inflammatory Diseases. Front. Immunol. 2021, 12, 739918. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Miyazaki, I.; Sogawa, N.; Miyoshi, K.; Asanuma, M. Neuroprotective effects of metallothionein against rotenone-induced myenteric neurodegeneration in parkinsonian mice. Neurotox. Res. 2014, 26, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Haton, C.; Francois, A.; Vandamme, M.; Wysocki, J.; Griffiths, N.M.; Benderitter, M. Imbalance of the antioxidant network of mouse small intestinal mucosa after radiation exposure. Radiat. Res. 2007, 167, 445–453. [Google Scholar] [CrossRef]
- Zhang, Y.; James, M.; Middleton, F.A.; Davis, R.L. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 137B, 5–16. [Google Scholar] [CrossRef]
- Sahoo, K.; Sharma, A. Understanding the mechanistic roles of environmental heavy metal stressors in regulating ferroptosis: Adding new paradigms to the links with diseases. Apoptosis 2023. [Google Scholar] [CrossRef] [PubMed]
- Vellingiri, B.; Suriyanarayanan, A.; Abraham, K.S.; Venkatesan, D.; Iyer, M.; Raj, N.; Gopalakrishnan, A.V. Influence of heavy metals in Parkinson’s disease: An overview. J. Neurol. 2022, 269, 5798–5811. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Li, J.; Niu, S.; Xue, Y.; Tang, M. Neurotoxicity of metal-containing nanoparticles and implications in glial cells. J. Appl. Toxicol. 2021, 41, 65–81. [Google Scholar] [CrossRef]
- Nabi, M.; Tabassum, N. Role of Environmental Toxicants on Neurodegenerative Disorders. Front. Toxicol. 2022, 4, 837579. [Google Scholar] [CrossRef]
- Zhang, N.; Xiong, G.; Liu, Z. Toxicity of metal-based nanoparticles: Challenges in the nano era. Front. Bioeng. Biotechnol. 2022, 10, 1001572. [Google Scholar] [CrossRef]
- Juarez-Rebollar, D.; Rios, C.; Nava-Ruiz, C.; Mendez-Armenta, M. Metallothionein in Brain Disorders. Oxid. Med. Cell. Longev. 2017, 2017, 5828056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waalkes, M.P.; Harvey, M.J.; Klaassen, C.D. Relative in vitro affinity of hepatic metallothionein for metals. Toxicol. Lett. 1984, 20, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.; Philcox, J.C.; Carey, L.C.; Rofe, A.M. Metallothionein: The multipurpose protein. Cell. Mol. Life Sci. 2002, 59, 627–647. [Google Scholar] [CrossRef]
- Gotz, M.E.; Double, K.; Gerlach, M.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. Ann. N.Y. Acad. Sci. 2004, 1012, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Garza-Lombo, C.; Posadas, Y.; Quintanar, L.; Gonsebatt, M.E.; Franco, R. Neurotoxicity Linked to Dysfunctional Metal Ion Homeostasis and Xenobiotic Metal Exposure: Redox Signaling and Oxidative Stress. Antioxid. Redox Signal. 2018, 28, 1669–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, R.M.; Wagener, A.; Fietzek, U.M.; Klopstock, T.; Mosharov, E.V.; Zucca, F.A.; Sulzer, D.; Zecca, L.; Burbulla, L.F. Interactions of dopamine, iron, and alpha-synuclein linked to dopaminergic neuron vulnerability in Parkinson’s disease and Neurodegeneration with Brain Iron Accumulation disorders. Neurobiol. Dis. 2022, 175, 105920. [Google Scholar] [CrossRef]
- Zecca, L.; Gallorini, M.; Schunemann, V.; Trautwein, A.X.; Gerlach, M.; Riederer, P.; Vezzoni, P.; Tampellini, D. Iron, neuromelanin and ferritin content in the substantia nigra of normal subjects at different ages: Consequences for iron storage and neurodegenerative processes. J. Neurochem. 2001, 76, 1766–1773. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased nigral iron content in postmortem parkinsonian brain. Lancet 1987, 2, 1219–1220. [Google Scholar] [CrossRef]
- Genoud, S.; Roberts, B.R.; Gunn, A.P.; Halliday, G.M.; Lewis, S.J.G.; Ball, H.J.; Hare, D.J.; Double, K.L. Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics 2017, 9, 1447–1455. [Google Scholar] [CrossRef] [Green Version]
