
Rescue of Dopamine Neurons from Iron-Dependent Ferroptosis by Doxycycline and Demeclocycline and Their Non-Antibiotic Derivatives
 , , ,
, , ,
Abstract
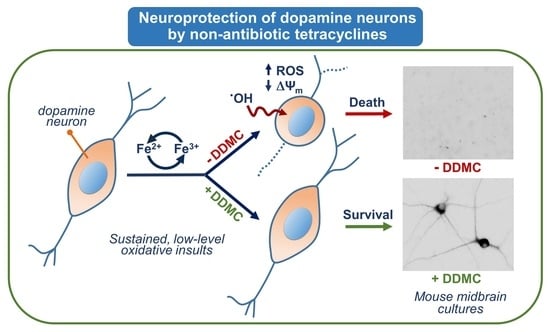
:
1. Introduction
2. Materials and Methods
2.1. Use of Animals
2.2. Midbrain Cell Culture Protocol
2.3. Treatments with Test TCs and Reference Compounds
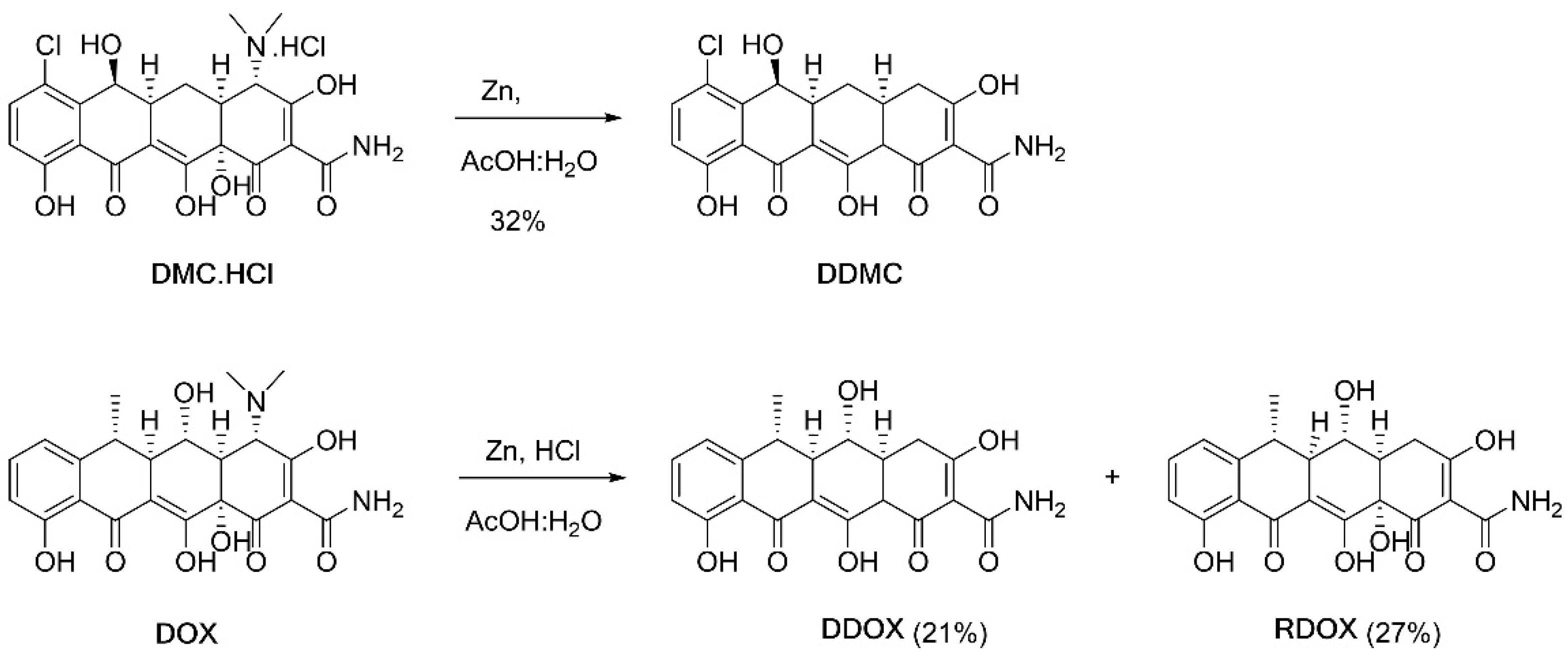
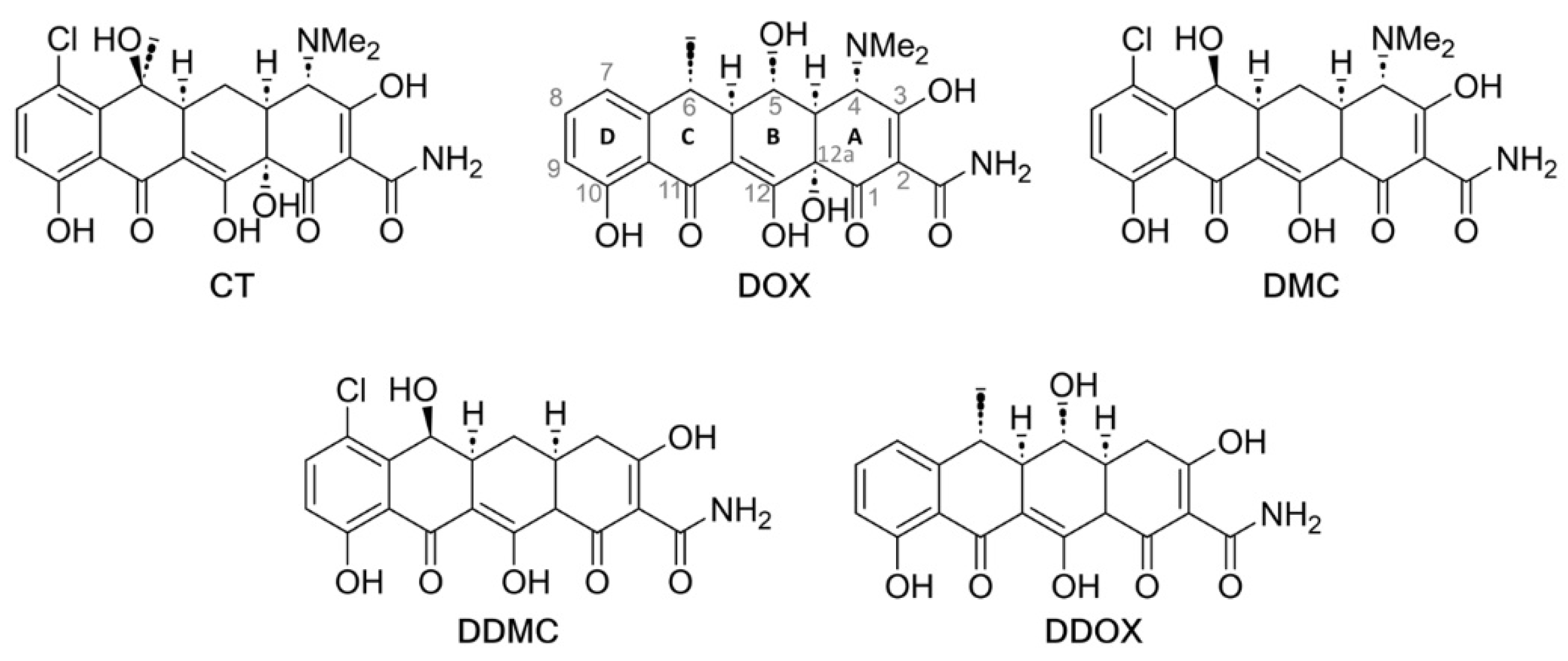
2.4. Synthesis and Spectrometric Characterization of the Two Novel TCs DDOX and DDMC
2.5. Evaluation of the Antimicrobial Activity of Test TCs
2.6. Novel Non-Antibiotic TCs Have the Capacity to Penetrate the Brain
2.7. Immunodetection of DA Neurons
2.8. Cell Counting Operations
2.9. Assessment of Concomitant Changes in ROS Levels and Mitochondrial Membrane Potential
2.10. Tritiated-DA Uptake
2.11. Statistical Analysis
3. Results
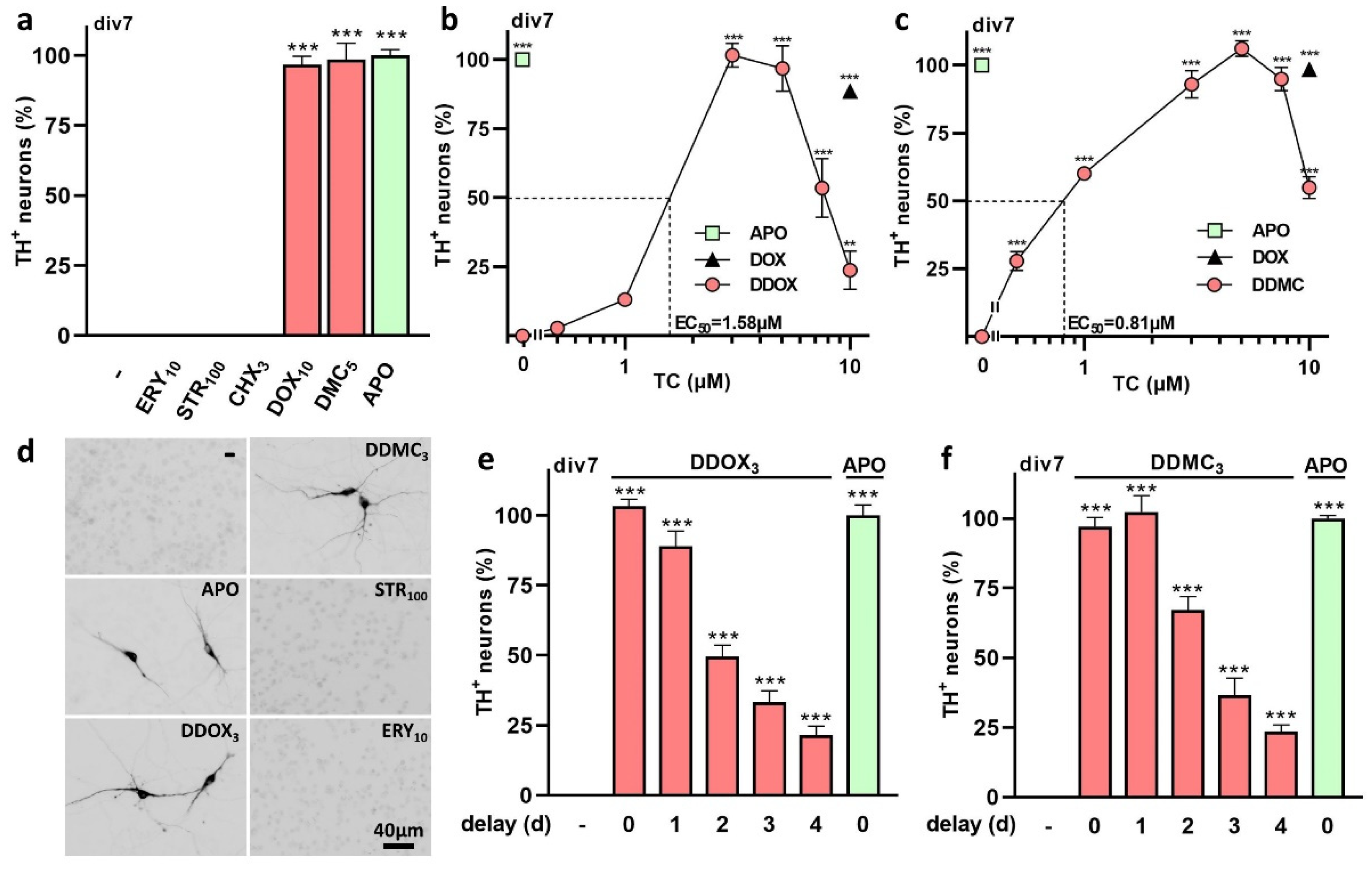
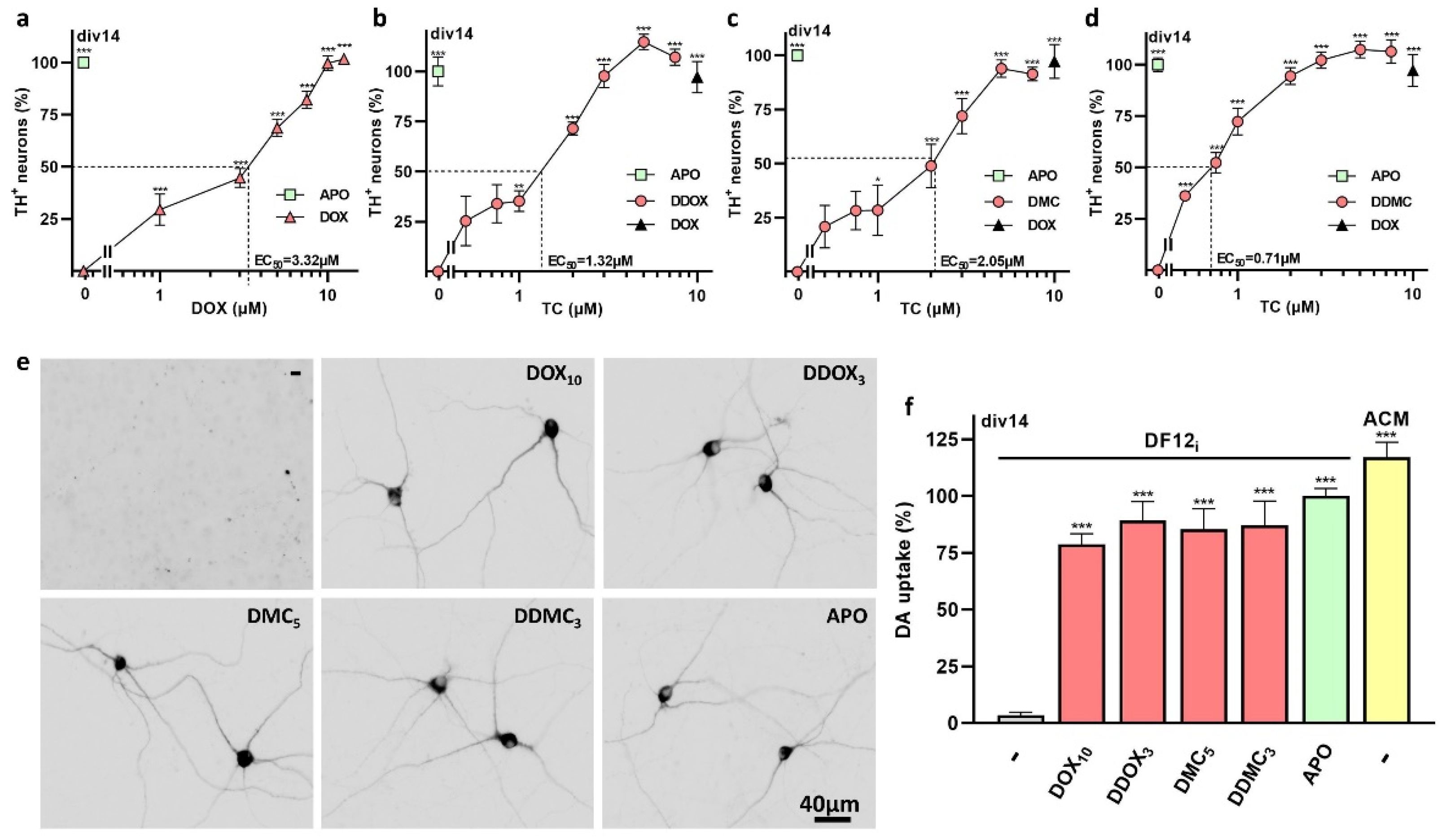
3.1. The TC Antibiotics DOX and DMC Prevent Iron-Dependent DA Cell Death in Midbrain Cultures
3.2. Protective Effects of TC Antibiotics Are Not Reproduced by Other Antimicrobial Molecules
3.3. Non-Antibiotic TC Derivatives Provide Robust Neuroprotection for DA Neurons
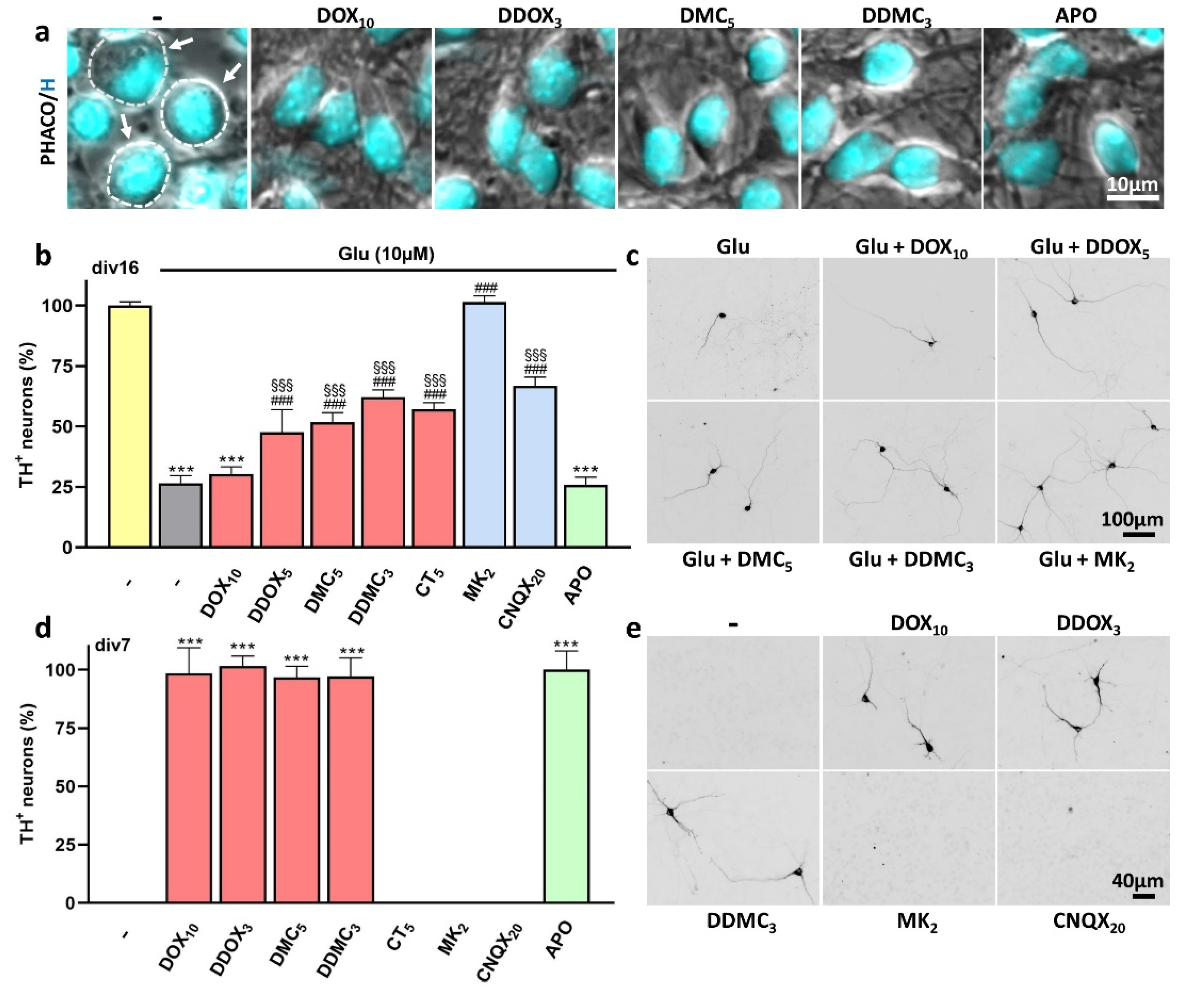
3.4. Protection by DOX and Other Protective TCs against Spontaneous DA Cell Death Does Not Occur through Inhibition of Excitotoxic Stress
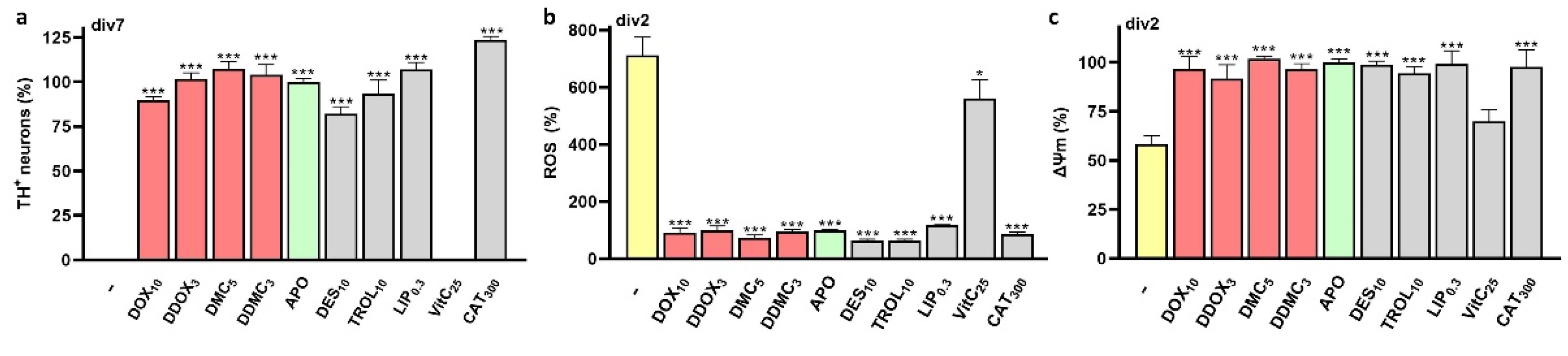
3.5. Neuroprotective TCs Prevent Intracellular ROS Production and Preserve Mitochondrial Health
3.6. TCs Prevent Iron-Catalyzed Oxidative Stress and Its Consequences
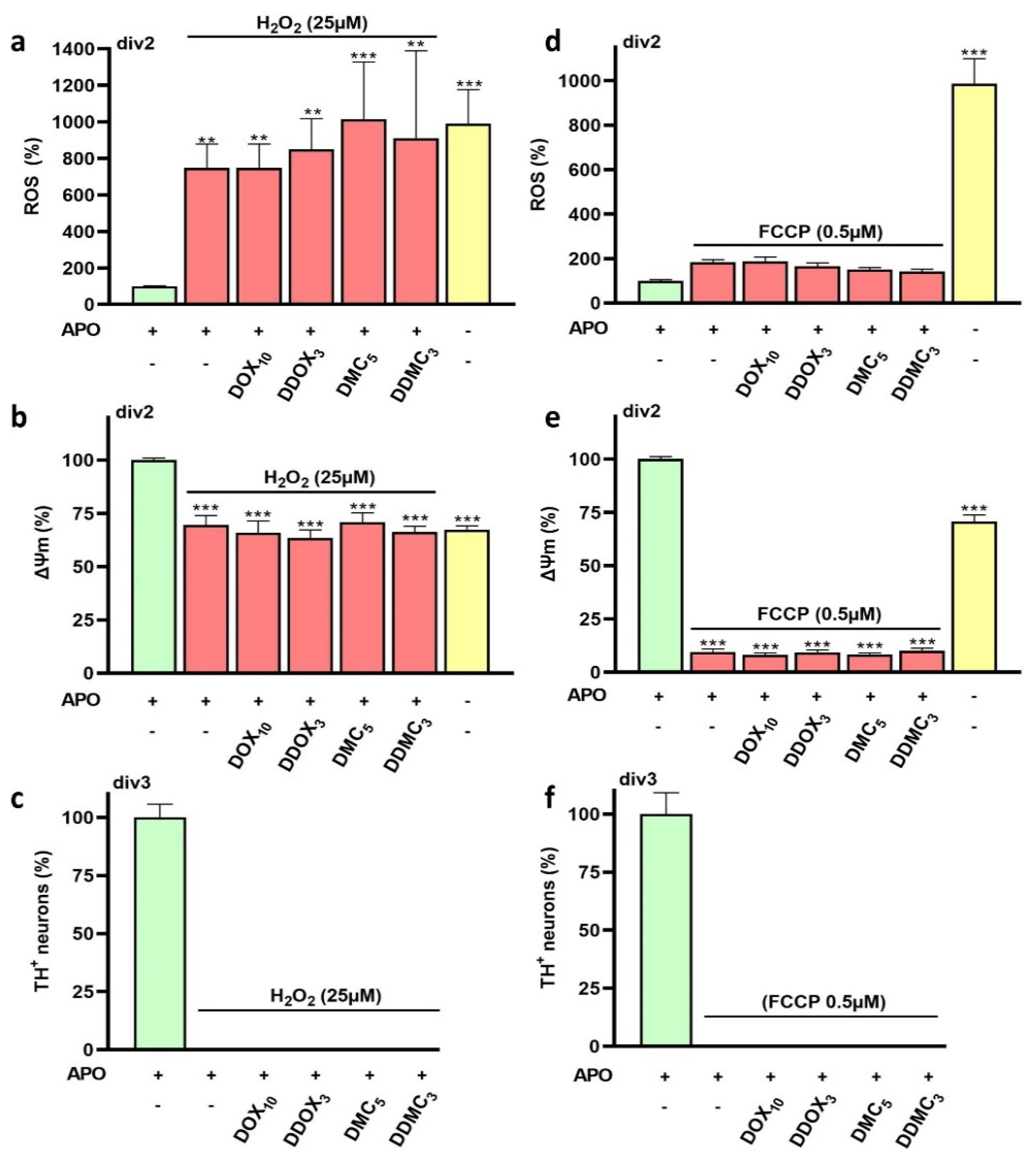
