
Cell Rearrangement and Oxidant/Antioxidant Imbalance in Huntington’s Disease




Abstract
:
1. Introduction
1.1. HD Epidemiology
1.2. HD Pathogenesis
2. Cells Rearrangement in HD
3. Oxidative Stress
Oxidative Stress in HD
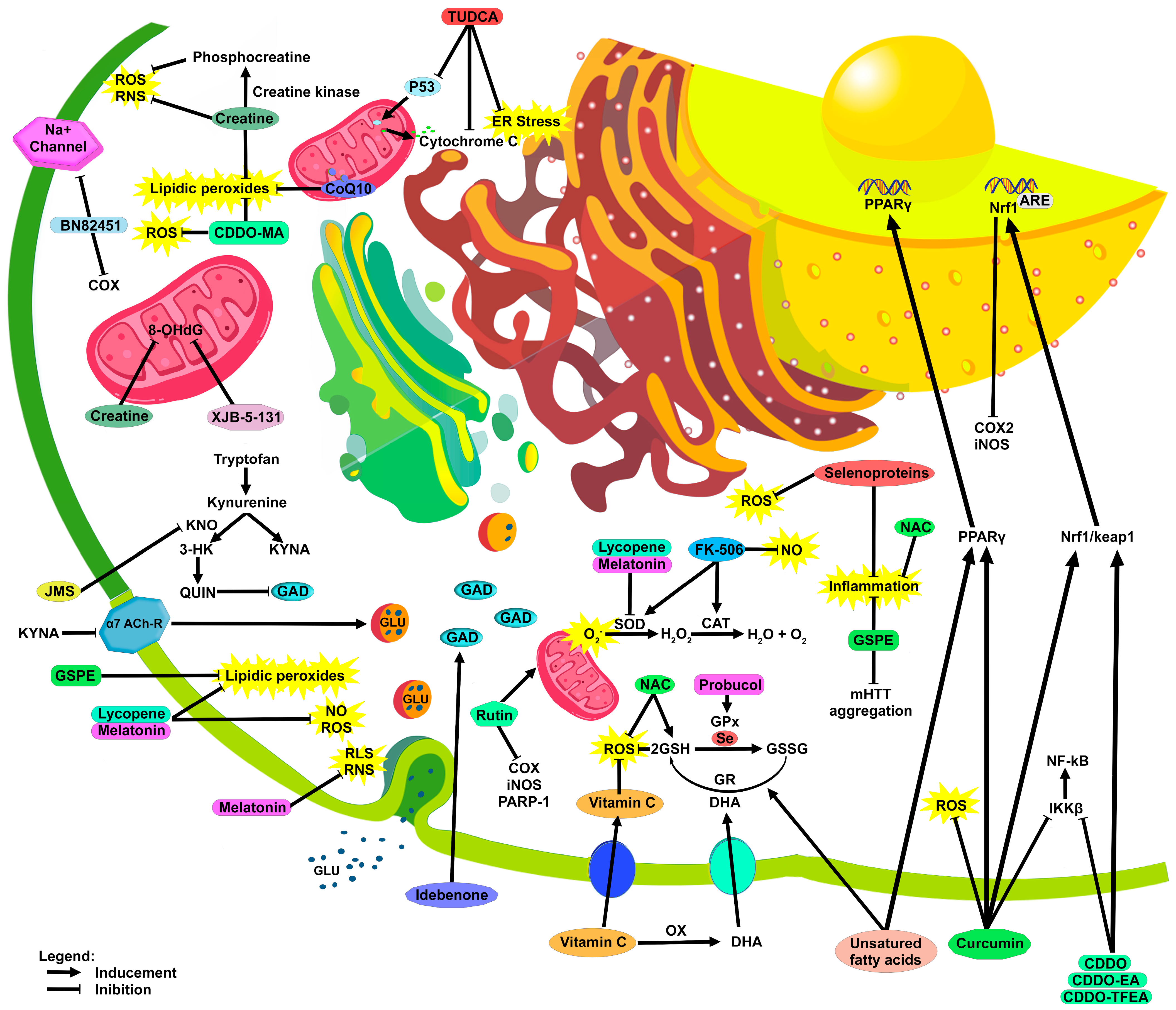
4. Antioxidant Defense and Antioxidant Compounds in HD
4.1. Vitamin C
4.2. Selenium
4.3. Unsaturated Fatty Acids
4.4. Creatine
4.5. Coenzyme Q10
4.6. Idebenone
4.7. Curcumin
4.8. Grape Seed Polyphenolic Extract
4.9. Lycopene
4.10. Melatonin
4.11. N-Acetylcysteine
4.12. Rutin
4.13. Tauroursodeoxycholic Acid
4.14. Tacrolimus
4.15. Synthetic Triterpenoids
4.16. XJB-5-131
4.17. Probucol
4.18. BN82451
4.19. Kynurenine-3-Monooxygenase Inhibitors
5. The Oxidant-Antioxidant Balance: A Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ross, C.A.; Tabrizi, S.J. Huntington’s Disease: From Molecular Pathogenesis to Clinical Treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Wexler, N.S.; Lorimer, J.; Porter, J.; Gomez, F.; Moskowitz, C.; Shackell, E.; Marder, K.; Penchaszadeh, G.; Roberts, S.A.; Gayán, J.; et al. Venezuelan Kindreds Reveal That Genetic and Environmental Factors Modulate Huntington’s Disease Age of Onset. Proc. Natl. Acad. Sci. USA 2004, 101, 3498–3503. [Google Scholar] [CrossRef] [PubMed]
- Telenius, H.; Kremer, B.; Goldberg, Y.P.; Theilmann, J.; Andrew, S.E.; Zeisler, J.; Adam, S.; Greenberg, C.; Ives, E.J.; Clarke, L.A.; et al. Somatic and Gonadal Mosaicism of the Huntington Disease Gene CAG Repeat in Brain and Sperm. Nat. Genet. 1994, 6, 409–414. [Google Scholar] [CrossRef]
- Kremer, H.P.; Roos, R.A.; Dingjan, G.; Marani, E.; Bots, G.T. Atrophy of the Hypothalamic Lateral Tuberal Nucleus in Huntington’s Disease. J. Neuropathol. Exp. Neurol. 1990, 49, 371–382. [Google Scholar] [CrossRef]
- Heinsen, H.; Rüb, U.; Gangnus, D.; Jungkunz, G.; Bauer, M.; Ulmar, G.; Bethke, B.; Schüler, M.; Böcker, F.; Eisenmenger, W.; et al. Nerve Cell Loss in the Thalamic Centromedian-Parafascicular Complex in Patients with Huntington’s Disease. Acta Neuropathol. 1996, 91, 161–168. [Google Scholar] [CrossRef]
- Petersén, A.; Castilho, R.F.; Hansson, O.; Wieloch, T.; Brundin, P. Oxidative Stress, Mitochondrial Permeability Transition and Activation of Caspases in Calcium Ionophore A23187-Induced Death of Cultured Striatal Neurons. Brain Res. 2000, 857, 20–29. [Google Scholar] [CrossRef]
- Kassubek, J.; Juengling, F.D.; Ecker, D.; Landwehrmeyer, G.B. Thalamic Atrophy in Huntington’s Disease Co-Varies with Cognitive Performance: A Morphometric MRI Analysis. Cereb. Cortex 2005, 15, 846–853. [Google Scholar] [CrossRef]
- Petersén, Å.; Gil, J.; Maat-Schieman, M.L.C.; Björkqvist, M.; Tanila, H.; Araújo, I.M.; Smith, R.; Popovic, N.; Wierup, N.; Norlén, P.; et al. Orexin Loss in Huntington’s Disease. Hum. Mol. Genet. 2005, 14, 39–47. [Google Scholar] [CrossRef]
- Reiner, A.; Albin, R.L.; Anderson, K.D.; D’Amato, C.J.; Penney, J.B.; Young, A.B. Differential Loss of Striatal Projection Neurons in Huntington Disease. Proc. Natl. Acad. Sci. USA 1988, 85, 5733–5737. [Google Scholar] [CrossRef]
- Rosas, H.D.; Koroshetz, W.J.; Chen, Y.I.; Skeuse, C.; Vangel, M.; Cudkowicz, M.E.; Caplan, K.; Marek, K.; Seidman, L.J.; Makris, N.; et al. Evidence for More Widespread Cerebral Pathology in Early HD: An MRI-Based Morphometric Analysis. Neurology 2003, 60, 1615–1620. [Google Scholar] [CrossRef]
- Montoya, A.; Price, B.H.; Menear, M.; Lepage, M. Brain Imaging and Cognitive Dysfunctions in Huntington’s Disease. J. Psychiatry Neurosci. 2006, 31, 21–29. [Google Scholar]
- Walker, F.O. Huntington’s Disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Phillips, O.; Squitieri, F.; Sanchez-Castaneda, C.; Elifani, F.; Griguoli, A.; Maglione, V.; Caltagirone, C.; Sabatini, U.; Di Paola, M. The Corticospinal Tract in Huntington’s Disease. Cereb. Cortex 2015, 25, 2670–2682. Available online: https://academic.oup.com/cercor/article/25/9/2670/2926079 (accessed on 4 January 2023). [CrossRef]
- Schwab, L.C.; Garas, S.N.; Drouin-Ouellet, J.; Mason, S.L.; Stott, S.R.; Barker, R.A. Dopamine and Huntington’s Disease. Expert Rev. Neurother. 2015, 15, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Lindroos, R.; Dorst, M.C.; Du, K.; Filipović, M.; Keller, D.; Ketzef, M.; Kozlov, A.K.; Kumar, A.; Lindahl, M.; Nair, A.G.; et al. Basal Ganglia Neuromodulation Over Multiple Temporal and Structural Scales—Simulations of Direct Pathway MSNs Investigate the Fast Onset of Dopaminergic Effects and Predict the Role of Kv4.2. Front. Neural Circuits 2018, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, C.; Murphy, K.P.S.; Parent, M.; Levine, M.S. Chapter 10—The Role of Dopamine in Huntington’s Disease. In Progress in Brain Research; Diana, M., Di Chiara, G., Spano, P., Eds.; Dopamine; Elsevier: Amsterdam, The Netherlands, 2014; Volume 211, pp. 235–254. [Google Scholar]
- André, V.M.; Cepeda, C.; Levine, M.S. Dopamine and Glutamate in Huntington’s Disease: A Balancing Act. CNS Neurosci. Ther. 2010, 16, 163–178. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Protective Effect of Hesperidin and Naringin against 3-Nitropropionic Acid Induced Huntington’s like Symptoms in Rats: Possible Role of Nitric Oxide. Behav. Brain Res. 2010, 206, 38–46. [Google Scholar] [CrossRef]
- Ransome, M.I.; Renoir, T.; Hannan, A.J. Hippocampal Neurogenesis, Cognitive Deficits and Affective Disorder in Huntington’s Disease. Neural Plast. 2012, 2012, e874387. [Google Scholar] [CrossRef]
- Giralt, A.; Saavedra, A.; Alberch, J.; Pérez-Navarro, E. Cognitive Dysfunction in Huntington’s Disease: Humans, Mouse Models and Molecular Mechanisms. J. Huntingt. Dis. 2012, 1, 155–173. [Google Scholar] [CrossRef]
- Thu, D.C.; Oorschot, D.E.; Tippett, L.J.; Nana, A.L.; Hogg, V.M.; Synek, B.J.; Luthi-Carter, R.; Waldvogel, H.J.; Faull, R.L.M. Cell Loss in the Motor and Cingulate Cortex Correlates with Symptomatology in Huntington’s Disease. Brain 2010, 133, 1094–1110. Available online: https://academic.oup.com/brain/article/133/4/1094/312364 (accessed on 4 January 2023). [CrossRef] [PubMed]
- Evans, S.J.W.; Douglas, I.; Rawlins, M.D.; Wexler, N.S.; Tabrizi, S.J.; Smeeth, L. Prevalence of Adult Huntington’s Disease in the UK Based on Diagnoses Recorded in General Practice Records. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Morrison, P.J.; Harding-Lester, S.; Bradley, A. Uptake of Huntington Disease Predictive Testing in a Complete Population. Clin. Genet. 2011, 80, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kay, C.; Hayden, M.R.; Leavitt, B.R. Chapter 3—Epidemiology of Huntington Disease. In Handbook of Clinical Neurology; Feigin, A.S., Anderson, K.E., Eds.; Huntington Disease; Elsevier: Amsterdam, The Netherlands, 2017; Volume 144, pp. 31–46. [Google Scholar]
- Kay, C.; Fisher, E.; Hayden, M.R. Epidemiology. In Huntington’s Disease; Bates, G., Tabrizi, S., Jones, L., Eds.; Oxford University Press: Oxford, UK, 2014; ISBN 978-0-19-992914-6. [Google Scholar]
- Morrison, P.J. Prevalence Estimates of Huntington Disease in Caucasian Populations Are Gross Underestimates. Mov. Disord. 2012, 27, 1707–1708, author reply 1708–1709. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Arroyo, M.A.; Moreno, S.; Valiente, A. Incidence and Mutation Rates of Huntington’s Disease in Spain: Experience of 9 Years of Direct Genetic Testing. J. Neurol. Neurosurg. Psychiatry 2005, 76, 337–342. [Google Scholar] [CrossRef]
- Almqvist, E.W.; Elterman, D.S.; MacLeod, P.M.; Hayden, M.R. High Incidence Rate and Absent Family Histories in One Quarter of Patients Newly Diagnosed with Huntington Disease in British Columbia. Clin. Genet. 2001, 60, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; MacGregor, J.M.; Beighton, P.H. The Prevalence of Huntington’s Chorea in South Africa. S. Afr. Med. J. 1980, 58, 193–196. [Google Scholar]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network Organization of the Huntingtin Proteomic Interactome in Mammalian Brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef]
- Velier, J.; Kim, M.; Schwarz, C.; Kim, T.W.; Sapp, E.; Chase, K.; Aronin, N.; DiFiglia, M. Wild-Type and Mutant Huntingtins Function in Vesicle Trafficking in the Secretory and Endocytic Pathways. Exp. Neurol. 1998, 152, 34–40. [Google Scholar] [CrossRef]
- Zucker, B.; Luthi-Carter, R.; Kama, J.A.; Dunah, A.W.; Stern, E.A.; Fox, J.H.; Standaert, D.G.; Young, A.B.; Augood, S.J. Transcriptional Dysregulation in Striatal Projection- and Interneurons in a Mouse Model of Huntington’s Disease: Neuronal Selectivity and Potential Neuroprotective Role of HAP1. Hum. Mol. Genet. 2005, 14, 179–189. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Strand, A.; Peters, N.L.; Solano, S.M.; Hollingsworth, Z.R.; Menon, A.S.; Frey, A.S.; Spektor, B.S.; Penney, E.B.; Schilling, G.; et al. Decreased Expression of Striatal Signaling Genes in a Mouse Model of Huntington’s Disease. Hum. Mol. Genet. 2000, 9, 1259–1271. [Google Scholar] [CrossRef]
- Reiner, A.; Dragatsis, I.; Zeitlin, S.; Goldowitz, D. Wild-Type Huntingtin Plays a Role in Brain Development and Neuronal Survival. Mol. Neurobiol. 2003, 28, 259–276. [Google Scholar] [CrossRef]
