
Hyperoxia Disrupts Lung Lymphatic Homeostasis in Neonatal Mice

 ,
, 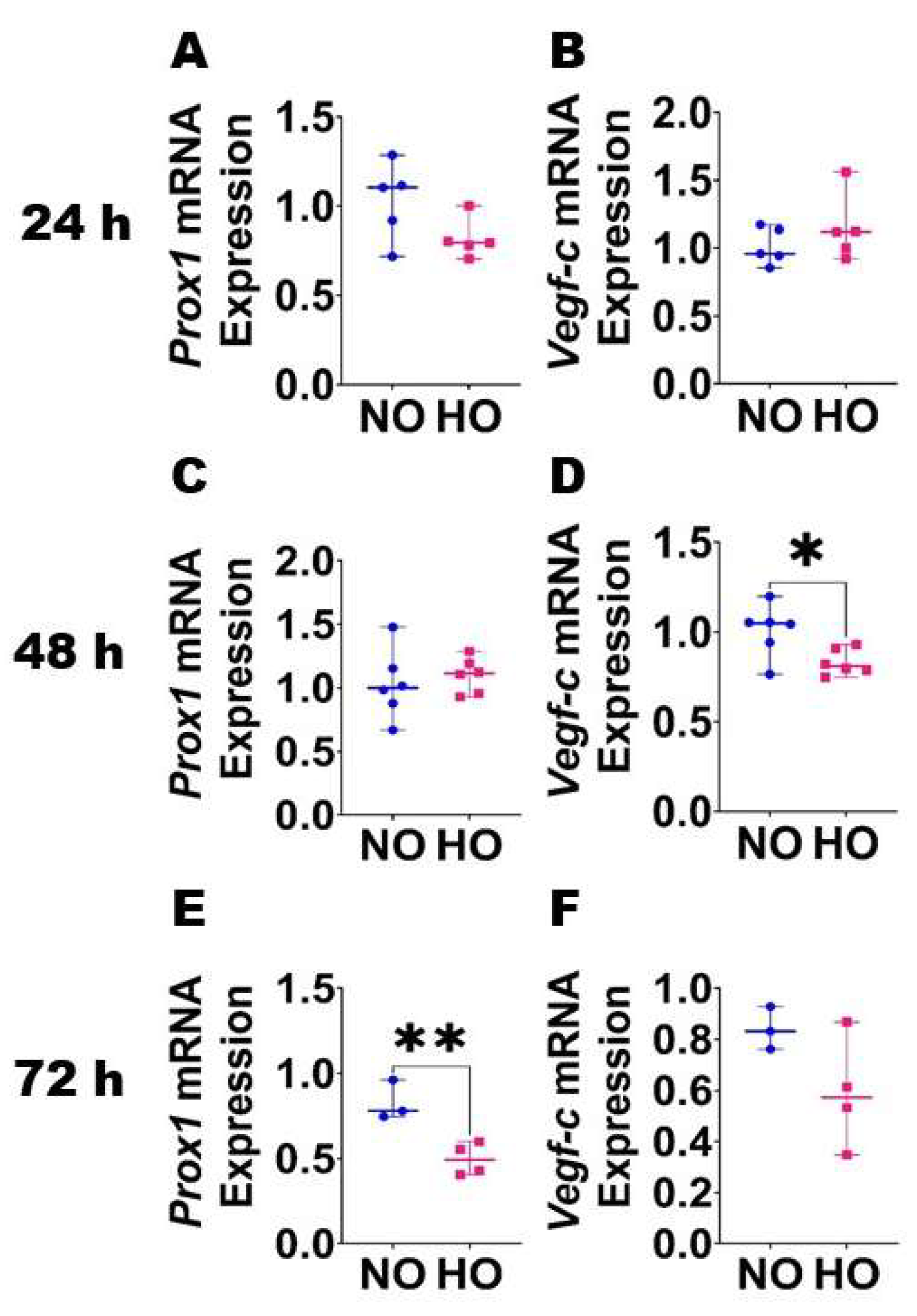{kind=link}
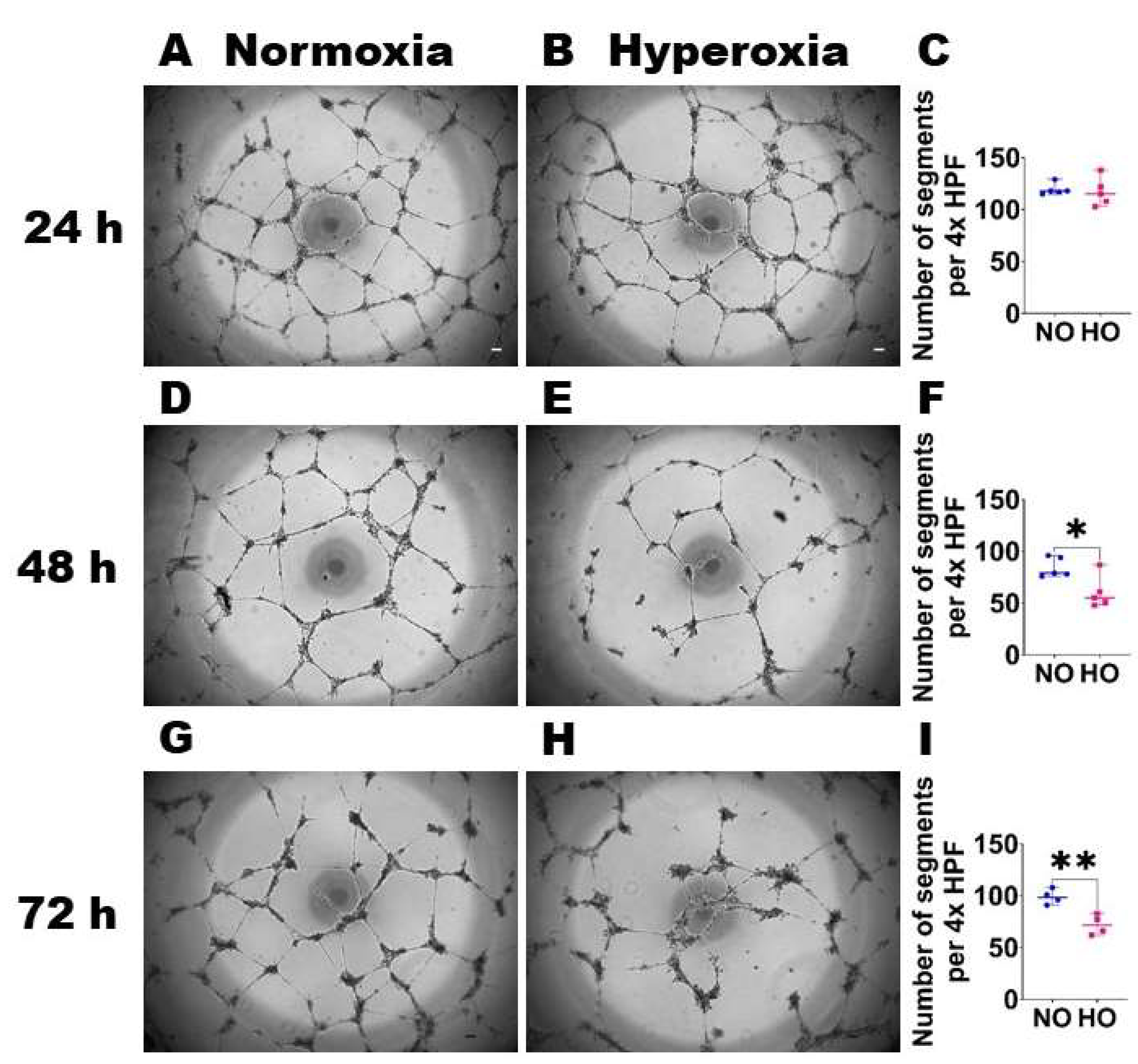{kind=link}
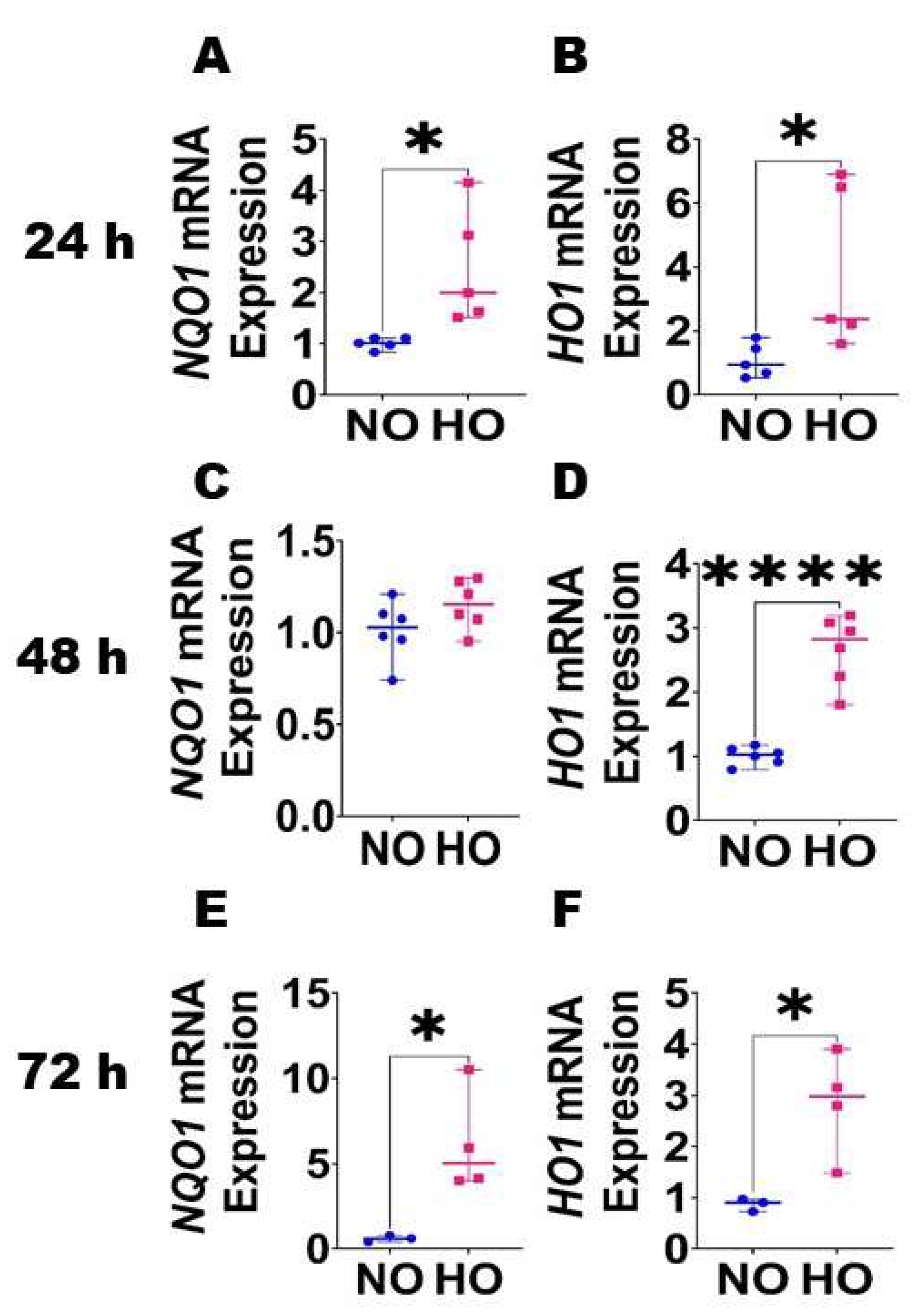{kind=link}
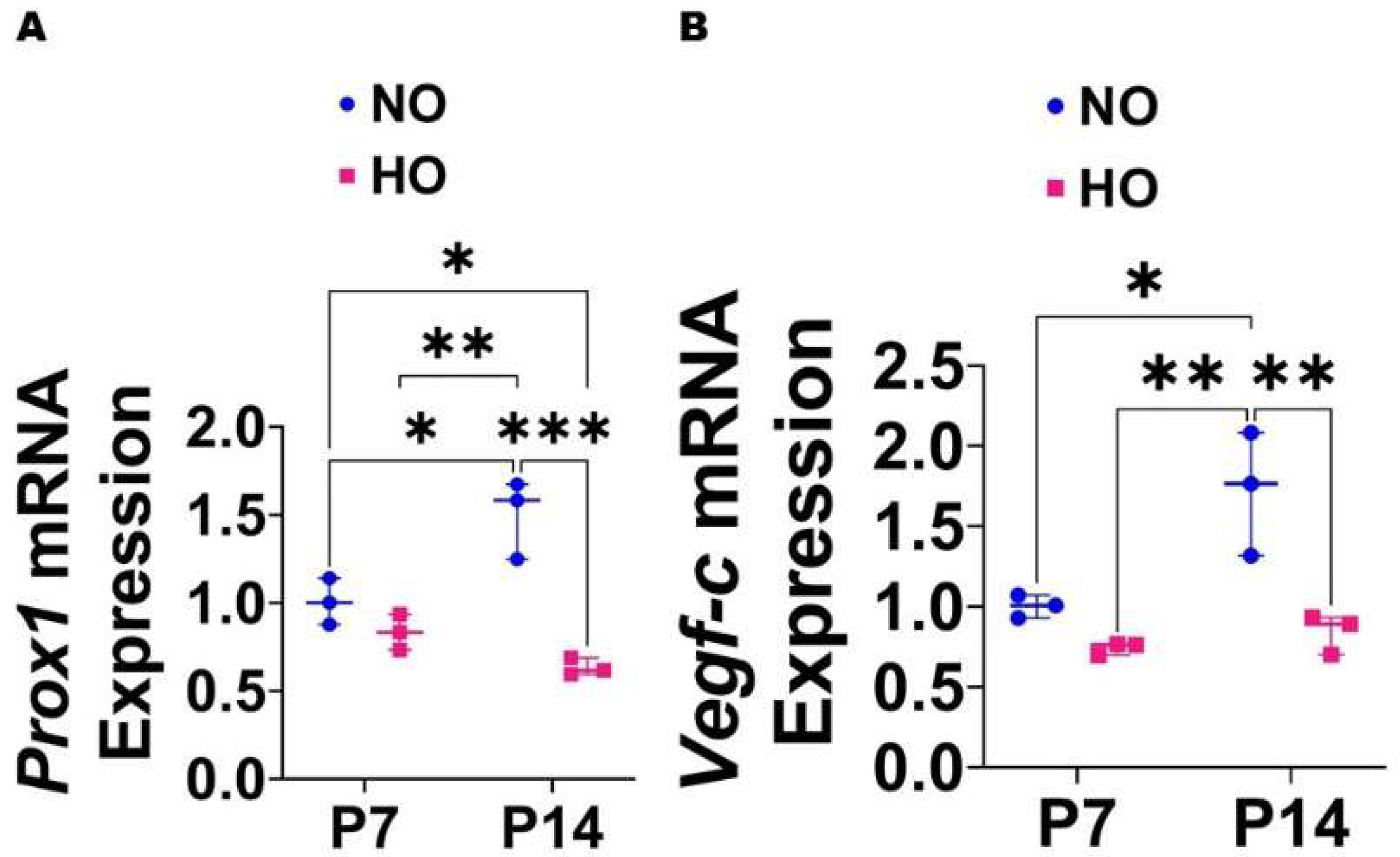{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Vitro Experiments
2.1.1. Cell Culture
2.1.2. Exposure of Cells to Hyperoxia
2.1.3. Real-Time RT-PCR Assays
2.1.4. Tubule Formation Assay
2.1.5. Statistical Analyses
2.2. In Vivo Experiments
2.2.1. Animals
2.2.2. Hyperoxia Experiments
2.2.3. Analyses of Lung Lymphatic Vascularization
2.2.4. Lung Tissue Extraction and Real-Time RT-PCR Assays
2.2.5. Analyses of Lung Lymphatic Function
2.2.6. Statistical Analyses
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jobe, A.H. Animal Models, Learning Lessons to Prevent and Treat Neonatal Chronic Lung Disease. Front. Med. 2015, 2, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husain, A.N.; Siddiqui, N.H.; Stocker, J.T. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum. Pathol. 1998, 29, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Coalson, J.J. Pathology of new bronchopulmonary dysplasia. Semin. Neonatol. SN 2003, 8, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Thébaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Prim. 2019, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Lapcharoensap, W.; Bennett, M.V.; Xu, X.; Lee, H.C.; Dukhovny, D. Hospitalization costs associated with bronchopulmonary dysplasia in the first year of life. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2019, 40, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.C.; Lorch, S.A. Health Care Costs of Major Morbidities associated with Prematurity in United States Children’s Hospitals. J. Pediatr. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Simpson, S.J.; Hall, G.L.; Wilson, A.C. Lung function following very preterm birth in the era of ‘new’ bronchopulmonary dysplasia. Respirol. (Carlton Vic.) 2015, 20, 535–540. [Google Scholar] [CrossRef]
- Islam, J.Y.; Keller, R.L.; Aschner, J.L.; Hartert, T.V.; Moore, P.E. Understanding the Short- and Long-Term Respiratory Outcomes of Prematurity and Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2015, 192, 134–156. [Google Scholar] [CrossRef] [Green Version]
- Twilhaar, E.S.; Wade, R.M.; de Kieviet, J.F.; van Goudoever, J.B.; van Elburg, R.M.; Oosterlaan, J. Cognitive Outcomes of Children Born Extremely or Very Preterm Since the 1990s and Associated Risk Factors: A Meta-analysis and Meta-regression. JAMA Pediatr. 2018, 172, 361–367. [Google Scholar] [CrossRef]
- Haggie, S.; Robinson, P.; Selvadurai, H.; Fitzgerald, D.A. Bronchopulmonary dysplasia: A review of the pulmonary sequelae in the post-surfactant era. J. Paediatr. Child Health 2020, 56, 680–689. [Google Scholar] [CrossRef]