3.7. Neuroprotective TCs Are Inoperative against Oxidative Insults or Mitochondrial Deficits Generated Acutely
3.8. The Protective Effects of TCs Are Still Observable When Low-Level Iron-Mediated Oxidative Stress Takes Place in Mature Midbrain Cultures
4. Discussion
4.1. DOX and DMC, but Not CT, Exert Robust Protective Effects for DA Neurons Enduring Low-Level Intensity Iron-Mediated Insults
4.2. Protective Effects of DOX and DMC Are Not Related to Their Antibiotic Activity
4.3. Non-Antibiotic TC Derivatives Are Strongly Neuroprotective for DA Neurons
4.4. A Delayed Treatment with TCs Provides Protection as Long as Neurodegeneration Is Ongoing
4.5. TCs Do Not Operate by Preventing Glutamate-Induced Excitotoxicity
4.6. Nature of the Cell Death Process Prevented by TCs
4.7. Mechanisms Contributing to ROS Inhibition by TCs
4.8. Neuroprotective TCs also Ensure Mitochondrial Health
4.9. Neuroprotective TCs Are Ineffective against Acute Oxidative or Mitochondrial Insults
4.10. The Rescuing Effects of TCs Are Still Observable in Mature Midbrain Cultures
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease. Progress Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanudjojo, B.; Shaikh, S.S.; Fenyi, A.; Bousset, L.; Agarwal, D.; Marsh, J.; Zois, C.; Heman-Ackah, S.; Fischer, R.; Sims, D.; et al. Phenotypic Manifestation of α-Synuclein Strains Derived from Parkinson’s Disease and Multiple System Atrophy in Human Dopaminergic Neurons. Nat. Commun. 2021, 12, 3817. [Google Scholar] [CrossRef] [PubMed]