- Ghosh, R.; Tabrizi, S.J. Clinical Features of Huntington’s Disease. In Polyglutamine Disorders. Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2018; Volume 1049, pp. 1–28. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Hoffner, G.; Island, M.-L.; Djian, P. Purification of Neuronal Inclusions of Patients with Huntington’s Disease Reveals a Broad Range of N-Terminal Fragments of Expanded Huntingtin and Insoluble Polymers. J. Neurochem. 2005, 95, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.K.; Schilling, G.; Peters, M.F.; Herring, W.J.; Sharp, A.H.; Kaminsky, Z.; Masone, J.; Khan, F.A.; Delanoy, M.; Borchelt, D.R.; et al. Truncated N-Terminal Fragments of Huntingtin with Expanded Glutamine Repeats Form Nuclear and Cytoplasmic Aggregates in Cell Culture. Hum. Mol. Genet. 1998, 7, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion Body Formation Reduces Levels of Mutant Huntingtin and the Risk of Neuronal Death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Slow, E.J.; Graham, R.K.; Osmand, A.P.; Devon, R.S.; Lu, G.; Deng, Y.; Pearson, J.; Vaid, K.; Bissada, N.; Wetzel, R.; et al. Absence of Behavioral Abnormalities and Neurodegeneration In Vivo despite Widespread Neuronal Huntingtin Inclusions. Proc. Natl. Acad. Sci. USA 2005, 102, 11402–11407. [Google Scholar] [CrossRef]
- Pieri, L.; Madiona, K.; Bousset, L.; Melki, R. Fibrillar α-Synuclein and Huntingtin Exon 1 Assemblies Are Toxic to the Cells. Biophys. J. 2012, 102, 2894–2905. [Google Scholar] [CrossRef]
- Nucifora, L.G.; Burke, K.A.; Feng, X.; Arbez, N.; Zhu, S.; Miller, J.; Yang, G.; Ratovitski, T.; Delannoy, M.; Muchowski, P.J.; et al. Identification of Novel Potentially Toxic Oligomers Formed In Vitro from Mammalian-Derived Expanded Huntingtin Exon-1 Protein. J. Biol. Chem. 2012, 287, 16017–16028. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, P.; Snapp, E.L. Formation and Toxicity of Soluble Polyglutamine Oligomers in Living Cells. PLoS ONE 2010, 5, e15245. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Inui, T.; Popiel, H.A.; Fujikake, N.; Hasegawa, K.; Urade, Y.; Goto, Y.; Naiki, H.; Toda, T. A Toxic Monomeric Conformer of the Polyglutamine Protein. Nat. Struct. Mol. Biol. 2007, 14, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Arrasate, M.; Brooks, E.; Libeu, C.P.; Legleiter, J.; Hatters, D.; Curtis, J.; Cheung, K.; Krishnan, P.; Mitra, S.; et al. Identifying Polyglutamine Protein Species in Situ That Best Predict Neurodegeneration. Nat. Chem. Biol. 2011, 7, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Sahl, S.J.; Weiss, L.E.; Duim, W.C.; Frydman, J.; Moerner, W.E. Cellular Inclusion Bodies of Mutant Huntingtin Exon 1 Obscure Small Fibrillar Aggregate Species. Sci. Rep. 2012, 2, 895. [Google Scholar] [CrossRef]
- Sathasivam, K.; Neueder, A.; Gipson, T.A.; Landles, C.; Benjamin, A.C.; Bondulich, M.K.; Smith, D.L.; Faull, R.L.M.; Roos, R.A.C.; Howland, D.; et al. Aberrant Splicing of HTT Generates the Pathogenic Exon 1 Protein in Huntington Disease. Proc. Natl. Acad. Sci. USA 2013, 110, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Sieradzan, K.A.; Mechan, A.O.; Jones, L.; Wanker, E.E.; Nukina, N.; Mann, D.M. Huntington’s Disease Intranuclear Inclusions Contain Truncated, Ubiquitinated Huntingtin Protein. Exp. Neurol. 1999, 156, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Leitman, J.; Ulrich Hartl, F.; Lederkremer, G.Z. Soluble Forms of PolyQ-Expanded Huntingtin Rather than Large Aggregates Cause Endoplasmic Reticulum Stress. Nat. Commun. 2013, 4, 2753. [Google Scholar] [CrossRef]
- Takahashi, T.; Kikuchi, S.; Katada, S.; Nagai, Y.; Nishizawa, M.; Onodera, O. Soluble Polyglutamine Oligomers Formed Prior to Inclusion Body Formation Are Cytotoxic. Hum. Mol. Genet. 2008, 17, 345–356. [Google Scholar] [CrossRef]
- Wang, C.-E.; Tydlacka, S.; Orr, A.L.; Yang, S.-H.; Graham, R.K.; Hayden, M.R.; Li, S.; Chan, A.W.S.; Li, X.-J. Accumulation of N-Terminal Mutant Huntingtin in Mouse and Monkey Models Implicated as a Pathogenic Mechanism in Huntington’s Disease. Hum. Mol. Genet. 2008, 17, 2738–2751. [Google Scholar] [CrossRef]
- Castiglioni, V.; Onorati, M.; Rochon, C.; Cattaneo, E. Induced Pluripotent Stem Cell Lines from Huntington’s Disease Mice Undergo Neuronal Differentiation While Showing Alterations in the Lysosomal Pathway. Neurobiol. Dis. 2012, 46, 30–40. [Google Scholar] [CrossRef]
- The HD iPSC Consortium. HD iPSC Consortium Induced Pluripotent Stem Cells from Patients with Huntington’s Disease Show CAG-Repeat-Expansion-Associated Phenotypes. Cell Stem Cell 2012, 11, 264–278. [Google Scholar] [CrossRef]
- Yang, W.; Dunlap, J.R.; Andrews, R.B.; Wetzel, R. Aggregated Polyglutamine Peptides Delivered to Nuclei Are Toxic to Mammalian Cells. Hum. Mol. Genet. 2002, 11, 2905–2917. [Google Scholar] [CrossRef]
- Monsellier, E.; Bousset, L.; Melki, R. α-Synuclein and Huntingtin Exon 1 Amyloid Fibrils Bind Laterally to the Cellular Membrane. Sci. Rep. 2016, 6, 19180. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Abounit, S.; Marzo, L.; Danckaert, A.; Chamoun, Z.; Roux, P.; Zurzolo, C. Transfer of Polyglutamine Aggregates in Neuronal Cells Occurs in Tunneling Nanotubes. J. Cell Sci. 2013, 126, 3678–3685. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.; Tenreiro, S.; Miller-Fleming, L.; Outeiro, T.F. Visualization of Cell-to-Cell Transmission of Mutant Huntingtin Oligomers. PLoS Curr. 2011, 3, RRN1210. [Google Scholar] [CrossRef]
- Babcock, D.T.; Ganetzky, B. Transcellular Spreading of Huntingtin Aggregates in the Drosophila Brain. Proc. Natl. Acad. Sci. USA 2015, 112, E5427–E5433. Available online: https://pubmed.ncbi.nlm.nih.gov/26351672/ (accessed on 4 January 2023). [CrossRef] [PubMed]
- Pearce, M.M.P.; Spartz, E.J.; Hong, W.; Luo, L.; Kopito, R.R. Prion-like Transmission of Neuronal Huntingtin Aggregates to Phagocytic Glia in the Drosophila Brain. Nat. Commun. 2015, 6, 6768. [Google Scholar] [CrossRef]
- Pecho-Vrieseling, E.; Rieker, C.; Fuchs, S.; Bleckmann, D.; Esposito, M.S.; Botta, P.; Goldstein, C.; Bernhard, M.; Galimberti, I.; Müller, M.; et al. Transneuronal Propagation of Mutant Huntingtin Contributes to Non-Cell Autonomous Pathology in Neurons. Nat. Neurosci. 2014, 17, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Legleiter, J.; Mitchell, E.; Lotz, G.P.; Sapp, E.; Ng, C.; DiFiglia, M.; Thompson, L.M.; Muchowski, P.J. Mutant Huntingtin Fragments Form Oligomers in a Polyglutamine Length-Dependent Manner In Vitro and In Vivo. J. Biol. Chem. 2010, 285, 14777–14790. [Google Scholar] [CrossRef]
- Ast, A.; Buntru, A.; Schindler, F.; Hasenkopf, R.; Schulz, A.; Brusendorf, L.; Klockmeier, K.; Grelle, G.; McMahon, B.; Niederlechner, H.; et al. MHTT Seeding Activity: A Marker of Disease Progression and Neurotoxicity in Models of Huntington’s Disease. Mol. Cell 2018, 71, 675–688.e6. [Google Scholar] [CrossRef]
- Cicchetti, F.; Lacroix, S.; Cisbani, G.; Vallières, N.; Saint-Pierre, M.; St-Amour, I.; Tolouei, R.; Skepper, J.N.; Hauser, R.A.; Mantovani, D.; et al. Mutant Huntingtin Is Present in Neuronal Grafts in Huntington Disease Patients. Ann. Neurol. 2014, 76, 31–42. [Google Scholar] [CrossRef]
- Beck, M.; Hurt, E. The Nuclear Pore Complex: Understanding Its Function through Structural Insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73–89. [Google Scholar] [CrossRef]
- Cavazza, T.; Vernos, I. The RanGTP Pathway: From Nucleo-Cytoplasmic Transport to Spindle Assembly and Beyond. Front. Cell Dev. Biol. 2015, 3, 82. [Google Scholar] [CrossRef] [PubMed]
- Hetzer, M.; Gruss, O.J.; Mattaj, I.W. The Ran GTPase as a Marker of Chromosome Position in Spindle Formation and Nuclear Envelope Assembly. Nat. Cell Biol. 2002, 4, E177–E184. [Google Scholar] [CrossRef] [PubMed]
- Hosp, F.; Vossfeldt, H.; Heinig, M.; Vasiljevic, D.; Arumughan, A.; Wyler, E.; Genetic and Environmental Risk for Alzheimer’s Disease GERAD1 Consortium; Landthaler, M.; Hubner, N.; Wanker, E.E.; et al. Quantitative Interaction Proteomics of Neurodegenerative Disease Proteins. Cell Rep. 2015, 11, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Riggs, A.D. DNA Methylation and Gene Function. Science 1980, 210, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, Recognition and Regulation of Non-CpG Methylation in the Adult Mammalian Brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 Gene Expression and Histone H3 (K9) Trimethylation in Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef]
- Stack, E.C.; Del Signore, S.J.; Luthi-Carter, R.; Soh, B.Y.; Goldstein, D.R.; Matson, S.; Goodrich, S.; Markey, A.L.; Cormier, K.; Hagerty, S.W. Modulation of Nucleosome Dynamics in Huntington’s Disease. Hum. Mol. Genet. 2007, 16, 1164–1175. Available online: https://pubmed.ncbi.nlm.nih.gov/17403718/ (accessed on 15 February 2023). [CrossRef]
- Thomas, E.A. DNA Methylation in Huntington’s Disease: Implications for Transgenerational Effects. Neurosci. Lett. 2016, 625, 34–39. [Google Scholar] [CrossRef]
- Jia, H.; Morris, C.D.; Williams, R.M.; Loring, J.F.; Thomas, E.A. HDAC Inhibition Imparts Beneficial Transgenerational Effects in Huntington’s Disease Mice via Altered DNA and Histone Methylation. Proc. Natl. Acad. Sci. USA 2015, 112, E56–E64. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.W.; Yildirim, F.; Yap, Y.S.; Dalin, S.; Matthews, B.J.; Velez, P.J.; Labadorf, A.; Housman, D.E.; Fraenkel, E. Extensive Changes in DNA Methylation Are Associated with Expression of Mutant Huntingtin. Proc. Natl. Acad. Sci. USA 2013, 110, 2354–2359. [Google Scholar] [CrossRef]