- Stokes, A.H.; Hastings, T.G.; Vrana, K.E. Cytotoxic and genotoxic potential of dopamine. J. Neurosci. Res. 1999, 55, 659–665. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Jansen van Rensburg, Z.; Abrahams, S.; Bardien, S.; Kenyon, C. Toxic Feedback Loop Involving Iron, Reactive Oxygen Species, alpha-Synuclein and Neuromelanin in Parkinson’s Disease and Intervention with Turmeric. Mol. Neurobiol. 2021, 58, 5920–5936. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alcocer, A.; Duarte-Jurado, A.P.; Soto-Dominguez, A.; Loera-Arias, M.J.; Villarreal-Silva, E.E.; Saucedo-Cardenas, O.; de Oca-Luna, R.M.; Garcia-Garcia, A.; Rodriguez-Rocha, H. Unscrambling the Role of Redox-Active Biometals in Dopaminergic Neuronal Death and Promising Metal Chelation-Based Therapy for Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 1256. [Google Scholar] [CrossRef] [PubMed]
- Febbraro, F.; Andersen, K.J.; Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. Chronic intranasal deferoxamine ameliorates motor defects and pathology in the alpha-synuclein rAAV Parkinson’s model. Exp. Neurol. 2013, 247, 45–58. [Google Scholar] [CrossRef]
- Dexter, D.T.; Statton, S.A.; Whitmore, C.; Freinbichler, W.; Weinberger, P.; Tipton, K.F.; Della Corte, L.; Ward, R.J.; Crichton, R.R. Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson’s disease after peripheral administration. J. Neural Transm. 2011, 118, 223–231. [Google Scholar] [CrossRef]
- Kaur, D.; Yantiri, F.; Rajagopalan, S.; Kumar, J.; Mo, J.Q.; Boonplueang, R.; Viswanath, V.; Jacobs, R.; Yang, L.; Beal, M.F.; et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: A novel therapy for Parkinson’s disease. Neuron 2003, 37, 899–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garcon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Luo, M.; Xiong, B.; Liu, X. Upregulation of Metallothionein 1 G (MT1G) Negatively Regulates Ferroptosis in Clear Cell Renal Cell Carcinoma by Reducing Glutathione Consumption. J. Oncol. 2022, 2022, 4000617. [Google Scholar] [CrossRef]
- Orihuela, R.; Fernandez, B.; Palacios, O.; Valero, E.; Atrian, S.; Watt, R.K.; Dominguez-Vera, J.M.; Capdevila, M. Ferritin and metallothionein: Dangerous liaisons. Chem. Commun. 2011, 47, 12155–12157. [Google Scholar] [CrossRef]
- Tsang, T.; Davis, C.I.; Brady, D.C. Copper biology. Curr. Biol. 2021, 31, R421–R427. [Google Scholar] [CrossRef] [PubMed]
- Amoros, R.; Murcia, M.; Gonzalez, L.; Soler-Blasco, R.; Rebagliato, M.; Iniguez, C.; Carrasco, P.; Vioque, J.; Broberg, K.; Levi, M.; et al. Maternal copper status and neuropsychological development in infants and preschool children. Int. J. Hyg. Environ. Health 2019, 222, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarnacka, B.; Jopowicz, A.; Maslinska, M. Copper, Iron, and Manganese Toxicity in Neuropsychiatric Conditions. Int. J. Mol. Sci. 2021, 22, 7820. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A.; et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 2011, 303, 95–99. [Google Scholar] [CrossRef]
- Pall, H.S.; Williams, A.C.; Blake, D.R.; Lunec, J.; Gutteridge, J.M.; Hall, M.; Taylor, A. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet 1987, 2, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Boll, M.C.; Alcaraz-Zubeldia, M.; Montes, S.; Rios, C. Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NO(x) content in the CSF. A different marker profile in four neurodegenerative diseases. Neurochem. Res. 2008, 33, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Letelier, M.E.; Faundez, M.; Jara-Sandoval, J.; Molina-Berrios, A.; Cortes-Troncoso, J.; Aracena-Parks, P.; Marin-Catalan, R. Mechanisms underlying the inhibition of the cytochrome P450 system by copper ions. J. Appl. Toxicol. 2009, 29, 695–702. [Google Scholar] [CrossRef]
- Paik, S.R.; Shin, H.J.; Lee, J.H. Metal-catalyzed oxidation of alpha-synuclein in the presence of Copper(II) and hydrogen peroxide. Arch. Biochem. Biophys. 2000, 378, 269–277. [Google Scholar] [CrossRef]
- Meloni, G.; Vasak, M. Redox activity of alpha-synuclein-Cu is silenced by Zn(7)-metallothionein-3. Free Radic. Biol. Med. 2011, 50, 1471–1479. [Google Scholar] [CrossRef]
- Chasapis, C.T.; Ntoupa, P.A.; Spiliopoulou, C.A.; Stefanidou, M.E. Recent aspects of the effects of zinc on human health. Arch. Toxicol. 2020, 94, 1443–1460. [Google Scholar] [CrossRef]