- Corti, O.; Lesage, S.; Brice, A. What Genetics Tells Us About the Causes and Mechanisms of Parkinson’s Disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s Disease: A Target for Neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Devos, D.; Hirsch, E.; Wyse, R. Seven Solutions for Neuroprotection in Parkinson’s Disease. Mov. Disord. 2021, 36, 306–316. [Google Scholar] [CrossRef]
- Valentín, S.; Morales, A.; Sánchez, J.L.; Rivera, A. Safety and Efficacy of Doxycycline in the Treatment of Rosacea. Clin. Cosmet. Investig. Derm. 2009, 2, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Lazzarini, M.; Martin, S.; Mitkovski, M.; Vozari, R.R.; Stühmer, W.; Bel, E.D. Doxycycline Restrains Glia and Confers Neuroprotection in a 6-OHDA Parkinson Model: Doxycycline in a 6-OHDA Parkinson Model. Glia 2013, 61, 1084–1100. [Google Scholar] [CrossRef]
- González-Lizárraga, F.; Socías, S.B.; Ávila, C.L.; Torres-Bugeau, C.M.; Barbosa, L.R.S.; Binolfi, A.; Sepúlveda-Díaz, J.E.; Del-Bel, E.; Fernandez, C.O.; Papy-Garcia, D.; et al. Repurposing Doxycycline for Synucleinopathies: Remodelling of α-Synuclein Oligomers towards Non-Toxic Parallel Beta-Sheet Structured Species. Sci. Rep. 2017, 7, 41755. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Meijide, A.; Parrales, V.; Vasili, E.; González-Lizárraga, F.; König, A.; Lázaro, D.F.; Lannuzel, A.; Haik, S.; Del Bel, E.; Chehín, R.; et al. Doxycycline Inhibits α-Synuclein-Associated Pathologies in Vitro and in Vivo. Neurobiol. Dis. 2021, 151, 105256. [Google Scholar] [CrossRef]
- La Vitola, P.; Artioli, L.; Cerovic, M.; Poletto, C.; Dacomo, L.; Leva, S.; Balducci, C.; Forloni, G. Repositioning Doxycycline for Treating Synucleinopathies: Evidence from a Pre-Clinical Mouse Model. Park. Relat. Disord. 2023, 106, 105229. [Google Scholar] [CrossRef]