- Horvath, S.; Langfelder, P.; Kwak, S.; Aaronson, J.; Rosinski, J.; Vogt, T.F.; Eszes, M.; Faull, R.L.M.; Curtis, M.A.; Waldvogel, H.J.; et al. Huntington’s Disease Accelerates Epigenetic Aging of Human Brain and Disrupts DNA Methylation Levels. Aging 2016, 8, 1485–1504. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.-P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human Aging-Associated DNA Hypermethylation Occurs Preferentially at Bivalent Chromatin Domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P.; et al. Age-Dependent DNA Methylation of Genes That Are Suppressed in Stem Cells Is a Hallmark of Cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef]
- Numata, S.; Ye, T.; Hyde, T.M.; Guitart-Navarro, X.; Tao, R.; Wininger, M.; Colantuoni, C.; Weinberger, D.R.; Kleinman, J.E.; Lipska, B.K. DNA Methylation Signatures in Development and Aging of the Human Prefrontal Cortex. Am. J. Hum. Genet. 2012, 90, 260–272. [Google Scholar] [CrossRef]
- Johansson, A.; Enroth, S.; Gyllensten, U. Continuous Aging of the Human DNA Methylome throughout the Human Lifespan. PLoS ONE 2013, 8, e67378. [Google Scholar] [CrossRef]
- Lu, A.T.; Narayan, P.; Grant, M.J.; Langfelder, P.; Wang, N.; Kwak, S.; Wilkinson, H.; Chen, R.Z.; Chen, J.; Simon Bawden, C.; et al. DNA Methylation Study of Huntington’s Disease and Motor Progression in Patients and in Animal Models. Nat. Commun. 2020, 11, 4529. [Google Scholar] [CrossRef]
- Choo, Y.S.; Johnson, G.V.W.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant Huntingtin Directly Increases Susceptibility of Mitochondria to the Calcium-Induced Permeability Transition and Cytochrome c Release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef]
- Panov, A.V.; Gutekunst, C.-A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early Mitochondrial Calcium Defects in Huntington’s Disease Are a Direct Effect of Polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef]
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of Mitochondrial Protein Import by Mutant Huntingtin. Nat. Neurosci. 2014, 17, 822–831. [Google Scholar] [CrossRef]
- Yablonska, S.; Ganesan, V.; Ferrando, L.M.; Kim, J.; Pyzel, A.; Baranova, O.V.; Khattar, N.K.; Larkin, T.M.; Baranov, S.V.; Chen, N.; et al. Mutant Huntingtin Disrupts Mitochondrial Proteostasis by Interacting with TIM23. Proc. Natl. Acad. Sci. USA 2019, 116, 16593–16602. [Google Scholar] [CrossRef]
- Jin, Y.N.; Johnson, G.V.W. The Interrelationship between Mitochondrial Dysfunction and Transcriptional Dysregulation in Huntington Disease. J. Bioenerg. Biomembr. 2010, 42, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Rivera-Sánchez, S.; Castro, M.D.R.; Acevedo-Torres, K.; Rane, A.; Torres-Ramos, C.A.; Nicholls, D.G.; Andersen, J.K.; Ayala-Torres, S. Mitochondrial DNA Damage Is Associated with Reduced Mitochondrial Bioenergetics in Huntington’s Disease. Free Radic. Biol. Med. 2012, 53, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Beal, M.F. The Energetics of Huntington’s Disease. Neurochem. Res. 2004, 29, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial Loss, Dysfunction and Altered Dynamics in Huntington’s Disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef]
- Johri, A.; Chandra, A.; Flint Beal, M. PGC-1α, Mitochondrial Dysfunction, and Huntington’s Disease. Free Radic. Biol. Med. 2013, 62, 37–46. [Google Scholar] [CrossRef]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H. Mitochondrial Defect in Huntington’s Disease Caudate Nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M.F. Oxidative Damage and Metabolic Dysfunction in Huntington’s Disease: Selective Vulnerability of the Basal Ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1alpha by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef]
- Intihar, T.A.; Martinez, E.A.; Gomez-Pastor, R. Mitochondrial Dysfunction in Huntington’s Disease; Interplay Between HSF1, P53 and PGC-1α Transcription Factors. Front. Cell Neurosci. 2019, 13, 103. Available online: https://pubmed.ncbi.nlm.nih.gov/30941017/ (accessed on 13 February 2023). [CrossRef] [PubMed]
- Orr, A.L.; Li, S.; Wang, C.-E.; Li, H.; Wang, J.; Rong, J.; Xu, X.; Mastroberardino, P.G.; Greenamyre, J.T.; Li, X.-J. N-Terminal Mutant Huntingtin Associates with Mitochondria and Impairs Mitochondrial Trafficking. J. Neurosci. 2008, 28, 2783–2792. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Dyer, R.B.; Badger, J.D.; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant Huntingtin Impairs Axonal Trafficking in Mammalian Neurons In Vivo and In Vitro. Mol. Cell Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Reddy, P.H. Abnormal Mitochondrial Dynamics, Mitochondrial Loss and Mutant Huntingtin Oligomers in Huntington’s Disease: Implications for Selective Neuronal Damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Shirendeb, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; Dufour, J.L.; Mao, P.; Reddy, P.H. Mutant Huntingtin’s Interaction with Mitochondrial Protein Drp1 Impairs Mitochondrial Biogenesis and Causes Defective Axonal Transport and Synaptic Degeneration in Huntington’s Disease. Hum. Mol. Genet. 2012, 21, 406–420. Available online: https://pubmed.ncbi.nlm.nih.gov/21997870/ (accessed on 4 January 2023). [CrossRef]
- Lin, J.-T.; Chang, W.-C.; Chen, H.-M.; Lai, H.-L.; Chen, C.-Y.; Tao, M.-H.; Chern, Y. Regulation of Feedback between Protein Kinase A and the Proteasome System Worsens Huntington’s Disease. Mol. Cell Biol. 2013, 33, 1073–1084. [Google Scholar] [CrossRef]
- Cortes, C.J.; La Spada, A.R. The Many Faces of Autophagy Dysfunction in Huntington’s Disease: From Mechanism to Therapy. Drug Discov. Today 2014, 19, 963–971. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Lindquist, S. Impaired ERAD and ER Stress Are Early and Specific Events in Polyglutamine Toxicity. Genes Dev. 2008, 22, 3308–3319. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER Stress Sensor and Cell Fate Executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 MRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in Endoplasmic Reticulum Stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER Stress-Induced Cell Death Mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Ohno, M. PERK as a Hub of Multiple Pathogenic Pathways Leading to Memory Deficits and Neurodegeneration in Alzheimer’s Disease. Brain Res. Bull. 2018, 141, 72–78. [Google Scholar] [CrossRef]
- Taalab, K.; Cheng, T.; Zhang, Y. Mapping Landslide Susceptibility and Types Using Random Forest. Big Earth Data 2018, 2, 159–178. [Google Scholar] [CrossRef]
- Reijonen, S.; Putkonen, N.; Nørremølle, A.; Lindholm, D.; Korhonen, L. Inhibition of Endoplasmic Reticulum Stress Counteracts Neuronal Cell Death and Protein Aggregation Caused by N-Terminal Mutant Huntingtin Proteins. Exp. Cell Res. 2008, 314, 950–960. [Google Scholar] [CrossRef]
- Cho, Y.M.; Jang, Y.-S.; Jang, Y.-M.; Chung, S.-M.; Kim, H.-S.; Lee, J.-H.; Jeong, S.-W.; Kim, I.-K.; Kim, J.J.; Kim, K.-S.; et al. Induction of Unfolded Protein Response during Neuronal Induction of Rat Bone Marrow Stromal Cells and Mouse Embryonic Stem Cells. Exp. Mol. Med. 2009, 41, 440–452. [Google Scholar] [CrossRef]
- Noh, J.-Y.; Lee, H.; Song, S.; Kim, N.S.; Im, W.; Kim, M.; Seo, H.; Chung, C.-W.; Chang, J.-W.; Ferrante, R.J.; et al. SCAMP5 Links Endoplasmic Reticulum Stress to the Accumulation of Expanded Polyglutamine Protein Aggregates via Endocytosis Inhibition. J. Biol. Chem. 2009, 284, 11318–11325. [Google Scholar] [CrossRef] [PubMed]
- Carnemolla, A.; Fossale, E.; Agostoni, E.; Michelazzi, S.; Calligaris, R.; De Maso, L.; Del Sal, G.; MacDonald, M.E.; Persichetti, F. Rrs1 Is Involved in Endoplasmic Reticulum Stress Response in Huntington Disease. J. Biol. Chem. 2009, 284, 18167–18173. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the UPR Transcription Factor XBP1 Protects against Huntington’s Disease through the Regulation of FoxO1 and Autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Leitman, J.; Barak, B.; Benyair, R.; Shenkman, M.; Ashery, U.; Hartl, F.U.; Lederkremer, G.Z. ER Stress-Induced EIF2-Alpha Phosphorylation Underlies Sensitivity of Striatal Neurons to Pathogenic Huntingtin. PLoS ONE 2014, 9, e90803. [Google Scholar] [CrossRef]
- Yang, H.; Liu, C.; Zhong, Y.; Luo, S.; Monteiro, M.J.; Fang, S. Huntingtin Interacts with the Cue Domain of Gp78 and Inhibits Gp78 Binding to Ubiquitin and P97/VCP. PLoS ONE 2010, 5, e8905. [Google Scholar] [CrossRef] [PubMed]
- Hyrskyluoto, A.; Bruelle, C.; Lundh, S.H.; Do, H.T.; Kivinen, J.; Rappou, E.; Reijonen, S.; Waltimo, T.; Petersén, Å.; Lindholm, D.; et al. Ubiquitin-Specific Protease-14 Reduces Cellular Aggregates and Protects against Mutant Huntingtin-Induced Cell Degeneration: Involvement of the Proteasome and ER Stress-Activated Kinase IRE1α. Hum. Mol. Genet. 2014, 23, 5928–5939. [Google Scholar] [CrossRef]
- Hernández, I.H.; Torres-Peraza, J.; Santos-Galindo, M.; Ramos-Morón, E.; Fernández-Fernández, M.R.; Pérez-Álvarez, M.J.; Miranda-Vizuete, A.; Lucas, J.J. The Neuroprotective Transcription Factor ATF5 Is Decreased and Sequestered into Polyglutamine Inclusions in Huntington’s Disease. Acta Neuropathol. 2017, 134, 839–850. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Johnson, G.V.W. Role of Mitochondrial Dysfunction in the Pathogenesis of Huntington’s Disease. Brain Res. Bull. 2009, 80, 242–247. [Google Scholar] [CrossRef]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s Disease. Biochim. Biophys. Acta 2010, 1802, 52–61. [Google Scholar] [CrossRef]
- Cho, I.-H. Effects of Panax Ginseng in Neurodegenerative Diseases. J. Ginseng. Res. 2012, 36, 342–353. [Google Scholar] [CrossRef]