- Pavic, M.; Turcic, P.; Ljubojevic, M. Forgotten partners and function regulators of inducible metallothioneins. Arh. Hig. Rada. Toksikol. 2019, 70, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Cui, W.; Tan, Y.; Luo, P.; Chen, Q.; Zhang, C.; Qu, W.; Miao, L.; Cai, L. Zinc is essential for the transcription function of Nrf2 in human renal tubule cells in vitro and mouse kidney in vivo under the diabetic condition. J. Cell. Mol. Med. 2014, 18, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Li, N.; Sheng, W.; Ji, X.; Liang, X.; Kong, B.; Yin, P.; Li, Y.; Zhang, X.; Liu, K. Toxicity of different zinc oxide nanomaterials and dose-dependent onset and development of Parkinson’s disease-like symptoms induced by zinc oxide nanorods. Environ. Int. 2021, 146, 106179. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Wu, J.; Xu, Y.; Ding, C.C.; Mestre, A.A.; Lin, C.C.; Yang, W.H.; Chi, J.T. Zinc transporter ZIP7 is a novel determinant of ferroptosis. Cell Death Dis. 2021, 12, 198. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, Y.; Ma, G. Modulation Effects of Fe(3+), Zn(2+), and Cu(2+) Ions on the Amyloid Fibrillation of alpha-Synuclein: Insights from a FTIR Investigation. Molecules 2022, 27, 8383. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Muzzioli, M.; Giacconi, R. Zinc and immunoresistance to infection in aging: New biological tools. Trends Pharmacol. Sci. 2000, 21, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Aschner, J.L.; Aschner, M. Nutritional aspects of manganese homeostasis. Mol. Aspects Med. 2005, 26, 353–362. [Google Scholar] [CrossRef]
- Chen, P.; Bornhorst, J.; Aschner, M. Manganese metabolism in humans. Front. Biosci. 2018, 23, 1655–1679. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Finley, E.J.; Gavin, C.E.; Aschner, M.; Gunter, T.E. Manganese neurotoxicity and the role of reactive oxygen species. Free Radic. Biol. Med. 2013, 62, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Martins, A.C., Jr.; Gubert, P.; Villas Boas, G.R.; Meirelles Paes, M.; Santamaria, A.; Lee, E.; Tinkov, A.A.; Bowman, A.B.; Aschner, M. Manganese-induced neurodegenerative diseases and possible therapeutic approaches. Expert Rev. Neurother. 2020, 20, 1109–1121. [Google Scholar] [CrossRef]
- Racette, B.A.; Searles Nielsen, S.; Criswell, S.R.; Sheppard, L.; Seixas, N.; Warden, M.N.; Checkoway, H. Dose-dependent progression of parkinsonism in manganese-exposed welders. Neurology. 2017, 88, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Guilarte, T.R.; Burton, N.C.; McGlothan, J.L.; Verina, T.; Zhou, Y.; Alexander, M.; Pham, L.; Griswold, M.; Wong, D.F.; Syversen, T.; et al. Impairment of nigrostriatal dopamine neurotransmission by manganese is mediated by pre-synaptic mechanism(s): Implications to manganese-induced parkinsonism. J. Neurochem. 2008, 107, 1236–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, J.A.; Li, Z.; Sridhar, S.; Khoshbouei, H. The effect of manganese on dopamine toxicity and dopamine transporter (DAT) in control and DAT transfected HEK cells. Neurotoxicology 2013, 35, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Harischandra, D.S.; Rokad, D.; Neal, M.L.; Ghaisas, S.; Manne, S.; Sarkar, S.; Panicker, N.; Zenitsky, G.; Jin, H.; Lewis, M.; et al. Manganese promotes the aggregation and prion-like cell-to-cell exosomal transmission of alpha-synuclein. Sci. Signal. 2019, 12, eaau4543. [Google Scholar] [CrossRef]
- Xu, B.; Huang, S.; Liu, Y.; Wan, C.; Gu, Y.; Wang, D.; Yu, H. Manganese promotes alpha-synuclein amyloid aggregation through the induction of protein phase transition. J. Biol. Chem. 2022, 298, 101469. [Google Scholar] [CrossRef] [PubMed]
- Erikson, K.M.; Suber, R.L.; Aschner, M. Glutamate/aspartate transporter (GLAST), taurine transporter and metallothionein mRNA levels are differentially altered in astrocytes exposed to manganese chloride, manganese phosphate or manganese sulfate. Neurotoxicology 2002, 23, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branca, J.J.V.; Fiorillo, C.; Carrino, D.; Paternostro, F.; Taddei, N.; Gulisano, M.; Pacini, A.; Becatti, M. Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System. Antioxidants 2020, 9, 492. [Google Scholar] [CrossRef]