- Ferreira Junior, N.C.; dos Santos Pereira, M.; Francis, N.; Ramirez, P.; Martorell, P.; González-Lizarraga, F.; Figadère, B.; Chehin, R.; Del Bel, E.; Raisman-Vozari, R.; et al. The Chemically-Modified Tetracycline COL-3 and Its Parent Compound Doxycycline Prevent Microglial Inflammatory Responses by Reducing Glucose-Mediated Oxidative Stress. Cells 2021, 10, 2163. [Google Scholar] [CrossRef]
- Rousseau, E.; Michel, P.P.; Hirsch, E.C. The Iron-Binding Protein Lactoferrin Protects Vulnerable Dopamine Neurons from Degeneration by Preserving Mitochondrial Calcium Homeostasis. Mol. Pharm. 2013, 84, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Riederer, P.; Monoranu, C.; Strobel, S.; Iordache, T.; Sian-Hülsmann, J. Iron as the concert master in the pathogenic orchestra playing in sporadic Parkinson’s disease. J. Neural. Transm. 2021, 128, 1577–1598. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Brandel, J.-P.; Galle, P.; Javoy-Agid, F.; Agid, Y. Iron and Aluminum Increase in the Substantia Nigra of Patients with Parkinson’s Disease: An X-Ray Microanalysis. J. Neurochem. 1991, 56, 446–451. [Google Scholar] [CrossRef]
- Michel, P.P.; Vyas, S.; Agid, Y. Toxic Effects of Iron for Cultured Mesencephalic Dopaminergic Neurons Derived from Rat Embryonic Brains. J. Neurochem. 1992, 59, 118–127. [Google Scholar] [CrossRef]
- Tourville, A.; Akbar, D.; Corti, O.; Prehn, J.H.M.; Melki, R.; Hunot, S.; Michel, P.P. Modelling α-Synuclein Aggregation and Neurodegeneration with Fibril Seeds in Primary Cultures of Mouse Dopaminergic Neurons. Cells 2022, 11, 1640. [Google Scholar] [CrossRef]