- Hurst, J.R.; Beckmann, J.; Ni, Y.; Bolton, C.E.; McEniery, C.M.; Cockcroft, J.R.; Marlow, N. Respiratory and Cardiovascular Outcomes in Survivors of Extremely Preterm Birth at 19 Years. Am. J. Respir. Crit. Care Med. 2020, 202, 422–432. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Balany, J.; Bhandari, V. Understanding the Impact of Infection, Inflammation, and Their Persistence in the Pathogenesis of Bronchopulmonary Dysplasia. Front. Med. 2015, 2, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallapur, S.G.; Jobe, A.H. Contribution of inflammation to lung injury and development. Arch. Dis. Childhood. Fetal Neonatal Ed. 2006, 91, F132–F135. [Google Scholar] [CrossRef] [PubMed]
- Savani, R.C. Modulators of inflammation in Bronchopulmonary Dysplasia. Semin. Perinatol. 2018, 42, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Abman, S.H. Lung vascular development: Implications for the pathogenesis of bronchopulmonary dysplasia. Annu. Rev. Physiol. 2005, 67, 623–661. [Google Scholar] [CrossRef] [Green Version]
- Thebaud, B.; Abman, S.H. Bronchopulmonary dysplasia: Where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am. J. Respir. Crit. Care Med. 2007, 175, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.; Baveja, R.; Liang, O.D.; Fernandez-Gonzalez, A.; Lee, C.; Mitsialis, S.A.; Kourembanas, S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Rong, M.; Platteau, A.; Hehre, D.; Smith, H.; Ruiz, P.; Whitsett, J.; Bancalari, E.; Wu, S. CTGF disrupts alveolarization and induces pulmonary hypertension in neonatal mice: Implication in the pathogenesis of severe bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L330–L340. [Google Scholar] [CrossRef] [Green Version]
- Leroy, S.; Caumette, E.; Waddington, C.; Hebert, A.; Brant, R.; Lavoie, P.M. A Time-Based Analysis of Inflammation in Infants at Risk of Bronchopulmonary Dysplasia. J. Pediatr. 2018, 192, 60–65.e61. [Google Scholar] [CrossRef]
- El-Chemaly, S.; Malide, D.; Zudaire, E.; Ikeda, Y.; Weinberg, B.A.; Pacheco-Rodriguez, G.; Rosas, I.O.; Aparicio, M.; Ren, P.; MacDonald, S.D.; et al. Abnormal lymphangiogenesis in idiopathic pulmonary fibrosis with insights into cellular and molecular mechanisms. Proc. Natl. Acad. Sci. USA 2009, 106, 3958–3963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Iwama, N.; Date, F.; Shibata, N.; Miki, H.; Yamauchi, K.; Sawai, T.; Sato, S.; Takahashi, T.; Ono, M. Macrophages participate in lymphangiogenesis in idiopathic diffuse alveolar damage through CCL19-CCR7 signal. Hum. Pathol. 2009, 40, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Tammela, T.; Ator, E.; Lyubynska, N.; Achen, M.G.; Hicklin, D.J.; Jeltsch, M.; Petrova, T.V.; Pytowski, B.; Stacker, S.A.; et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J. Clin. Investig. 