- Shukla, A.; Shukla, G.S.; Srimal, R.C. Cadmium-induced alterations in blood-brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum. Exp. Toxicol. 1996, 15, 400–405. [Google Scholar] [CrossRef]
- Ruczaj, A.; Brzoska, M.M. Environmental exposure of the general population to cadmium as a risk factor of the damage to the nervous system: A critical review of current data. J. Appl. Toxicol. 2023, 43, 66–88. [Google Scholar] [CrossRef]
- Ma, Y.; Su, Q.; Yue, C.; Zou, H.; Zhu, J.; Zhao, H.; Song, R.; Liu, Z. The Effect of Oxidative Stress-Induced Autophagy by Cadmium Exposure in Kidney, Liver, and Bone Damage, and Neurotoxicity. Int. J. Mol. Sci. 2022, 23, 13491. [Google Scholar] [CrossRef]
- Okuda, B.; Iwamoto, Y.; Tachibana, H.; Sugita, M. Parkinsonism after acute cadmium poisoning. Clin. Neurol. Neurosurg. 1997, 99, 263–265. [Google Scholar] [CrossRef]
- Gupta, R.; Shukla, R.K.; Pandey, A.; Sharma, T.; Dhuriya, Y.K.; Srivastava, P.; Singh, M.P.; Siddiqi, M.I.; Pant, A.B.; Khanna, V.K. Involvement of PKA/DARPP-32/PP1alpha and beta- arrestin/Akt/GSK-3beta Signaling in Cadmium-Induced DA-D2 Receptor-Mediated Motor Dysfunctions: Protective Role of Quercetin. Sci. Rep. 2018, 8, 2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Robinson, J.F.; Sidhu, J.S.; Hong, S.; Faustman, E.M. A system-based comparison of gene expression reveals alterations in oxidative stress, disruption of ubiquitin-proteasome system and altered cell cycle regulation after exposure to cadmium and methylmercury in mouse embryonic fibroblast. Toxicol. Sci. 2010, 114, 356–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S. Positive and negative regulators of the metallothionein gene (review). Mol. Med. Rep. 2015, 12, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaassen, C.D.; Liu, J.; Choudhuri, S. Metallothionein: An intracellular protein to protect against cadmium toxicity. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 267–294. [Google Scholar] [CrossRef] [Green Version]
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-Dependent Antioxidant Enzymes: Actions and Properties of Selenoproteins. Antioxidants 2018, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Adani, G.; Filippini, T.; Michalke, B.; Vinceti, M. Selenium and Other Trace Elements in the Etiology of Parkinson’s Disease: A Systematic Review and Meta-Analysis of Case-Control Studies. Neuroepidemiology 2020, 54, 1–23. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Sun, H.L.; Wang, T.; Liu, X.X.; Liu, C.; Li, W.J.; Li, X. Selenium level does not differ in blood but increased in cerebrospinal fluid in Parkinson’s disease: A meta-analysis. Int. J. Neurosci. 2021, 131, 95–101. [Google Scholar] [CrossRef]
- Ellwanger, J.H.; Molz, P.; Dallemole, D.R.; Pereira dos Santos, A.; Muller, T.E.; Cappelletti, L.; Goncalves da Silva, M.; Franke, S.I.; Pra, D.; Pegas Henriques, J.A. Selenium reduces bradykinesia and DNA damage in a rat model of Parkinson’s disease. Nutrition 2015, 31, 359–365. [Google Scholar] [CrossRef]
- Sun, C.; Du, Z.; Liu, X.; Yang, Y.; Zhou, S.; Li, C.; Cao, X.; Zhao, Q.; Wong, K.; Chen, W.; et al. Selenium Forms and Dosages Determined Their Biological Actions in Mouse Models of Parkinson’s Disease. Nutrients 2022, 15, 11. [Google Scholar] [CrossRef]
- Yildizhan, K.; Naziroglu, M. Protective role of selenium on MPP(+) and homocysteine-induced TRPM2 channel activation in SH-SY5Y cells. J. Recept. Signal Transduct. Res. 2022, 42, 399–408. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Xie, H.; Bai, X.; Zhao, J.; Cui, L.; Zhang, J.; Li, B.; Li, Y.F. MALDI-TOF-MS and XAS analysis of complexes formed by metallothionein with mercury and/or selenium. Biometals 2021, 34, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Shindo, Y.; Oka, K. Magnesium Is a Key Player in Neuronal Maturation and Neuropathology. Int. J. Mol. Sci. 2019, 20, 3439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajput, K.; Afridi, H.I.; Kazi, T.G.; Talpur, F.N.; Baig, J.A. Sodium, Potassium, Calcium, and Magnesium in the Scalp Hair and Blood Samples Related to the Clinical Stages of the Parkinson’s Disease. Biol. Trace Elem. Res. 2021, 199, 2582–2589. [Google Scholar] [CrossRef]