- Michel, P.P.; Ruberg, M.; Agid, Y. Rescue of Mesencephalic Dopamine Neurons by Anticancer Drug Cytosine Arabinoside. J. Neurochem. 1997, 69, 1499–1507. [Google Scholar] [CrossRef]
- dos Santos Pereira, M.; Abreu, G.H.D.; Rocca, J.; Hamadat, S.; Raisman-Vozari, R.; Michel, P.P.; Del Bel, E. Contributive Role of TNF-α to L-DOPA-Induced Dyskinesia in a Unilateral 6-OHDA Lesion Model of Parkinson’s Disease. Front. Pharmacol. 2021, 11, 617085. [Google Scholar] [CrossRef] [PubMed]
- Tomas-Grau, R.; González-Lizárraga, F.; Ploper, D.; Avila, C.L.; Socías, S.B.; Besnault, P.; Tourville, A.; Mella, R.M.; Villacé, P.; Salado, C.; et al. Neuroprotective Effects of a Novel Demeclocycline Derivative Lacking Antibiotic Activity: From a Hit to a Promising Lead Compound. Cells 2022, 11, 2759. [Google Scholar] [CrossRef] [PubMed]
- Mawabo, I.K.; Noumedem, J.A.K.; Kuiate, J.R.; Kuete, V. Tetracycline Improved the Efficiency of Other Antimicrobials against Gram-Negative Multidrug-Resistant Bacteria. J. Infect. Public Health 2015, 8, 226–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SFM Antibiogram Committee. Comité de l’Antibiogramme de La Société Française de Microbiologie Report 2003. Int. J. Antimicrob. Agents 2003, 21, 364–391. [Google Scholar] [CrossRef]
- Joshi, D.C.; Bakowska, J.C. Determination of Mitochondrial Membrane Potential and Reactive Oxygen Species in Live Rat Cortical Neurons. JoVE 2011, 51, e2704. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- EFSA Panel on Additives and Products or Substances used in Animal Feed (FEEDAP); Guidance on the assessment of bacterial susceptibility to antimicrobials of human and veterinary importance. EFSA J. 2012, 10, 2740. [CrossRef]