2005, 115, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggenberger, R.; Ullmann, S.; Proulx, S.T.; Pytowski, B.; Alitalo, K.; Detmar, M. Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J. Exp. Med. 2010, 207, 2255–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara, A.R.; Cosgrove, G.P.; Janssen, W.J.; Huie, T.J.; Burnham, E.L.; Heinz, D.E.; Curran-Everett, D.; Sahin, H.; Schwarz, M.I.; Cool, C.D.; et al. Increased lymphatic vessel length is associated with the fibroblast reticulum and disease severity in usual interstitial pneumonia and nonspecific interstitial pneumonia. Chest 2012, 142, 1569–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Wilder, J.; Rietz, C.; Gigliotti, A.; Tang, X.; Shi, Y.; Guilmette, R.; Wang, H.; George, G.; Nilo de Magaldi, E.; et al. Radiation-induced impairment in lung lymphatic vasculature. Lymphat. Res. Biol. 2014, 12, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Meinecke, A.K.; Nagy, N.; Lago, G.D.; Kirmse, S.; Klose, R.; Schrodter, K.; Zimmermann, A.; Helfrich, I.; Rundqvist, H.; Theegarten, D.; et al. Aberrant mural cell recruitment to lymphatic vessels and impaired lymphatic drainage in a murine model of pulmonary fibrosis. Blood 2012, 119, 5931–5942. [Google Scholar] [CrossRef]
- Ebina, M. Remodeling of airway walls in fatal asthmatics decreases lymphatic distribution; beyond thickening of airway smooth muscle layers. Allergol. Int. 2008, 57, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Baluk, P.; Yao, L.C.; Feng, J.; Romano, T.; Jung, S.S.; Schreiter, J.L.; Yan, L.; Shealy, D.J.; McDonald, D.M. TNF-alpha drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J. Clin. Invest. 2009, 119, 2954–2964. [Google Scholar] [CrossRef]
- Yao, L.C.; Baluk, P.; Feng, J.; McDonald, D.M. Steroid-resistant lymphatic remodeling in chronically inflamed mouse airways. Am. J. Pathol. 2010, 176, 1525–1541. [Google Scholar] [CrossRef] [Green Version]
- Hardavella, G.; Tzortzaki, E.G.; Siozopoulou, V.; Galanis, P.; Vlachaki, E.; Avgousti, M.; Stefanou, D.; Siafakas, N.M. Lymphangiogenesis in COPD: Another link in the pathogenesis of the disease. Respir. Med. 2012, 106, 687–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Andersson, C.K.; Graham, G.J.; Lofdahl, C.G.; Erjefalt, J.S. Increased number and altered phenotype of lymphatic vessels in peripheral lung compartments of patients with COPD. Respir. Res. 2013, 14, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.J.; Berkelhamer, S.K.; Kim, G.A.; Taylor, J.M.; O’Shea, K.M.; Steinhorn, R.H.; Farrow, K.N. Disrupted Pulmonary Artery cGMP Signaling in Mice with Hyperoxia-Induced Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 50, 369–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, R.T.; Shrestha, A.K.; Reynolds, C.L.; Barrios, R.; Shivanna, B. Long-term pulmonary and cardiovascular morbidities of neonatal hyperoxia exposure in