- Shen, Y.; Dai, L.; Tian, H.; Xu, R.; Li, F.; Li, Z.; Zhou, J.; Wang, L.; Dong, J.; Sun, L. Treatment Of Magnesium-L-Threonate Elevates The Magnesium Level In The Cerebrospinal Fluid And Attenuates Motor Deficits And Dopamine Neuron Loss In A Mouse Model Of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2019, 15, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Kotani, M.; Kim, K.H.; Ishizaki, N.; Funaba, M.; Matsui, T. Magnesium and calcium deficiencies additively increase zinc concentrations and metallothionein expression in the rat liver. Br. J. Nutr. 2013, 109, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 1978, 14, 633–643. [Google Scholar]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-induced neurotoxicity: The role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef]
- Choi, H.J.; Kim, S.W.; Lee, S.Y.; Hwang, O. Dopamine-dependent cytotoxicity of tetrahydrobiopterin: A possible mechanism for selective neurodegeneration in Parkinson’s disease. J. Neurochem. 2003, 86, 143–152. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Dopaminergic neuron-specific oxidative stress caused by dopamine itself. Acta Med. Okayama 2008, 62, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Foppoli, C.; Coccia, R.; Cini, C.; Rosei, M.A. Catecholamines oxidation by xanthine oxidase. Biochim. Biophys. Acta. 1997, 1334, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Hastings, T.G. Enzymatic oxidation of dopamine: The role of prostaglandin H synthase. J. Neurochem. 1995, 64, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Korytowski, W.; Sarna, T.; Kalyanaraman, B.; Sealy, R.C. Tyrosinase-catalyzed oxidation of dopa and related catechol(amine)s: A kinetic electron spin resonance investigation using spin-stabilization and spin label oximetry. Biochim. Biophys. Acta 1987, 924, 383–392. [Google Scholar] [CrossRef]
- Rosei, M.A.; Blarzino, C.; Foppoli, C.; Mosca, L.; Coccia, R. Lipoxygenase-catalyzed oxidation of catecholamines. Biochem. Biophys. Res. Commun. 1994, 200, 344–350. [Google Scholar] [CrossRef]
- Kuhn, D.M.; Arthur, R.E., Jr.; Thomas, D.M.; Elferink, L.A. Tyrosine hydroxylase is inactivated by catechol-quinones and converted to a redox-cycling quinoprotein: Possible relevance to Parkinson’s disease. J. Neurochem. 1999, 73, 1309–1317. [Google Scholar] [CrossRef]
- LaVoie, M.J.; Ostaszewski, B.L.; Weihofen, A.; Schlossmacher, M.G.; Selkoe, D.J. Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 2005, 11, 1214–1221. [Google Scholar] [CrossRef]
- Whitehead, R.E.; Ferrer, J.V.; Javitch, J.A.; Justice, J.B. Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J. Neurochem. 2001, 76, 1242–1251. [Google Scholar] [CrossRef]
- Xu, Y.; Stokes, A.H.; Roskoski, R., Jr.; Vrana, K.E. Dopamine, in the presence of tyrosinase, covalently modifies and inactivates tyrosine hydroxylase. J. Neurosci. Res. 1998, 54, 691–697. [Google Scholar] [CrossRef]
- Berman, S.B.; Hastings, T.G. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: Implications for Parkinson’s disease. J. Neurochem. 1999, 73, 1127–1137. [Google Scholar] [CrossRef]
- Kuhn, D.M.; Francescutti-Verbeem, D.M.; Thomas, D.M. Dopamine quinones activate microglia and induce a neurotoxic gene expression profile: Relationship to methamphetamine-induced nerve ending damage. Ann. N. Y. Acad. Sci. 2006, 1074, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Zafar, K.S.; Siegel, D.; Ross, D. A potential role for cyclized quinones derived from dopamine, DOPA, and 3,4-dihydroxyphenylacetic acid in proteasomal inhibition. Mol. Pharmacol. 2006, 70, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, K.A.; Rochet, J.C.; Bieganski, R.M.; Lansbury, P.T., Jr. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001, 294, 1346–1349. [Google Scholar] [CrossRef]