- Lim, D.; Fedrizzi, L.; Tartari, M.; Zuccato, C.; Cattaneo, E.; Brini, M.; Carafoli, E. Calcium Homeostasis and Mitochondrial Dysfunction in Striatal Neurons of Huntington Disease. J. Biol. Chem. 2008, 283, 5780–5789. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Cleeter, M.W.; Xuereb, J.; Taanman, J.W.; Cooper, J.M.; Schapira, A.H. Biochemical Abnormalities and Excitotoxicity in Huntington’s Disease Brain. Ann. Neurol. 1999, 45, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Squitieri, F.; Falleni, A.; Cannella, M.; Orobello, S.; Fulceri, F.; Lenzi, P.; Fornai, F. Abnormal Morphology of Peripheral Cell Tissues from Patients with Huntington Disease. J. Neural Transm. 2010, 117, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Antioxidants in Huntington’s Disease. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2012, 1822, 664–674. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Functions and Dysfunctions of Mitochondrial Dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Song, I.; Huganir, R.L. Regulation of AMPA Receptors during Synaptic Plasticity. Trends Neurosci. 2002, 25, 578–588. [Google Scholar] [CrossRef]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [CrossRef]
- Chang, C.-R.; Blackstone, C. Dynamic Regulation of Mitochondrial Fission through Modification of the Dynamin-Related Protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef]
- Allen, G.F.G.; Toth, R.; James, J.; Ganley, I.G. Loss of Iron Triggers PINK1/Parkin-Independent Mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 Is Able to Induce Mitophagy via LC3 Binding, Regardless of PARKIN and P62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. The Regulation of Autophagosome Dynamics by Huntingtin and HAP1 Is Disrupted by Expression of Mutant Huntingtin, Leading to Defective Cargo Degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin Functions as a Scaffold for Selective Macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Winderickx, J.; Franssens, V.; Liu, B. A Mitochondria-Associated Oxidative Stress Perspective on Huntington’s Disease. Front. Mol. Neurosci. 2018, 11, 329. [Google Scholar] [CrossRef]
- Beckman, J.S.; Koppenol, W.H. Nitric Oxide, Superoxide, and Peroxynitrite: The Good, the Bad, and Ugly. Am. J. Physiol.-Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The Role of Antioxidants in the Chemistry of Oxidative Stress: A Review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Prousek, J. Fenton Chemistry in Biology and Medicine. Pure Appl. Chem. 2007, 79, 2325–2338. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Valle NR, D.; Huang, P. Redox Regulation of Cell Survival. Antioxid. Redox Signal. 2008, 10, 1343–1373. Available online: https://www.liebertpub.com/doi/10.1089/ars.2007.1957 (accessed on 13 January 2023). [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of Peroxisomes in ROS/RNS-Metabolism: Implications for Human Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef]
- Higdon, A.; Diers, A.R.; Oh, J.Y.; Landar, A.; Darley-Usmar, V.M. Cell Signalling by Reactive Lipid Species: New Concepts and Molecular Mechanisms. Biochem. J. 2012, 442, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Hauck, A.K.; Bernlohr, D.A. Oxidative Stress and Lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative Stress, Mitochondrial Dysfunction and Neurodegenerative Diseases; a Mechanistic Insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Okamoto, S.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K.; et al. Balance between Synaptic versus Extrasynaptic NMDA Receptor Activity Influences Inclusions and Neurotoxicity of Mutant Huntingtin. Nat. Med. 2009, 15, 1407–1413. [Google Scholar] [CrossRef]
- Smith-Dijak, A.I.; Sepers, M.D.; Raymond, L.A. Alterations in Synaptic Function and Plasticity in Huntington Disease. J. Neurochem. 2019, 150, 346–365. [Google Scholar] [CrossRef]
- Valle, I.; Álvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1α Regulates the Mitochondrial Antioxidant Defense System in Vascular Endothelial Cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Hennessey, T.; Johri, A.; Tiwari, S.K.; Mishra, D.; Agarwal, S.; Kim, Y.S.; Beal, M.F. Transducer of Regulated CREB-Binding Proteins (TORCs) Transcription and Function Is Impaired in Huntington’s Disease. Hum. Mol. Genet. 2012, 21, 3474–3488. [Google Scholar] [CrossRef]
- McConoughey, S.J.; Basso, M.; Niatsetskaya, Z.V.; Sleiman, S.F.; Smirnova, N.A.; Langley, B.C.; Mahishi, L.; Cooper, A.J.L.; Antonyak, M.A.; Cerione, R.A.; et al. Inhibition of Transglutaminase 2 Mitigates Transcriptional Dysregulation in Models of Huntington Disease. EMBO Mol. Med. 2010, 2, 349–370. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68. [Google Scholar] [CrossRef]
- Polidori, M.C.; Mecocci, P.; Browne, S.E.; Senin, U.; Beal, M.F. Oxidative Damage to Mitochondrial DNA in Huntington’s Disease Parietal Cortex. Neurosci. Lett. 1999, 272, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-L.; Weissman, L.; Bohr, V.A.; Mattson, M.P. Mitochondrial DNA Damage and Repair in Neurodegenerative Disorders. DNA Repair 2008, 7, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Kolobkova, Y.A.; Vigont, V.A.; Shalygin, A.V.; Kaznacheyeva, E.V. Huntington’s Disease: Calcium Dyshomeostasis and Pathology Models. Acta Nat. 2017, 9, 34–46. [Google Scholar] [CrossRef]
- Seredenina, T.; Luthi-Carter, R. What Have We Learned from Gene Expression Profiles in Huntington’s Disease? Neurobiol. Dis. 2012, 45, 83–98. [Google Scholar] [CrossRef]
- Hipp, M.S.; Patel, C.N.; Bersuker, K.; Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Brandeis, M.; Kopito, R.R. Indirect Inhibition of 26S Proteasome Activity in a Cellular Model of Huntington’s Disease. J. Cell Biol. 2012, 196, 573–587. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo Recognition Failure Is Responsible for Inefficient Autophagy in Huntington’s Disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef]
- Bartzokis, G.; Lu, P.H.; Tishler, T.A.; Fong, S.M.; Oluwadara, B.; Finn, J.P.; Huang, D.; Bordelon, Y.; Mintz, J.; Perlman, S. Myelin Breakdown and Iron Changes in Huntington’s Disease: Pathogenesis and Treatment Implications. Neurochem. Res. 2007, 32, 1655–1664. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P.; Schapira, A.H.V.; Marsden, C.D. Alterations in Levels of Iron, Ferritin, and Other Trace Metals in Neurodegenerative Diseases Affecting the Basal Ganglia. Ann. Neurol. 1992, 32, S94–S100. [Google Scholar] [CrossRef]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal Cells Rearrangement during Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human Astrocytes: Structure and Functions in the Healthy Brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef]
- Wake, H.; Fields, R.D. Physiological Function of Microglia. Neuron Glia Biol. 2011, 7, 1–3. [Google Scholar] [CrossRef]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Ben Haim, L.; Carrillo-de Sauvage, M.-A.; Ceyzériat, K.; Escartin, C. Elusive Roles for Reactive Astrocytes in Neurodegenerative Diseases. Front. Cell. Neurosci. 2015, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Magistretti, P.J. The Role of Astroglia in Neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar] [CrossRef]
- Phatnani, H.; Maniatis, T. Astrocytes in Neurodegenerative Disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of Endogenous Neuroprotective Effects of Astrocytes in Brain Injury. Oxidative Med. Cell. Longev. 2018, 2018, e6501031. [Google Scholar] [CrossRef]
- Hsieh, H.-L.; Yang, C.-M. Role of Redox Signaling in Neuroinflammation and Neurodegenerative Diseases. BioMed Res. Int. 2013, 2013, e484613. [Google Scholar] [CrossRef]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef]
- Khakh, B.S.; Beaumont, V.; Cachope, R.; Munoz-Sanjuan, I.; Goldman, S.A.; Grantyn, R. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci. 2017, 40, 422–437. [Google Scholar] [CrossRef]
- Gray, M. Astrocytes in Huntington’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 355–381. [Google Scholar] [CrossRef]
- Valadão, P.A.C.; Santos, K.B.S.; Ferreira e Vieira, T.H.; Macedo e Cordeiro, T.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammation in Huntington’s Disease: A Few New Twists on an Old Tale. J. Neuroimmunol. 2020, 348, 577380. [Google Scholar] [CrossRef]
- Pawate, S.; Shen, Q.; Fan, F.; Bhat, N.R. Redox Regulation of Glial Inflammatory Response to Lipopolysaccharide and Interferonγ. J. Neurosci. Res. 2004, 77, 540–551. [Google Scholar] [CrossRef]
- Maritim, A.C.; Sanders, R.A.; Watkins, J.B. Diabetes, Oxidative Stress, and Antioxidants: A Review. J. Biochem. Mol. Toxicol. 2003, 17, 24–38. Available online: https://onlinelibrary.wiley.com/doi/10.1002/jbt.10058 (accessed on 13 January 2023). [CrossRef]
- Fang, Y.-Z.; Yang, S.; Wu, G. Free Radicals, Antioxidants, and Nutrition. Nutrition 2002, 18, 872–879. [Google Scholar] [CrossRef]
- Finkel, T. Oxidant Signals and Oxidative Stress. Curr. Opin. Cell Biol. 2003, 15, 247–254. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide Anion Radical (O2-.), Superoxide Dismutases, and Related Matters. J. Biol. Chem. 1997, 272, 18515–18517. [Google Scholar] [CrossRef]
- Percy, M.E. Catalase: An Old Enzyme with a New Role? Can. J. Biochem. Cell Biol. 1984, 62, 1006–1014. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive Species and Antioxidants. Redox Biology Is a Fundamental Theme of Aerobic Life. Plant Physiol. 2006, 141, 312–322. Available online: https://pubmed.ncbi.nlm.nih.gov/16760481/ (accessed on 13 January 2023). [CrossRef] [PubMed]