mice. Int. J. Biochem. Cell Biol. 2018, 94, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.L.; Zhang, S.; Shrestha, A.K.; Barrios, R.; Shivanna, B. Phenotypic assessment of pulmonary hypertension using high-resolution echocardiography is feasible in neonatal mice with experimental bronchopulmonary dysplasia and pulmonary hypertension: A step toward preventing chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 1597–1605. [Google Scholar] [CrossRef] [Green Version]
- Elsaie, A.; Menon, R.T.; Shrestha, A.K.; Gowda, S.H.; Varghese, N.P.; Barrios, R.J.; Blanco, C.L.; Konduri, G.G.; Shivanna, B. Endothelial Adenosine Monophosphate-Activated Protein Kinase-Alpha1 Deficiency Potentiates Hyperoxia-Induced Experimental Bronchopulmonary Dysplasia and Pulmonary Hypertension. Antioxidants 2021, 10, 1913. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.T.; Thapa, S.; Shrestha, A.K.; Barrios, R.; Shivanna, B. Extracellular Signal-Regulated Kinase 1 Alone Is Dispensable for Hyperoxia-Mediated Alveolar and Pulmonary Vascular Simplification in Neonatal Mice. Antioxidants 2022, 11, 1130. [Google Scholar] [CrossRef]
- Shivanna, B.; Jiang, W.; Wang, L.; Couroucli, X.I.; Moorthy, B. Omeprazole attenuates hyperoxic lung injury in mice via aryl hydrocarbon receptor activation and is associated with increased expression of cytochrome P4501A enzymes. J. Pharm. Exp. 2011, 339, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Arnaoutova, I.; Kleinman, H.K. In Vitro angiogenesis: Endothelial cell tube formation on gelled basement membrane extract. Nat. Protoc. 2010, 5, 628–635. [Google Scholar] [CrossRef]
- Ribatti, D.; Guidolin, D.; Conconi, M.T.; Nico, B.; Baiguera, S.; Parnigotto, P.P.; Vacca, A.; Nussdorfer, G.G. Vinblastine inhibits the angiogenic response induced by adrenomedullin in vitro and in vivo. Oncogene 2003, 22, 6458–6461. [Google Scholar] [CrossRef] [Green Version]
- Shivanna, B.; Zhang, W.; Jiang, W.; Welty, S.E.; Couroucli, X.I.; Wang, L.; Moorthy, B. Functional deficiency of aryl hydrocarbon receptor augments oxygen toxicity-induced alveolar simplification in newborn mice. Toxicol. Appl. Pharm. 2013, 267, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Shivanna, B.; Chu, C.; Welty, S.E.; Jiang, W.; Wang, L.; Couroucli, X.I.; Moorthy, B. Omeprazole attenuates hyperoxic injury in H441 cells via the aryl hydrocarbon receptor. Free. Radic. Biol. Med. 2011, 51, 1910–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Outtz Reed, H.; Wang, L.; Sonett, J.; Chen, M.; Yang, J.; Li, L.; Aradi, P.; Jakus, Z.; D’Armiento, J.M.; Hancock, W.W.; et al. Lymphatic impairment leads to pulmonary tertiary lymphoid organ formation and alveolar damage. J. Clin. Investig. 2019, 129, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.J.; Pryhuber, G.S.; Huyck, H.; Watkins, R.H.; Metlay, L.A.; Maniscalco, W.M. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 164, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Hasan, J.; Beharry, K.D.; Valencia, A.M.; Strauss, A.; Modanlou, H.D. Soluble vascular endothelial growth factor receptor 1 in tracheal aspirate fluid of preterm neonates at birth may be predictive of bronchopulmonary dysplasia/chronic lung disease. Pediatrics 2009, 123, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Thébaud, B.; Ladha, F.; Michelakis, E.D.; Sawicka, M.; Thurston, G.; Eaton, F.; Hashimoto, K.; Harry, G.; Haromy, A.; Korbutt, G.; et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: Evidence that angiogenesis participates in alveolarization. Circulation 2005, 112, 2477–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, M.; Das, P.; Pawar, A.; Aghai, Z.H.; Kaskinen, A.; Zhuang, Z.W.; Ambalavanan, N.; Pryhuber, G.; Andersson, S.; Bhandari, V. Hyperoxia causes miR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nat. Commun. 2017, 8, 1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniscalco, W.M.; Watkins, R.H.; Pryhuber, G.S.; Bhatt, A.; Shea, C.; Huyck, H. Angiogenic factors and alveolar vasculature: Development and alterations by injury in very premature baboons. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L811–L823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigle, J.T.; Oliver, G. Prox1 function is required for the development of the murine lymphatic system. Cell 1999, 98, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- François, M.; Caprini, A.; Hosking, B.; Orsenigo, F.; Wilhelm, D.; Browne, C.; Paavonen, K.; Karnezis, T.; Shayan, R.; Downes, M.; et al. Sox18 induces development of the lymphatic vasculature in mice. Nature 2008, 456, 643–647. [Google Scholar] [CrossRef]
- Wigle, J.T.; Harvey, N.; Detmar, M.; Lagutina, I.; Grosveld, G.; Gunn, M.D.; Jackson, D.G.; Oliver, G. An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. Embo. J. 2002, 21, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2004, 5, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Ylä-Herttuala, S.; Alitalo, K. Gene transfer as a tool to induce therapeutic vascular growth. Nat. Med. 2003, 9, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Wada, J.; Sato, Y. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat. Rev. Nephrol. 2020, 16, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Dashkevich, A.; Hagl, C.; Beyersdorf, F.; Nykänen, A.I.; Lemström, K.B. VEGF Pathways in the Lymphatics of Healthy and Diseased Heart. Microcirculation 2016, 23, 5–14. [Google Scholar] [CrossRef]
- Chen, J.C.; Chang, Y.W.; Hong, C.C.; Yu, Y.H.; Su, J.L. The role of the VEGF-C/VEGFRs axis in tumor progression and therapy. Int. J. Mol. Sci. 2012, 14, 88–107. [Google Scholar] [CrossRef]