- Sato, M.; Bremner, I. Oxygen free radicals and metallothionein. Free Radic. Biol. Med. 1993, 14, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Sharma, S. Metallothioneins 1 and 2 attenuate peroxynitrite-induced oxidative stress in Parkinson disease. Exp. Biol. Med. 2006, 231, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Asanuma, M.; Hozumi, H.; Miyoshi, K.; Sogawa, N. Protective effects of metallothionein against dopamine quinone-induced dopaminergic neurotoxicity. FEBS Lett. 2007, 581, 5003–5008. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, M.A.; Eibl, J.K.; Crispo, J.A.; Ross, G.M. Covalent arylation of metallothionein by oxidized dopamine products: A possible mechanism for zinc-mediated enhancement of dopaminergic neuron survival. Neurotox. Res. 2008, 14, 317–328. [Google Scholar] [CrossRef]
- Sharma, S.; Ebadi, M. Significance of metallothioneins in aging brain. Neurochem. Int. 2014, 65, 40–48. [Google Scholar] [CrossRef]
- Sharma, S.; Moon, C.S.; Khogali, A.; Haidous, A.; Chabenne, A.; Ojo, C.; Jelebinkov, M.; Kurdi, Y.; Ebadi, M. Biomarkers in Parkinson’s disease (recent update). Neurochem. Int. 2013, 63, 201–229. [Google Scholar] [CrossRef]
- Sharma, S.; Rais, A.; Sandhu, R.; Nel, W.; Ebadi, M. Clinical significance of metallothioneins in cell therapy and nanomedicine. Int. J. Nanomed. 2013, 8, 1477–1488. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.J. Metallothionein redox cycle and function. Exp. Biol. Med. 2006, 231, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.M.; Toulouse, A.; Connor, T.J.; Nolan, Y.M. Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology 2012, 62, 2154–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Pan, Y.T.; Zhang, Z.Y.; Yang, H.; Yu, S.Y.; Zheng, Y.; Ma, J.H.; Wang, X.M. Systemic activation of NLRP3 inflammasome and plasma alpha-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J. Neuroinflamm. 2020, 17, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.G.; Mohney, R.P.; Wilson, B.; Jeohn, G.H.; Liu, B.; Hong, J.S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef]
- Castano, A.; Herrera, A.J.; Cano, J.; Machado, A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J. Neurochem. 1998, 70, 1584–1592. [Google Scholar] [CrossRef] [Green Version]
- Aloe, L.; Fiore, M. TNF-alpha expressed in the brain of transgenic mice lowers central tyroxine hydroxylase immunoreactivity and alters grooming behavior. Neurosci. Lett. 1997, 238, 65–68. [Google Scholar] [CrossRef]
- Penkowa, M.; Florit, S.; Giralt, M.; Quintana, A.; Molinero, A.; Carrasco, J.; Hidalgo, J. Metallothionein reduces central nervous system inflammation, neurodegeneration, and cell death following kainic acid-induced epileptic seizures. J. Neurosci. Res. 2005, 79, 522–534. [Google Scholar] [CrossRef]
- Carrasco, J.; Hernandez, J.; Bluethmann, H.; Hidalgo, J. Interleukin-6 and tumor necrosis factor-alpha type 1 receptor deficient mice reveal a role of IL-6 and TNF-alpha on brain metallothionein-I and -III regulation. Brain Res. Mol. Brain Res. 1998, 57, 221–234. [Google Scholar] [CrossRef]
- Lee, D.K.; Carrasco, J.; Hidalgo, J.; Andrews, G.K. Identification of a signal transducer and activator of transcription (STAT) binding site in the mouse metallothionein-I promoter involved in interleukin-6-induced gene expression. Biochem. J. 1999, 337 Pt 1, 59–65. [Google Scholar] [CrossRef]
- Sawada, J.; Kikuchi, Y.; Shibutani, M.; Mitsumori, K.; Inoue, K.; Kasahara, T. Induction of metallothionein in astrocytes by cytokines and heavy metals. Biol. Signal. 1994, 3, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Giralt, M.; Penkowa, M.; Lago, N.; Molinero, A.; Hidalgo, J. Metallothionein-1+2 protect the CNS after a focal brain injury. Exp. Neurol. 2002, 173, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Penkowa, M.; Carrasco, J.; Giralt, M.; Moos, T.; Hidalgo, J. CNS wound healing is severely depressed in metallothionein I- and II-deficient mice. J. Neurosci. 1999, 19, 2535–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosz, M.; Olbert, M.; Wyszogrodzka, G.; Mlyniec, K.; Librowski, T. Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-kappaB signaling. Inflammopharmacology. 2017, 25, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hongxia, L.; Yuxiao, T.; Zhilei, S.; Yan, S.; Yicui, Q.; Jiamin, S.; Xin, X.; Jianxin, Y.; Fengfeng, M.; Hui, S. Zinc inhibited LPS-induced inflammatory responses by upregulating A20 expression in microglia BV2 cells. J. Affect. Disord. 2019, 249, 136–142. [Google Scholar] [CrossRef]