- Halliwell, B. Free Radicals and Antioxidants—Quo Vadis? Trends Pharmacol. Sci. 2011, 32, 125–130. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Rebec, G.V.; Barton, S.J.; Marseilles, A.M.; Collins, K. Ascorbate Treatment Attenuates the Huntington Behavioral Phenotype in Mice. Neuroreport 2003, 14, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias-Pinto, A.; Moll, P.; Solís-Maldonado, M.; Acuña, A.I.; Riveros, A.; Miró, M.P.; Papic, E.; Beltrán, F.A.; Cepeda, C.; Concha, I.I.; et al. Beyond the Redox Imbalance: Oxidative Stress Contributes to an Impaired GLUT3 Modulation in Huntington’s Disease. Free Radic. Biol. Med. 2015, 89, 1085–1096. [Google Scholar] [CrossRef]
- Acuña, A.I.; Esparza, M.; Kramm, C.; Beltrán, F.A.; Parra, A.V.; Cepeda, C.; Toro, C.A.; Vidal, R.L.; Hetz, C.; Concha, I.I.; et al. A Failure in Energy Metabolism and Antioxidant Uptake Precede Symptoms of Huntington’s Disease in Mice. Nat. Commun. 2013, 4, 2917. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, A.; Salvatierra-Sánchez, R.; Vázquez-Román, B.; Santiago-López, D.; Villeda-Hernández, J.; Galván-Arzate, S.; E Jimenez-Capdeville, M.; Ali, S.F. Protective Effects of the Antioxidant Selenium on Quinolinic Acid-induced Neurotoxicity in Rats: In Vitro and In Vivo Studies. J. Neurochem. 2004, 86, 479–488. Available online: https://onlinelibrary.wiley.com/doi/full/10.1046/j.1471-4159.2003.01857.x (accessed on 13 January 2023). [CrossRef]
- Bortolatto, C.F.; Jesse, C.R.; Wilhelm, E.A.; Chagas, P.M.; Nogueira, C.W. Organoselenium Bis Selenide Attenuates 3-Nitropropionic Acid-Induced Neurotoxicity in Rats. Neurotox. Res. 2013, 23, 214–224. [Google Scholar] [CrossRef]
- Lu, Z.; Marks, E.; Chen, J.; Moline, J.; Barrows, L.; Raisbeck, M.; Volitakis, I.; Cherny, R.A.; Chopra, V.; Bush, A.I.; et al. Altered Selenium Status in Huntington’s Disease: Neuroprotection by Selenite in the N171-82Q Mouse Model. Neurobiol. Dis. 2014, 71, 34–42. [Google Scholar] [CrossRef]
- Tasset, I.; Sánchez, F.; Túnez, I. The molecular bases of Huntington’s disease: The role played by oxidative stress. Rev. Neurol. 2009, 49, 424–429. [Google Scholar]
- Túnez, I.; Sánchez-López, F.; Agüera, E.; Fernández-Bolaños, R.; Sánchez, F.M.; Tasset-Cuevas, I. Important Role of Oxidative Stress Biomarkers in Huntington’s Disease. J. Med. Chem. 2011, 54, 5602–5606. [Google Scholar] [CrossRef] [PubMed]
- Morales-Martínez, A.; Sánchez-Mendoza, A.; Martínez-Lazcano, J.C.; Pineda-Farías, J.B.; Montes, S.; El-Hafidi, M.; Martínez-Gopar, P.E.; Tristán-López, L.; Pérez-Neri, I.; Zamorano-Carrillo, A.; et al. Essential Fatty Acid-Rich Diets Protect against Striatal Oxidative Damage Induced by Quinolinic Acid in Rats. Nutr. Neurosci. 2016, 20, 388–395. Available online: https://www.tandfonline.com/doi/full/10.1080/1028415X.2016.1147683 (accessed on 13 January 2023). [CrossRef] [PubMed]
- Vaddadi, K.S. Use of Gamma-Linolenic Acid in the Treatment of Schizophrenia and Tardive Dyskinesia. Prostaglandins Leukot. Essent. Fat. Acids 1992, 46, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Vaddadi, K.S.; Soosai, E.; Chiu, E.; Dingjan, P. A Randomised, Placebo-Controlled, Double Blind Study of Treatment of Huntington’s Disease with Unsaturated Fatty Acids. Neuroreport 2002, 13, 29–33. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Andreassen, O.A.; Jenkins, B.G.; Dedeoglu, A.; Kuemmerle, S.; Kubilus, J.K.; Kaddurah-Daouk, R.; Hersch, S.M.; Beal, M.F. Neuroprotective Effects of Creatine in a Transgenic Mouse Model of Huntington’s Disease. J. Neurosci. 2000, 20, 4389–4397. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Jenkins, B.G.; Ferrante, K.L.; Thomas, M.; Friedlich, A.; Browne, S.E.; Schilling, G.; Borchelt, D.R.; et al. Creatine Increase Survival and Delays Motor Symptoms in a Transgenic Animal Model of Huntington’s Disease. Neurobiol. Dis. 2001, 8, 479–491. [Google Scholar] [CrossRef]
- Shear, D.A.; Haik, K.L.; Dunbar, G.L. Creatine Reduces 3-Nitropropionic-Acid-Induced Cognitive and Motor Abnormalities in Rats. Neuroreport 2000, 11, 1833–1837. [Google Scholar] [CrossRef]
- Hersch, S.M.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L.; et al. Creatine in Huntington Disease Is Safe, Tolerable, Bioavailable in Brain and Reduces Serum 8OH2′dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef]
- Beal, M.F. Neuroprotective Effects of Creatine. Amino Acids 2011, 40, 1305–1313. [Google Scholar] [CrossRef]
- Hersch, S.M.; Schifitto, G.; Oakes, D.; Bredlau, A.-L.; Meyers, C.M.; Nahin, R.; Rosas, H.D.; For the Huntington Study Group CREST-E Investigators and Coordinators. The CREST-E Study of Creatine for Huntington Disease: A Randomized Controlled Trial. Neurology 2017, 89, 594–601. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic Effects of Coenzyme Q10 and Remacemide in Transgenic Mouse Models of Huntington’s Disease. J. Neurosci. 2002, 22, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Flint Beal, M. Combination Therapy with Coenzyme Q10 and Creatine Produces Additive Neuroprotective Effects in Models of Parkinson’s and Huntington’s Diseases. J. Neurochem. 2009, 109, 1427–1439. [Google Scholar] [CrossRef]
- Huntington Study Group Pre2CARE Investigators. Safety and Tolerability of High-dosage Coenzyme Q10 in Huntington’s Disease and Healthy Subjects. Mov. Disord. 2010, 25, 1924–1928. Available online: https://movementdisorders.onlinelibrary.wiley.com/doi/10.1002/mds.22408 (accessed on 13 January 2023). [CrossRef]
- Miyamoto, M.; Coyle, J.T. Idebenone Attenuates Neuronal Degeneration Induced by Intrastriatal Injection of Excitotoxins. Exp. Neurol. 1990, 108, 38–45. [Google Scholar] [CrossRef]
- Ranen, N.G.; Peyser, C.E.; Coyle, J.T.; Bylsma, F.W.; Sherr, M.; Day, L.; Folstein, M.F.; Brandt, J.; Ross, C.A.; Folstein, S.E. A Controlled Trial of Idebenone in Huntington’s Disease. Mov. Disord. 1996, 11, 549–554. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, P. Neuroprotective Activity of Curcumin in Combination with Piperine against Quinolinic Acid Induced Neurodegeneration in Rats. Pharmacology 2016, 97, 151–160. [Google Scholar] [CrossRef]
- Chongtham, A.; Agrawal, N. Curcumin Modulates Cell Death and Is Protective in Huntington’s Disease Model. Sci. Rep. 2016, 6, 18736. [Google Scholar] [CrossRef] [PubMed]
- Aditi, K.; Singh, A.; Shakarad, M.N.; Agrawal, N. Management of Altered Metabolic Activity in Drosophila Model of Huntington’s Disease by Curcumin. Exp. Biol. Med. 2022, 247, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Elifani, F.; Amico, E.; Pepe, G.; Capocci, L.; Castaldo, S.; Rosa, P.; Montano, E.; Pollice, A.; Madonna, M.; Filosa, S.; et al. Curcumin Dietary Supplementation Ameliorates Disease Phenotype in an Animal Model of Huntington’s Disease. Hum. Mol. Genet. 2019, 28, 4012–4021. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pfleger, C.M.; Friedman, L.; Vittorino, R.; Zhao, W.; Qian, X.; Conley, L.; Ho, L.; Pasinetti, G.M. Potential Application of Grape Derived Polyphenols in Huntington’s Disease. Transl. Neurosci. 2010, 1, 95–100. [Google Scholar] [CrossRef]
- Kumar, P.; Kalonia, H.; Kumar, A. Lycopene Modulates Nitric Oxide Pathways against 3-Nitropropionic Acid-Induced Neurotoxicity. Life Sci. 2009, 85, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, A. Effect of Lycopene and Epigallocatechin-3-Gallate against 3-Nitropropionic Acid Induced Cognitive Dysfunction and Glutathione Depletion in Rat: A Novel Nitric Oxide Mechanism. Food Chem. Toxicol. 2009, 47, 2522–2530. [Google Scholar] [CrossRef]
- Jain, D.; Gangshettiwar, A. Combination of Lycopene, Quercetin and Poloxamer 188 Alleviates Anxiety and Depression in 3-Nitropropionic Acid-Induced Huntington’s Disease in Rats. J. Intercult. Ethnopharmacol. 2014, 3, 186–191. [Google Scholar] [CrossRef]
- Túnez, I.; Montilla, P.; Del Carmen Muñoz, M.; Feijóo, M.; Salcedo, M. Protective Effect of Melatonin on 3-Nitropropionic Acid-Induced Oxidative Stress in Synaptosomes in an Animal Model of Huntington’s Disease. J. Pineal Res. 2004, 37, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Antunes Wilhelm, E.; Ricardo Jesse, C.; Folharini Bortolatto, C.; Wayne Nogueira, C. Correlations between Behavioural and Oxidative Parameters in a Rat Quinolinic Acid Model of Huntington’s Disease: Protective Effect of Melatonin. Eur. J. Pharmacol. 2013, 701, 65–72. [Google Scholar] [CrossRef]
- Uz, T.; Giusti, P.; Franceschini, D.; Kharlamov, A.; Manev, H. Protective Effect of Melatonin against Hippocampal Dna Damage Induced by Intraperitoneal Administration of Kainate to Rats. Neuroscience 1996, 73, 631–636. [Google Scholar] [CrossRef]
- Fontaine, M.A.; Geddes, J.W.; Banks, A.; Butterfield, D.A. Effect of Exogenous and Endogenous Antioxidants on 3-Nitropionic Acid-Induced In Vivo Oxidative Stress and Striatal Lesions: Insights into Huntington’s Disease. J. Neurochem. 2000, 75, 1709–1715. [Google Scholar] [CrossRef]
- Sandhir, R.; Sood, A.; Mehrotra, A.; Kamboj, S.S. N-Acetylcysteine Reverses Mitochondrial Dysfunctions and Behavioral Abnormalities in 3-Nitropropionic Acid-Induced Huntington’s Disease. Neurodegener. Dis. 2012, 9, 145–157. [Google Scholar] [CrossRef]
- Wright, D.J.; Gray, L.J.; Finkelstein, D.I.; Crouch, P.J.; Pow, D.; Pang, T.Y.; Li, S.; Smith, Z.M.; Francis, P.S.; Renoir, T.; et al. N-Acetylcysteine Modulates Glutamatergic Dysfunction and Depressive Behavior in Huntington’s Disease. Hum. Mol. Genet. 2016, 25, 2923–2933. [Google Scholar] [CrossRef] [PubMed]
- Wjpps|ABSTRACT. Available online: https://www.wjpps.com/Wjpps_controller/abstract_id/2546 (accessed on 13 January 2023).