- Schneider-Poetsch, T.; Ju, J.; Eyler, D.E.; Dang, Y.; Bhat, S.; Merrick, W.C.; Green, R.; Shen, B.; Liu, J.O. Inhibition of Eukaryotic Translation Elongation by Cycloheximide and Lactimidomycin. Nat. Chem. Biol. 2010, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Rungta, R.L.; Choi, H.B.; Tyson, J.R.; Malik, A.; Dissing-Olesen, L.; Lin, P.J.C.; Cain, S.M.; Cullis, P.R.; Snutch, T.P.; MacVicar, B.A. The Cellular Mechanisms of Neuronal Swelling Underlying Cytotoxic Edema. Cell 2015, 161, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Lavaur, J.; Le Nogue, D.; Lemaire, M.; Pype, J.; Farjot, G.; Hirsch, E.C.; Michel, P.P. The Noble Gas Xenon Provides Protection and Trophic Stimulation to Midbrain Dopamine Neurons. J. Neurochem. 2017, 142, 14–28. [Google Scholar] [CrossRef] [Green Version]
- Douhou, A.; Troadec, J.-D.; Ruberg, M.; Raisman-Vozari, R.; Michel, P.P. Survival Promotion of Mesencephalic Dopaminergic Neurons by Depolarizing Concentrations of K+ Requires Concurrent Inactivation of NMDA or AMPA/Kainate Receptors: Rescue of Dopamine Neurons by Depolarization. J. Neurochem. 2001, 78, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [Green Version]
- Perry, E.A.; Bennett, C.F.; Luo, C.; Balsa, E.; Jedrychowski, M.; O’Malley, K.E.; Latorre-Muro, P.; Ladley, R.P.; Reda, K.; Wright, P.M.; et al. Tetracyclines Promote Survival and Fitness in Mitochondrial Disease Models. Nat. Metab. 2021, 3, 33–42. [Google Scholar] [CrossRef]