- Ni, A.; Ernst, C. Evidence That Substantia Nigra Pars Compacta Dopaminergic Neurons Are Selectively Vulnerable to Oxidative Stress Because They Are Highly Metabolically Active. Front. Cell. Neurosci. 2022, 16, 826193. [Google Scholar] [CrossRef]
- Pacelli, C.; Giguere, N.; Bourque, M.J.; Levesque, M.; Slack, R.S.; Trudeau, L.E. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [Green Version]
- Piccinin, E.; Sardanelli, A.M.; Seibel, P.; Moschetta, A.; Cocco, T.; Villani, G. PGC-1s in the Spotlight with Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 3487. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Shiba-Fukushima, K.; Inoshita, T.; Hattori, N.; Imai, Y. PINK1-mediated phosphorylation of Parkin boosts Parkin activity in Drosophila. PLoS Genet. 2014, 10, e1004391. [Google Scholar] [CrossRef]
- Springer, W.; Kahle, P.J. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy. 2011, 7, 266–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, L.M.; Libedinsky, A.; Elorza, A.A. Role of Copper on Mitochondrial Function and Metabolism. Front. Mol. Biosci. 2021, 8, 711227. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Georgiev, O.; Schaffner, W. The parkin mutant phenotype in the fly is largely rescued by metal-responsive transcription factor (MTF-1). Mol. Cell. Biol. 2011, 31, 2151–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.C.; Son, M.; Kang, S.; Im, S.; Piao, Y.; Lim, K.S.; Song, M.Y.; Park, K.S.; Kim, Y.H.; Pak, Y.K. Cell-penetrating artificial mitochondria-targeting peptide-conjugated metallothionein 1A alleviates mitochondrial damage in Parkinson’s disease models. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.G.; Weiss, J.H. Zn(2+)-induced disruption of neuronal mitochondrial function: Synergism with Ca(2+), critical dependence upon cytosolic Zn(2+) buffering, and contributions to neuronal injury. Exp. Neurol. 2018, 302, 181–195. [Google Scholar] [CrossRef]
- Cerri, S.; Milanese, C.; Mastroberardino, P.G. Endocytic iron trafficking and mitochondria in Parkinson’s disease. Int. J. Biochem. Cell Biol. 2019, 110, 70–74. [Google Scholar] [CrossRef]
- Pizarro-Galleguillos, B.M.; Kunert, L.; Bruggemann, N.; Prasuhn, J. Iron- and Neuromelanin-Weighted Neuroimaging to Study Mitochondrial Dysfunction in Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 13678. [Google Scholar] [CrossRef]
- Prasuhn, J.; Gottlich, M.; Gerkan, F.; Kourou, S.; Ebeling, B.; Kasten, M.; Hanssen, H.; Klein, C.; Bruggemann, N. Relationship between brain iron deposition and mitochondrial dysfunction in idiopathic Parkinson’s disease. Mol. Med. 2022, 28, 28. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging. 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Travagli, R.A.; Browning, K.N.; Camilleri, M. Parkinson disease and the gut: New insights into pathogenesis and clinical relevance. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 673–685. [Google Scholar] [CrossRef]
- Fares, M.B.; Jagannath, S.; Lashuel, H.A. Reverse engineering Lewy bodies: How far have we come and how far can we go? Nat. Rev. Neurosci. 2021, 22, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Uemura, M.T.; Luk, K.C.; Lee, V.M.; Trojanowski, J.Q. Cell-to-Cell Transmission of Tau and alpha-Synuclein. Trends Mol. Med. 2020, 26, 936–952. [Google Scholar] [CrossRef] [PubMed]
- Du, X.Y.; Xie, X.X.; Liu, R.T. The Role of alpha-Synuclein Oligomers in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8645. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, S.R.; Shin, H.J.; Lee, J.H.; Chang, C.S.; Kim, J. Copper(II)-induced self-oligomerization of alpha-synuclein. Biochem. J. 1999, 340 Pt 3, 821–828. [Google Scholar] [CrossRef]
- Davies, P.; Wang, X.; Sarell, C.J.; Drewett, A.; Marken, F.; Viles, J.H.; Brown, D.R. The synucleins are a family of redox-active copper binding proteins. Biochemistry 2011, 50, 37–47. [Google Scholar] [CrossRef]
- Wang, C.; Liu, L.; Zhang, L.; Peng, Y.; Zhou, F. Redox reactions of the alpha-synuclein-Cu(2+) complex and their effects on neuronal cell viability. Biochemistry 2010, 49, 8134–8142. [Google Scholar] [CrossRef] [Green Version]