- Suganya, S.N.; Sumathi, T. Effect of Rutin against a Mitochondrial Toxin, 3-Nitropropionicacid Induced Biochemical, Behavioral and Histological Alterations-a Pilot Study on Huntington’s Disease Model in Rats. Metab. Brain Dis. 2017, 32, 471–481. [Google Scholar] [CrossRef]
- Keene, C.D.; Rodrigues, C.M.; Eich, T.; Linehan-Stieers, C.; Abt, A.; Kren, B.T.; Steer, C.J.; Low, W.C. A Bile Acid Protects against Motor and Cognitive Deficits and Reduces Striatal Degeneration in the 3-Nitropropionic Acid Model of Huntington’s Disease. Exp. Neurol. 2001, 171, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Keene, C.D.; Rodrigues, C.M.P.; Eich, T.; Chhabra, M.S.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic Acid, a Bile Acid, Is Neuroprotective in a Transgenic Animal Model of Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10671–10676. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kalonia, H.; Kumar, A. Possible Nitric Oxide Modulation in Protective Effect of FK-506 against 3-Nitropropionic Acid-Induced Behavioral, Oxidative, Neurochemical, and Mitochondrial Alterations in Rat Brain. Drug Chem. Toxicol. 2010, 33, 377–392. [Google Scholar] [CrossRef]
- Rosenstock, T.R.; de Brito, O.M.; Lombardi, V.; Louros, S.; Ribeiro, M.; Almeida, S.; Ferreira, I.L.; Oliveira, C.R.; Rego, A.C. FK506 Ameliorates Cell Death Features in Huntington’s Disease Striatal Cell Models. Neurochem. Int. 2011, 59, 600–609. [Google Scholar] [CrossRef]
- Stack, C.; Ho, D.; Wille, E.; Calingasan, N.Y.; Williams, C.; Liby, K.; Sporn, M.; Dumont, M.; Beal, M.F. Triterpenoids CDDO-Ethyl Amide and CDDO-Trifluoroethyl Amide Improve the Behavioral Phenotype and Brain Pathology in a Transgenic Mouse Model of Huntington’s Disease. Free Radic. Biol. Med. 2010, 49, 147–158. [Google Scholar] [CrossRef]
- Xun, Z.; Rivera-Sánchez, S.; Ayala-Peña, S.; Lim, J.; Budworth, H.; Skoda, E.M.; Robbins, P.D.; Niedernhofer, L.J.; Wipf, P.; McMurray, C.T. Targeting of XJB-5-131 to Mitochondria Suppresses Oxidative DNA Damage and Motor Decline in a Mouse Model of Huntington’s Disease. Cell Rep. 2012, 2, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, A.; Holt, A.; Brown, C.; Cosme, C.; Wipf, P.; Gomez-Marin, A.; Castro, M.R.; Ayala-Peña, S.; McMurray, C.T. Mitochondrial Targeting of XJB-5-131 Attenuates or Improves Pathophysiology in HdhQ150 Animals with Well-Developed Disease Phenotypes. Hum. Mol. Genet. 2016, 25, 1792–1802. [Google Scholar] [CrossRef]
- Polyzos, A.A.; Wood, N.I.; Williams, P.; Wipf, P.; Morton, A.J.; McMurray, C.T. XJB-5-131-Mediated Improvement in Physiology and Behaviour of the R6/2 Mouse Model of Huntington’s Disease Is Age- and Sex- Dependent. PLoS ONE 2018, 13, e0194580. [Google Scholar] [CrossRef] [PubMed]
- Colle, D.; Santos, D.B.; Moreira, E.L.G.; Hartwig, J.M.; dos Santos, A.A.; Zimmermann, L.T.; Hort, M.A.; Farina, M. Probucol Increases Striatal Glutathione Peroxidase Activity and Protects against 3-Nitropropionic Acid-Induced Pro-Oxidative Damage in Rats. PLoS ONE 2013, 8, e67658. [Google Scholar] [CrossRef]
- de Paula Nascimento-Castro, C.; Wink, A.C.; da Fônseca, V.S.; Bianco, C.D.; Winkelmann-Duarte, E.C.; Farina, M.; Rodrigues, A.L.S.; Gil-Mohapel, J.; de Bem, A.F.; Brocardo, P.S. Antidepressant Effects of Probucol on Early-Symptomatic YAC128 Transgenic Mice for Huntington’s Disease. Neural Plast. 2018, 2018, 4056383. [Google Scholar] [CrossRef]
- Klivenyi, P.; Ferrante, R.J.; Gardian, G.; Browne, S.; Chabrier, P.-E.; Beal, M.F. Increased Survival and Neuroprotective Effects of BN82451 in a Transgenic Mouse Model of Huntington’s Disease. J. Neurochem. 2003, 86, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Zwilling, D.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.-Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-Monooxygenase Inhibition in Blood Ameliorates Neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef]
- Hussein, M.; Fathy, W.; Hassan, A.; Elkareem, R.A.; Marzouk, S.; Kamal, Y.S. Zinc Deficiency Correlates with Severity of Diabetic Polyneuropathy. Brain Behav. 2021, 11, e2349. [Google Scholar] [CrossRef] [PubMed]
- Office of Dietary Supplements—Zinc. Available online: https://ods.od.nih.gov/factsheets/Zinc-HealthProfessional/ (accessed on 15 July 2022).