- Santa-Cecília, F.V.; Socias, B.; Ouidja, M.O.; Sepulveda-Diaz, J.E.; Acuña, L.; Silva, R.L.; Michel, P.P.; Del-Bel, E.; Cunha, T.M.; Raisman-Vozari, R. Doxycycline Suppresses Microglial Activation by Inhibiting the P38 MAPK and NF-KB Signaling Pathways. Neurotox. Res. 2016, 29, 447–459. [Google Scholar] [CrossRef]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef]
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative Stress in Parkinson’s Disease: A Mechanism of Pathogenic and Therapeutic Significance. Ann. N. Y. Acad. Sci. 2008, 1147, 93–104. [Google Scholar] [CrossRef]
- Guerreiro, S.; Ponceau, A.; Toulorge, D.; Martin, E.; Alvarez-Fischer, D.; Hirsch, E.C.; Michel, P.P. Protection of midbrain dopaminergic neurons by the end-product of purine metabolism uric acid: Potentiation by low-level depolarization. J. Neurochem. 2009, 109, 1118–1128. [Google Scholar] [CrossRef]
- Braun, A.R.; Liao, E.E.; Horvath, M.; Kalra, P.; Acosta, K.; Young, M.C.; Kochen, N.N.; Lo, C.H.; Brown, R.; Evans, M.D.; et al. Potent Inhibitors of Toxic Alpha-Synuclein Identified via Cellular Time-Resolved FRET Biosensors. NPJ Park. Dis. 2021, 7, 52. [Google Scholar] [CrossRef]
- Epe, B.; Woolley, P. The Binding of 6-Demethylchlortetracycline to 70S, 50S and 30S Ribosomal Particles: A Quantitative Study by Fluorescence Anisotropy. EMBO J. 1984, 3, 121–126. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. MicroBiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Atkinson, G.C.; Thakor, N.S.; Allas, Ü.; Lu, C.; Chan, K.-Y.; Tenson, T.; Schulten, K.; Wilson, K.S.; Hauryliuk, V.; et al. Mechanism of Tetracycline Resistance by Ribosomal Protection Protein Tet(O). Nat. Commun. 