- Stump, B.; Cui, Y.; Kidambi, P.; Lamattina, A.M.; El-Chemaly, S. Lymphatic Changes in Respiratory Diseases: More than Just Remodeling of the Lung? Am. J. Respir. Cell Mol. Biol. 2017, 57, 272–279. [Google Scholar] [CrossRef]
- Scheuer, T.; Sharkovska, Y.; Tarabykin, V.; Marggraf, K.; Brockmöller, V.; Bührer, C.; Endesfelder, S.; Schmitz, T. Neonatal Hyperoxia Perturbs Neuronal Development in the Cerebellum. Mol. Neurobiol. 2018, 55, 3901–3915. [Google Scholar] [CrossRef]
- Endesfelder, S.; Zaak, I.; Weichelt, U.; Bührer, C.; Schmitz, T. Caffeine protects neuronal cells against injury caused by hyperoxia in the immature brain. Free. Radic. Biol. Med. 2014, 67, 221–234. [Google Scholar] [CrossRef]
- Endesfelder, S.; Makki, H.; von Haefen, C.; Spies, C.D.; Bührer, C.; Sifringer, M. Neuroprotective effects of dexmedetomidine against hyperoxia-induced injury in the developing rat brain. PLoS ONE 2017, 12, e0171498. [Google Scholar] [CrossRef] [Green Version]
- Menon, R.T.; Shrestha, A.K.; Shivanna, B. Hyperoxia exposure disrupts adrenomedullin signaling in newborn mice: Implications for lung development in premature infants. Biochem. Biophys. Res. Commun. 2017, 487, 666–671. [Google Scholar] [CrossRef]
- Janér, J.; Lassus, P.; Haglund, C.; Paavonen, K.; Alitalo, K.; Andersson, S. Pulmonary vascular endothelial growth factor-C in development and lung injury in preterm infants. Am. J. Respir. Crit. Care Med. 2006, 174, 326–330. [Google Scholar] [CrossRef]
- Simpson, D.A.; Murphy, G.M.; Bhaduri, T.; Gardiner, T.A.; Archer, D.B.; Stitt, A.W. Expression of the VEGF gene family during retinal vaso-obliteration and hypoxia. Biochem. Biophys. Res. Commun. 1999, 262, 333–340. [Google Scholar] [CrossRef]
- Gerber, H.P.; Condorelli, F.; Park, J.; Ferrara, N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J. Biol. Chem. 1997, 272, 23659–23667. [Google Scholar] [CrossRef] [Green Version]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. Embo. J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehgal, A.; Gwini, S.M.; Menahem, S.; Allison, B.J.; Miller, S.L.; Polglase, G.R. Preterm growth restriction and bronchopulmonary dysplasia: The vascular hypothesis and related physiology. J. Physiol. 2019, 597, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.; Peisl, A.; Seedorf, G.; Nowlin, T.; Kim, C.; Bosco, J.; Kenniston, J.; Keefe, D.; Abman, S.H. Anti-sFlt-1 Therapy Preserves Lung Alveolar and Vascular Growth in Antenatal Models of Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2018, 197, 776–787. [Google Scholar] [CrossRef]
- Tang, J.R.; Markham, N.E.; Lin, Y.J.; McMurtry, I.F.; Maxey, A.; Kinsella, J.P.; Abman, S.H. Inhaled nitric oxide attenuates pulmonary hypertension and improves lung growth in infant rats after neonatal treatment with a VEGF receptor inhibitor. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L344–L351. [Google Scholar] [CrossRef]