- Castro, M.A.; Beltrán, F.A.; Brauchi, S.; Concha, I.I. A Metabolic Switch in Brain: Glucose and Lactate Metabolism Modulation by Ascorbic Acid. J. Neurochem. 2009, 110, 423–440. [Google Scholar] [CrossRef]
- Castro, M.; Caprile, T.; Astuya, A.; Millán, C.; Reinicke, K.; Vera, J.C.; Vásquez, O.; Aguayo, L.G.; Nualart, F. High-Affinity Sodium-Vitamin C Co-Transporters (SVCT) Expression in Embryonic Mouse Neurons. J. Neurochem. 2001, 78, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Pozo, M.; Cortés, C.; García, M.D.L.A.; Concha, I.I.; Nualart, F. Intracellular Ascorbic Acid Inhibits Transport of Glucose by Neurons, but Not by Astrocytes. J. Neurochem. 2007, 102, 773–782. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.; Mendiratta, S. Protection and Recycling of α-Tocopherol in Human Erythrocytes by Intracellular Ascorbic Acid. Arch. Biochem. Biophys. 1998, 349, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Majewska, M.D.; Bell, J.A. Ascorbic Acid Protects Neurons from Injury Induced by Glutamate and NMDA. Neuroreport 1990, 1, 194–196. [Google Scholar] [CrossRef]
- Solovyev, N.D. Importance of Selenium and Selenoprotein for Brain Function: From Antioxidant Protection to Neuronal Signalling. J. Inorg. Biochem. 2015, 153, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Berr, C.; Portet, F.; Carriere, I.; Akbaraly, T.N.; Feart, C.; Gourlet, V.; Combe, N.; Barberger-Gateau, P.; Ritchie, K. Olive Oil and Cognition: Results from the Three-City Study. Dement. Geriatr. Cogn. Disord. 2009, 28, 357–364. [Google Scholar] [CrossRef]
- Quiles, J.L.; Ochoa, J.J.; Ramirez-Tortosa, C.; Battino, M.; Huertas, J.R.; Martín, Y.; Mataix, J. Dietary Fat Type (Virgin Olive vs. Sunflower Oils) Affects Age-Related Changes in DNA Double-Strand-Breaks, Antioxidant Capacity and Blood Lipids in Rats. Exp. Gerontol. 2004, 39, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Fitó, M.; de la Torre, R.; Farré-Albaladejo, M.; Khymenetz, O.; Marrugat, J.; Covas, M.-I. Bioavailability and Antioxidant Effects of Olive Oil Phenolic Compounds in Humans: A Review. Ann. Ist. Super Sanita 2007, 43, 375–381. [Google Scholar]
- González-Correa, J.A.; Navas, M.D.; Lopez-Villodres, J.A.; Trujillo, M.; Espartero, J.L.; De La Cruz, J.P. Neuroprotective Effect of Hydroxytyrosol and Hydroxytyrosol Acetate in Rat Brain Slices Subjected to Hypoxia–Reoxygenation. Neurosci. Lett. 2008, 446, 143–146. [Google Scholar] [CrossRef]
- Covas, M.-I.; Nyyssönen, K.; Poulsen, H.E.; Kaikkonen, J.; Zunft, H.-J.F.; Kiesewetter, H.; Gaddi, A.; de la Torre, R.; Mursu, J.; Bäumler, H.; et al. The Effect of Polyphenols in Olive Oil on Heart Disease Risk Factors: A Randomized Trial. Ann. Intern. Med. 2006, 145, 333–341. [Google Scholar] [CrossRef]
- Díaz, R.J.; Yago, M.D.; Martínez-Victoria, E.; Naranjo, J.A.; Martínez, M.A.; Mañas, M. Comparison of the Effects of Dietary Sunflower Oil and Virgin Olive Oil on Rat Exocrine Pancreatic Secretion In Vivo. Lipids 2003, 38, 1119–1126. [Google Scholar] [CrossRef]
- Hagen, T.M.; Ingersoll, R.T.; Lykkesfeldt, J.; Liu, J.; Wehr, C.M.; Vinarsky, V.; Bartholomew, J.C.; Ames, A.B. (R)-Alpha-Lipoic Acid-Supplemented Old Rats Have Improved Mitochondrial Function, Decreased Oxidative Damage, and Increased Metabolic Rate. FASEB J. 1999, 13, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Pearson, J.; Rogers, D.A.; Bissada, N.; Vogl, A.W.; Hayden, M.R.; Leavitt, B.R. Loss of Wild-Type Huntingtin Influences Motor Dysfunction and Survival in the YAC128 Mouse Model of Huntington Disease. Hum. Mol. Genet. 2005, 14, 1379–1392. [Google Scholar] [CrossRef]
- Brocardo, P.S.; Gil-Mohapel, J.M. Therapeutic Strategies for Huntingtons Disease: From the Bench to the Clinic. Curr. Psychopharmacol. 2012, 1, 137–154. [Google Scholar] [CrossRef]
- Stefani, G.P.; Nunes, R.B.; Dornelles, A.Z.; Alves, J.P.; Piva, M.O.; Domenico, M.D.; Rhoden, C.R.; Lago, P.D. Effects of Creatine Supplementation Associated with Resistance Training on Oxidative Stress in Different Tissues of Rats. J. Int. Soc. Sport. Nutr. 2014, 11, 11. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A.; Beal, M.F. Potential for Creatine and Other Therapies Targeting Cellular Energy Dysfunction in Neurological Disorders. Ann. Neurol. 2001, 49, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Blamire, A.M.; Manners, D.N.; Rajagopalan, B.; Styles, P.; Schapira, A.H.V.; Warner, T.T. Creatine Therapy for Huntington’s Disease: Clinical and MRS Findings in a 1-Year Pilot Study. Neurology 2003, 61, 141–142. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Blamire, A.M.; Manners, D.N.; Rajagopalan, B.; Styles, P.; Schapira, A.H.V.; Warner, T.T. High-Dose Creatine Therapy for Huntington Disease: A 2-Year Clinical and MRS Study. Neurology 2005, 64, 1655–1656. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group. A Randomized, Placebo-Controlled Trial of Coenzyme Q10 and Remacemide in Huntington’s Disease. Neurology 2001, 57, 397–404. [Google Scholar] [CrossRef]
- Feigin, A.; Kieburtz, K.; Como, P.; Hickey, C.; Abwender, D.; Zimmerman, C.; Steinberg, K.; Shoulson, I. Assessment of Coenzyme Q10 Tolerability in Huntington’s Disease. Mov. Disord. 1996, 11, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Kieburtz, K.; Feigin, A.; McDermott, M.; Como, P.; Abwender, D.; Zimmerman, C.; Hickey, C.; Orme, C.; Claude, K.; Sotack, J.; et al. A Controlled Trial of Remacemide Hydrochloride in Huntington’s Disease. Mov. Disord. 1996, 11, 273–277. [Google Scholar] [CrossRef]
- Schilling, G.; Coonfield, M.L.; Ross, C.A.; Borchelt, D.R. Coenzyme Q10 and Remacemide Hydrochloride Ameliorate Motor Deficits in a Huntington’s Disease Transgenic Mouse Model. Neurosci. Lett. 2001, 315, 149–153. [Google Scholar] [CrossRef]
- Young, J.M.; Florkowski, C.M.; Molyneux, S.L.; McEwan, R.G.; Frampton, C.M.; George, P.M.; Scott, R.S. Effect of Coenzyme Q(10) Supplementation on Simvastatin-Induced Myalgia. Am. J. Cardiol. 2007, 100, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Essa, M.M.; Moghadas, M.; Ba-Omar, T.; Walid Qoronfleh, M.; Guillemin, G.J.; Manivasagam, T.; Justin-Thenmozhi, A.; Ray, B.; Bhat, A.; Chidambaram, S.B.; et al. Protective Effects of Antioxidants in Huntington’s Disease: An Extensive Review. Neurotox. Res. 2019, 35, 739–774. [Google Scholar] [CrossRef]
- Second Affiliated Hospital, School of Medicine, Zhejiang University. Non-Randomized Control Clinical Trial to Evaluate the Efficacy and Safety of Symptomatic Drug Therapy for Mild to Moderate Huntington’s Disease Patients; clinicaltrials.gov: Bethesda, MD, USA, 2021.
- D’Egidio, F.; Lombardozzi, G.; Kacem Ben Haj M’Barek, H.E.; Mastroiacovo, G.; Alfonsetti, M.; Cimini, A. The Influence of Dietary Supplementations on Neuropathic Pain. Life 2022, 12, 1125. [Google Scholar] [CrossRef]
- Ho, L.; Pasinetti, G.M. Polyphenolic Compounds for Treating Neurodegenerative Disorders Involving Protein Misfolding. Expert Rev. Proteom. 2010, 7, 579–589. [Google Scholar] [CrossRef]
- Huang, F.; Ning, H.; Xin, Q.-Q.; Huang, Y.; Wang, H.; Zhang, Z.-H.; Xu, D.-X.; Ichihara, G.; Ye, D.-Q. Melatonin Pretreatment Attenuates 2-Bromopropane-Induced Testicular Toxicity in Rats. Toxicology 2009, 256, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Stimming, E.F. Melatonin for Huntington’s Disease (HD) Gene Carriers with HD Related Sleep Disturbance—A Pilot Study; clinicaltrials.gov: Bethesda, MD, USA, 2022.
- Moussaieff, A.; Mechoulam, R. Boswellia Resin: From Religious Ceremonies to Medical Uses: A Review of In-Vitro, In-Vivo and Clinical Trials. J. Pharm. Pharmacol. 2009, 61, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.; Nguyen, H. St. John’s Wort. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Borlongan, C.V.; Koutouzis, T.K.; Sanberg, P.R. 3-Nitropropionic Acid Animal Model and Huntington’s Disease. Neurosci. Biobehav. Rev. 1997, 21, 289–293. [Google Scholar] [CrossRef]
- Loy, C. A Randomised Controlled Trial, Of N-Acetyl Cysteine (NAC), for Premanifest Huntingtin Gene Expansion Carriers; National Library of Medicine: Bethesda, MD, USA, 2022.
- Katsube, T.; Imawaka, N.; Kawano, Y.; Yamazaki, Y.; Shiwaku, K.; Yamane, Y. Antioxidant Flavonol Glycosides in Mulberry (Morus Alba L.) Leaves Isolated Based on LDL Antioxidant Activity. Food Chem. 2006, 97, 25–31. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, K.W.; Kim, D.Y.; Park, H.H.; Kwon, I.B.; Lee, H.J. Optimal Recovery of High-Purity Rutin Crystals from the Whole Plant of Fagopyrum Esculentum Moench (Buckwheat) by Extraction, Fractionation, and Recrystallization. Bioresour. Technol. 2005, 96, 1709–1712. [Google Scholar] [CrossRef]
- Aleksandrov, P.N.; Speranskaia, T.V.; Bobkov, I.G.; Zagorevskiĭ, V.A.; Zykov, D.A. Effect of rutin and esculamine on models of aseptic inflammation. Farmakol. Toksikol. 1986, 49, 84–86. [Google Scholar]
- Cruz, T.; Gálvez, J.; Ocete, M.A.; Crespo, M.E.; Sánchez de Medina, L.-H., F.; Zarzuelo, A. Oral Administration of Rutoside Can Ameliorate Inflammatory Bowel Disease in Rats. Life Sci. 1998, 62, 687–695. [Google Scholar] [CrossRef]
- Chen, S.; Gong, J.; Liu, F.; Mohammed, U. Naturally Occurring Polyphenolic Antioxidants Modulate IgE-Mediated Mast Cell Activation. Immunology 2000, 100, 471–480. [Google Scholar] [CrossRef]
- Abd-El-Fattah, A.A.; El-Sawalhi, M.M.; Rashed, E.R.; El-Ghazaly, M.A. Possible Role of Vitamin E, Coenzyme Q10 and Rutin in Protection against Cerebral Ischemia/Reperfusion Injury in Irradiated Rats. Int. J. Radiat. Biol. 2010, 86, 1070–1078. [Google Scholar] [CrossRef]
- Koda, T.; Kuroda, Y.; Imai, H. Protective Effect of Rutin against Spatial Memory Impairment Induced by Trimethyltin in Rats. Nutr. Res. 2008, 28, 629–634. [Google Scholar] [CrossRef]
- Fernández, S.P.; Wasowski, C.; Loscalzo, L.M.; Granger, R.E.; Johnston, G.A.R.; Paladini, A.C.; Marder, M. Central Nervous System Depressant Action of Flavonoid Glycosides. Eur. J. Pharmacol. 2006, 539, 168–176. [Google Scholar] [CrossRef]
- Zangerolamo, L.; Vettorazzi, J.F.; Rosa, L.R.O.; Carneiro, E.M.; Barbosa, H.C.L. The Bile Acid TUDCA and Neurodegenerative Disorders: An Overview. Life Sci. 2021, 272, 119252. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely Potent Triterpenoid Inducers of the Phase 2 Response: Correlations of Protection against Oxidant and Inflammatory Stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef] [PubMed]
- Suh, N.; Wang, Y.; Honda, T.; Gribble, G.W.; Dmitrovsky, E.; Hickey, W.F.; Maue, R.A.; Place, A.E.; Porter, D.M.; Spinella, M.J.; et al. A Novel Synthetic Oleanane Triterpenoid, 2-Cyano-3,12-Dioxoolean-1,9-Dien-28-Oic Acid, with Potent Differentiating, Antiproliferative, and Anti-Inflammatory Activity1. Cancer Res. 1999, 59, 336–341. [Google Scholar]
- Yates, M.S.; Tauchi, M.; Katsuoka, F.; Flanders, K.C.; Liby, K.T.; Honda, T.; Gribble, G.W.; Johnson, D.A.; Johnson, J.A.; Burton, N.C.; et al. Pharmacodynamic Characterization of Chemopreventive Triterpenoids as Exceptionally Potent Inducers of Nrf2-Regulated Genes. Mol. Cancer Ther. 2007, 6, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Honda, Y.; Favaloro, F.G.; Gribble, G.W.; Suh, N.; Place, A.E.; Rendi, M.H.; Sporn, M.B. A Novel Dicyanotriterpenoid, 2-Cyano-3,12-Dioxooleana-1,9(11)-Dien-28-Onitrile, Active at Picomolar Concentrations for Inhibition of Nitric Oxide Production. Bioorg. Med. Chem. Lett. 2002, 12, 1027–1030. [Google Scholar] [CrossRef]