- McLeary, F.A.; Rcom-H’cheo-Gauthier, A.N.; Kinder, J.; Goulding, M.; Khoo, T.K.; Mellick, G.D.; Chung, R.S.; Pountney, D.L. Dexamethasone Inhibits Copper-Induced Alpha-Synuclein Aggregation by a Metallothionein-Dependent Mechanism. Neurotox. Res. 2018, 33, 229–238. [Google Scholar] [CrossRef]
- Calvo, J.S.; Mulpuri, N.V.; Dao, A.; Qazi, N.K.; Meloni, G. Membrane insertion exacerbates the alpha-Synuclein-Cu(II) dopamine oxidase activity: Metallothionein-3 targets and silences all alpha-synuclein-Cu(II) complexes. Free Radic. Biol. Med. 2020, 158, 149–161. [Google Scholar] [CrossRef]
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Interaction of alpha-synuclein with divalent metal ions reveals key differences: A link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [Google Scholar] [CrossRef] [PubMed]
- Racette, B.A.; Criswell, S.R.; Lundin, J.I.; Hobson, A.; Seixas, N.; Kotzbauer, P.T.; Evanoff, B.A.; Perlmutter, J.S.; Zhang, J.; Sheppard, L.; et al. Increased risk of parkinsonism associated with welding exposure. Neurotoxicology 2012, 33, 1356–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, M.E.; Sugimoto, K.; Kubo, M.; Iwaki, H.; Tsujii, T.; Kyaw, W.T.; Nishikawa, N.; Nagai, M.; Tanaka, J.; Nomoto, M. Zonisamide up-regulated the mRNAs encoding astrocytic anti-oxidative and neurotrophic factors. Eur. J. Pharmacol. 2012, 689, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Hirai, K.; Tokuda, E. Effects of pergolide mesilate on metallothionein mRNAs expression in a mouse model for Parkinson disease. Biol. Pharm. Bull. 2009, 32, 1813–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Tarbutton, G.L.; Levin, J.L.; Plotkin, G.M.; Lowry, L.K.; Nalbone, J.T.; Shepherd, S. Pesticide/environmental exposures and Parkinson’s disease in East Texas. J. Agromed. 2008, 13, 37–48. [Google Scholar] [CrossRef]
- Bassotti, G.; Villanacci, V.; Antonelli, E.; Morelli, A.; Salerni, B. Enteric glial cells: New players in gastrointestinal motility? Lab. Investig. 2007, 87, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Jessen, K.R.; Mirsky, R. Glial cells in the enteric nervous system contain glial fibrillary acidic protein. Nature 1980, 286, 736–737. [Google Scholar] [CrossRef]
- Abdo, H.; Derkinderen, P.; Gomes, P.; Chevalier, J.; Aubert, P.; Masson, D.; Galmiche, J.P.; Vanden Berghe, P.; Neunlist, M.; Lardeux, B. Enteric glial cells protect neurons from oxidative stress in part via reduced glutathione. FASEB J. 2010, 24, 1082–1094. [Google Scholar] [CrossRef]
- Miyazaki, I.; Isooka, N.; Imafuku, F.; Sun, J.; Kikuoka, R.; Furukawa, C.; Asanuma, M. Chronic Systemic Exposure to Low-Dose Rotenone Induced Central and Peripheral Neuropathology and Motor Deficits in Mice: Reproducible Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3254. [Google Scholar] [CrossRef]
- Foligne, B.; George, F.; Standaert, A.; Garat, A.; Poiret, S.; Peucelle, V.; Ferreira, S.; Sobry, H.; Muharram, G.; Lucau-Danila, A.; et al. High-dose dietary supplementation with zinc prevents gut inflammation: Investigation of the role of metallothioneins and beyond by transcriptomic and metagenomic studies. FASEB J. 2020, 34, 12615–12633. [Google Scholar] [CrossRef]
- Diwakarla, S.; McQuade, R.M.; Constable, R.; Artaiz, O.; Lei, E.; Barnham, K.J.; Adlard, P.A.; Cherny, R.A.; Di Natale, M.R.; Wu, H.; et al. ATH434 Reverses Colorectal Dysfunction in the A53T Mouse Model of Parkinson’s Disease. J. Parkinsons. Dis. 2021, 11, 1821–1832. [Google Scholar] [CrossRef] [PubMed]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease—The gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyazaki, I.; Asanuma, M. Multifunctional Metallothioneins as a Target for Neuroprotection in Parkinson’s Disease. Antioxidants 2023, 12, 894. https://doi.org/10.3390/antiox12040894
Miyazaki I, Asanuma M. Multifunctional Metallothioneins as a Target for Neuroprotection in Parkinson’s Disease. Antioxidants. 2023; 12(4):894. https://doi.org/10.3390/antiox12040894
Chicago/Turabian StyleMiyazaki, Ikuko, and Masato Asanuma. 2023. "Multifunctional Metallothioneins as a Target for Neuroprotection in Parkinson’s Disease" Antioxidants 12, no. 4: 894. https://doi.org/10.3390/antiox12040894