- Kunig, A.M.; Balasubramaniam, V.; Markham, N.E.; Seedorf, G.; Gien, J.; Abman, S.H. Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L1068–L1078. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.J.; Markham, N.E.; Balasubramaniam, V.; Tang, J.R.; Maxey, A.; Kinsella, J.P.; Abman, S.H. Inhaled nitric oxide enhances distal lung growth after exposure to hyperoxia in neonatal rats. Pediatr. Res. 2005, 58, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appuhn, S.V.; Siebert, S.; Myti, D.; Wrede, C.; Surate Solaligue, D.E.; Pérez-Bravo, D.; Brandenberger, C.; Schipke, J.; Morty, R.E.; Grothausmann, R.; et al. Capillary Changes Precede Disordered Alveolarization in a Mouse Model of Bronchopulmonary Dysplasia. Am. J. Respir. Cell Mol. Biol. 2021, 65, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.T.; Shrestha, A.K.; Reynolds, C.L.; Barrios, R.; Caron, K.M.; Shivanna, B. Adrenomedullin Is Necessary to Resolve Hyperoxia-Induced Experimental Bronchopulmonary Dysplasia and Pulmonary Hypertension in Mice. Am. J. Pathol. 2020, 190, 711–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakus, Z.; Gleghorn, J.P.; Enis, D.R.; Sen, A.; Chia, S.; Liu, X.; Rawnsley, D.R.; Yang, Y.; Hess, P.R.; Zou, Z.; et al. Lymphatic function is required prenatally for lung inflation at birth. J. Exp. Med. 2014, 211, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, R.M.; Herman, A.; Ikegami, M.; Greenberg, J.M.; Akeson, A.L. Lymphatic ontogeny and effect of hypoplasia in developing lung. Mech. Dev. 2011, 128, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.C.; Testini, C.; Tvorogov, D.; Anisimov, A.; Vargas, S.O.; Baluk, P.; Pytowski, B.; Claesson-Welsh, L.; Alitalo, K.; McDonald, D.M. Pulmonary lymphangiectasia resulting from vascular endothelial growth factor-C overexpression during a critical period. Circ. Res. 2014, 114, 806–822. [Google Scholar] [CrossRef] [PubMed]
- McNellis, E.M.; Mabry, S.M.; Taboada, E.; Ekekezie, I.I. Altered pulmonary lymphatic development in infants with chronic lung disease. Biomed. Res. Int. 2014, 2014, 109891. [Google Scholar] [CrossRef] [Green Version]
- Baker, C.D.; Seedorf, G.J.; Wisniewski, B.L.; Black, C.P.; Ryan, S.L.; Balasubramaniam, V.; Abman, S.H. Endothelial colony-forming cell conditioned media promote angiogenesis in vitro and prevent pulmonary hypertension in experimental bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L73–L81. [Google Scholar] [CrossRef] [Green Version]
- Perveen, S.; Patel, H.; Arif, A.; Younis, S.; Codipilly, C.N.; Ahmed, M. Role of EC-SOD overexpression in preserving pulmonary angiogenesis inhibited by oxidative stress. PLoS ONE 2012, 7, e51945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MasoudiMotlagh, M.; Sepehr, R.; Sheibani, N.; Sorenson, C.M.; Ranji, M. Optical cryoimaging of mitochondrial redox state in bronchopulmonary-dysplasia injury models in mice lungs. Quant. Imaging. Med. Surg. 2015, 5, 159–162. [Google Scholar] [CrossRef]