- Wipf, P.; Polyzos, A.A.; McMurray, C.T. A Double-Pronged Sword: XJB-5-131 Is a Suppressor of Somatic Instability and Toxicity in Huntington’s Disease. J. Huntingt. Dis. 2022, 11, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Chabrier, P.E.; Auguet, M. Pharmacological Properties of BN82451: A Novel Multitargeting Neuroprotective Agent. CNS Drug. Rev. 2007, 13, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Matson, W.R.; Storey, E.; Milbury, P.; Ryan, E.A.; Ogawa, T.; Bird, E.D. Kynurenic Acid Concentrations Are Reduced in Huntington’s Disease Cerebral Cortex. J. Neurol. Sci. 1992, 108, 80–87. Available online: https://pubmed.ncbi.nlm.nih.gov/1385624/ (accessed on 13 January 2023). [CrossRef]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The Kynurenine Pathway Modulates Neurodegeneration in a Drosophila Model of Huntington’s Disease. Curr. Biol. 2011, 21, 961–966. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Dean, O.; Giorlando, F.; Berk, M. N-Acetylcysteine in Psychiatry: Current Therapeutic Evidence and Potential Mechanisms of Action. J. Psychiatry Neurosci. 2011, 36, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The Chemistry and Biological Activities of N-Acetylcysteine. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2013, 1830, 4117–4129. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Antioxidant | Mechanism of Action | Model | Effects | References |
|---|---|---|---|---|
| Vitamin C | In the presence of ROS, Vitamin C quickly oxidizes to dihydroxy ascorbic acid, acting as a regulator of redox balance in neurons | R6/2 mice | Restoration of behavioral response | [188] |
| Modulation of neuronal glucose uptake | [189] | |||
| STHdhQ cells | An early impairment of ascorbic acid uptake in HD neurons. | [190] | ||
| Modulation of neuronal glucose uptake | [189] | |||
| Selenium | Effects are mainly exerted due to the antioxidant function of selenoproteins, which help maintain the intracellular redox status and prevent cellular damage from free radicals | Quinolinic acid rat model of HD | Selenium partially protects against quinolinic acid-induced toxicity | [191] |
| 3-NP-induced HD-like rat model | Protective effects against HD-like signs induced by 3-NP in rats | [192] | ||
| N171-82Q mice | Sodium selenite supplementation decreased brain weight loss and GSSG levels, but also increased motor function | [193] | ||
| Unsaturated Fatty Acids | They display antimicrobial, antioxidant, and anti-inflammatory properties | 3-NP-induced HD-like rat model | EVOO act as a powerful brain antioxidant | [194] |
| Decreased lipid peroxidation and GSH recovery | [195] | |||
| Quinolinic acid rat model of HD | EVOO and fish-oil counteracted oxidative damage, increasing PPARg expression, and restoring rat behavior | [196] | ||
| HD patients | Restoration of the motor function | [197,198] | ||
| Creatine | It is effective against superoxide, peroxynitrite, and hydroxyl radicals | R6/2 mice | A role of metabolic dysfunction in a transgenic mouse model of HD was supported, suggesting a novel therapeutic strategy to slow the pathological process | [199] |
| N171-82Q mice | Creatine may exert therapeutic effects in HD | [200] | ||
| 3-NP-induced HD-like rat model | Creatine counteracted motor impairment and cognitive abnormalities | [201] | ||
| HD patients | Creatine supplementation significantly reduced elevated serum levels of 8-OHdG back to baseline levels seen in controls | [202] | ||
| Slowing of the ongoing cortical atrophy | [203] | |||
| Creatine monohydrate is not beneficial for slowing functional decline | [204] | |||
| Coenzyme Q10 (CoQ10) | It acts as an electron carrier in ETC 1 and reduces singlet oxygen, prevents oxidation of bases in mitochondria and formation of lipid peroxyl radicals and protein oxidation | R6/2 mice | CoQ10 or remacemide significantly extended survival and delayed the development of motor deficits, weight loss, cerebral atrophy, and neuronal intra-nuclear inclusions | [205] |
| The combination of CoQ10 and creatine improved motor performance and promoted survival in mice | [206] | |||
| Creatine promoted survival of mice, improved the motor function, and counteracted the striatal atrophy | [203] | |||
| 3-NP-induced HD-like rat model | The combination treatment blocked 3-NP-induced impairment of glutathione homeostasis, reduced lipid peroxidation and DNA oxidative damage in the striatum | [206] | ||
| HD patients | Dosages of 2400 mg/day may provide the best balance between tolerability and blood level achieved | [207] | ||
| Idebenone | It has antioxidant properties similar to CoQ10. It is a substrate of NQO 1 and 2 | Kainic acid-induced rat model | Restoration of GAD immunoreactivity | [208] |
| HD patients | No impact for idebenone on HD patient conditions compared to the placebo controls | [209] | ||
| Curcumin | It is involved in several functions, among which are antioxidant, neuroprotective, and anti-inflammatory activities. Regulates various pathways related to cell survivor, caspases, tumor suppression, and others | Quinolinic acid model of HD in rats | Combination of curmcumin and piperine showed strong antioxidant and protective effect against quinolinic acid-induced behavioral and neurological alteration in rats | [210] |
| Drosophila HD model | Suppression of cell death | [211] | ||
| Metabolic anomalies amelioration, ROS levels reduction, and motor impairment counteraction have been obtained after curcumin administration | [212] | |||
| R6/2 mice | Overall amelioration of the HD phenotype | [213] | ||
| Grape Seed Polyphenol Extract (GSPE) | It limits lipid peroxidation and reduces inflammation | R6/2 mice | GSPE might be able to modulate the onset and/or progression of HD | [214] |
| PC-12 HD | Reduced levels of carbonyl heightened by mHtt expression and inhibited formation of mHtt aggregate | |||
| Lycopene | It exerts antioxidant effects by quenching singlet oxygen | 3-NP-induced HD-like rat model | Pretreatment improved behavioral symptoms, counteracted oxidative damage, restored the activity of mitochondrial enzymes, but also reduction in lipid peroxidation, NO and SOD levels, and behavioral impairment | [215] |
| Administration of lycopene in combination with other compounds lead to modulation of NO, resulting in restoration of behavioral and biochemical improvement | [216] | |||
| Combination treatment of lycopene and quercetin with and without poloxamer 188 in HD alleviate anxiety and depression than single drug therapy | [217] | |||
| Melatonin | It shows strong antioxidant activity by scavenging ROS/ RNS and inhibiting NOS but also protects lipids in membranes, proteins in cytosol, DNA in nucleus and mitochondria from free radical damage | 3-NP-induced HD-like rat model | Melatonin prevents the deleterious effects induced by 3-NP | [218] |
| Quinolinic acid model of HD in rats | Melatonin helps in neurotoxicity caused by quinolinic acid in rats | [219] | ||
| Kainic acid-induced rat model | Melatonin counteracted the oxidative damage promoting neuroprotection | [220] | ||
| N-Acetylcysteine | It reduces oxidative stress by decreasing ROS and lipid peroxidation, and by restoration of antioxidant enzymes | 3-NP-induced HD-like rat model | NAC treatment may be a useful therapeutic strategy against 3-NP neurotoxicity and HD | [221] |
| NAC treatment counteracted mitochondrial dysfunctions and neurobehavioral deficits | [222] | |||
| R6/1 mice | NAC rescued glutamatergic dysfunction and depressive-like behavior in HD | [223] | ||
| Rutin | It shows antioxidant, anti-inflammatory, neuroprotective, and immunomodulatory properties. | 3-NP-induced HD-like rat model | Rutin improved the behavioral alterations and restored the activities of mitochondrial complex enzymes in rat model | [224] |
| Rutin may have an important role in protecting the striatum from oxidative stress caused by 3-NP | [225] | |||
| Tauroursodeoxycholic acid (TUDCA) | A hydrophilic bile acid, has anti-inflammatory and cytoprotective effects, antioxidant effects; mechanism of action not fully defined yet | 3-NP-induced HD-like rat model | TUDCA administration counteracted apoptosis and reduced lesion volumes, but also protected against cognitive impairment and motor deficit | [226] |
| R6/2 mice | Systemic administration of TUDCA significantly counteracted striatal neuropathology, reduced apoptosis, and improved locomotor and sensorimotor deficits | [227] | ||
| Tacrolimus (FK-506) | It has anti-inflammatory and antioxidant properties via modulation of NOS and HO activities | 3-NP-induced HD-like rat model | Treatment with tacrolimus improved behavioral anomalies and restored the levels of oxidative stress markers and antioxidant enzymes such as SOD and CAT | [228] |
| Primary rat striatal neurons | Treatment with tacrolimus counteracted effects of 3-NP exposure, inducing neuroprotection and reducing apoptosis | [229] | ||
| STHdhQ cells | ||||
| Synthetic triterpenoids | Triterpenoids, particularly the analogs of CDDO, have antioxidant and anti-inflammatory properties acting via Nrf2/ARE pathway | 3-NP-induced HD-like rat model | CDDO-MA can be neuroprotective in experimental model of HD | [206] |
| N171-82Q mice | CDDO-EA and CDDO-TFEA upregulated Nrf2/ARE induced genes in the brain and peripheral tissues, reduced oxidative stress, improved motor impairment and increased longevity, also rescuing striatal atrophy in the brain and vacuolation in the brown adipose tissue | [230] | ||
| XJB-5-131 | Synthetic compound with antioxidant properties and electron scavenging ability | HD150KI mice (test) C57BL/6 mice (controls) | XJB-5-131 can suppress weight loss, ameliorate mitochondrial activity, and improve motor performance | [231] |
| HdhQ150 mice | XJB-5-131 administration led to reduced neuronal atrophy and improved motor function, but also reduced level of 8-OHdG in mitochondria and nucleus of striatal cells derived from the mice | [232] | ||
| R6/2 mice | Chronic treatment with XJB-5-131 ameliorated behavioral e physiological deficits in an age- and sex dependent manner | [233] | ||
| Probucol | Lipid-lowering agent with antioxidant and anti-inflammatory properties | 3-NP-induced HD-like rat model | Improvement in motor function and oxidative stress condition with stimulation of GPx activity | [234] |
| YAC128 transgenic HD mouse model | Chronic administration of probucol reduced the occurrence of depressive-like behaviors | [235] | ||
| BN82451 | It reduces excitotoxicity, oxidative stress, and inflammation and is also a mitochondrial protective agent | R6/2 mice | BN82451 promoted survival, improved motor function, reduced morphology loss, and reduced levels of aggregates positive to ubiquitin | [236] |
| Kynurenine-3-monooxy inhibitor: JM6 | Inhibition of KMO 2 in blood increases kynurenic acid in brain and reduces extracellular glutamate and free radical production | R6/2 mice | Administration of JM6 resulted in increased kynurenic acid levels and reduced extracellular glutamate in the brain, prolonged lifespan and prevented synaptic loss | [237] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Egidio, F.; Castelli, V.; Cimini, A.; d’Angelo, M. Cell Rearrangement and Oxidant/Antioxidant Imbalance in Huntington’s Disease. Antioxidants 2023, 12, 571. https://doi.org/10.3390/antiox12030571
D’Egidio F, Castelli V, Cimini A, d’Angelo M. Cell Rearrangement and Oxidant/Antioxidant Imbalance in Huntington’s Disease. Antioxidants. 2023; 12(3):571. https://doi.org/10.3390/antiox12030571
Chicago/Turabian StyleD’Egidio, Francesco, Vanessa Castelli, Annamaria Cimini, and Michele d’Angelo. 2023. "Cell Rearrangement and Oxidant/Antioxidant Imbalance in Huntington’s Disease" Antioxidants 12, no. 3: 571. https://doi.org/10.3390/antiox12030571