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Kensler, T.W.; Yamamoto, M.; Zhang, L.Y.; Kleeberger, S.R. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 175–182. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends. Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Fernandez-Gonzalez, A.; Alex Mitsialis, S.; Liu, X.; Kourembanas, S. Vasculoprotective effects of heme oxygenase-1 in a murine model of hyperoxia-induced bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L775–L784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Biswasa, C.; Lin, Q.S.; La, P.; Namba, F.; Zhuang, T.; Muthu, M.; Dennery, P.A. Heme oxygenase-1 regulates postnatal lung repair after hyperoxia: Role of beta-catenin/hnRNPK signaling. Redox. Biol. 2013, 1, 234–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ataei Ataabadi, E.; Golshiri, K.; Jüttner, A.A.; de Vries, R.; Van den Berg-Garrelds, I.; Nagtzaam, N.M.A.; Khan, H.N.; Leijten, F.P.J.; Brandt, R.M.C.; Dik, W.A.; et al. Soluble guanylate cyclase activator BAY 54-6544 improves vasomotor function and survival in an accelerated ageing mouse model. Aging Cell 2022, 21, e13683. [Google Scholar] [CrossRef] [PubMed]
- Cen, M.; Ouyang, W.; Zhang, W.; Yang, L.; Lin, X.; Dai, M.; Hu, H.; Tang, H.; Liu, H.; Xia, J.; et al. MitoQ protects against hyperpermeability of endothelium barrier in acute lung injury via a Nrf2-dependent mechanism. Redox. Biol. 2021, 41, 101936. [Google Scholar] [CrossRef]
- Harris, N.R.; Nielsen, N.R.; Pawlak, J.B.; Aghajanian, A.; Rangarajan, K.; Serafin, D.S.; Farber, G.; Dy, D.M.; Nelson-Maney, N.P.; Xu, W.; et al. VE-Cadherin Is Required for Cardiac Lymphatic Maintenance and Signaling. Circ. Res. 2022, 130, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; De la Cruz, E.; Gu, X.; Balint, L.; Oxendine-Burns, M.; Terrones, T.; Ma, W.; Kuo, H.H.; Lantz, C.; Bansal, T.; et al. Lymphoangiocrine signals promote cardiac growth and repair. Nature 2020, 588, 705–711. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shankar, N.; Thapa, S.; Shrestha, A.K.; Sarkar, P.; Gaber, M.W.; Barrios, R.; Shivanna, B. Hyperoxia Disrupts Lung Lymphatic Homeostasis in Neonatal Mice. Antioxidants 2023, 12, 620. https://doi.org/10.3390/antiox12030620
Shankar N, Thapa S, Shrestha AK, Sarkar P, Gaber MW, Barrios R, Shivanna B. Hyperoxia Disrupts Lung Lymphatic Homeostasis in Neonatal Mice. Antioxidants. 2023; 12(3):620. https://doi.org/10.3390/antiox12030620
Chicago/Turabian StyleShankar, Nithyapriya, Shyam Thapa, Amrit Kumar Shrestha, Poonam Sarkar, M. Waleed Gaber, Roberto Barrios, and Binoy Shivanna. 2023. "Hyperoxia Disrupts Lung Lymphatic Homeostasis in Neonatal Mice" Antioxidants 12, no. 3: 620. https://doi.org/10.3390/antiox12030620