2013, 4, 1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortison, J.D.; Schenone, M.; Myers, J.A.; Zhang, Z.; Chen, L.; Ciarlo, C.; Comer, E.; Natchiar, S.K.; Carr, S.A.; Klaholz, B.P.; et al. Tetracyclines Modify Translation by Targeting Key Human rRNA Substructures. Cell Chem. Biol. 2018, 25, 1506–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solis, G.M.; Kardakaris, R.; Valentine, E.R.; Bar-Peled, L.; Chen, A.L.; Blewett, M.M.; McCormick, M.A.; Williamson, J.R.; Kennedy, B.; Cravatt, B.F.; et al. Translation Attenuation by Minocycline Enhances Longevity and Proteostasis in Old Post-Stress-Responsive Organisms. eLife 2018, 7, e40314. [Google Scholar] [CrossRef] [PubMed]
- de León, A.; Gibon, J.; Barker, P.A. NGF-Dependent and BDNF-Dependent DRG Sensory Neurons Deploy Distinct Degenerative Signaling Mechanisms. eNeuro 2021, 8, ENEURO.0277-20.2020. [Google Scholar] [CrossRef]
- Anthony, W.E.; Wang, B.; Sukhum, K.V.; D’Souza, A.W.; Hink, T.; Cass, C.; Seiler, S.; Reske, K.A.; Coon, C.; Dubberke, E.R.; et al. Acute and persistent effects of commonly used antibiotics on the gut microbiome and resistome in healthy adults. Cell Rep. 2022, 39, 110649. [Google Scholar] [CrossRef]
- Golub, L.M.; Ramamurthy, N.S.; McNamara, T.F.; Greenwald, R.A.; Rifkin, B.R. Tetracyclines Inhibit Connective Tissue Breakdown: New Therapeutic Implications for an Old Family of Drugs. Crit. Rev. Oral Biol. Med. 1991, 2, 297–321. [Google Scholar] [CrossRef]
- Bezard, E.; Dovero, S.; Prunier, C.; Ravenscroft, P.; Chalon, S.; Guilloteau, D.; Crossman, A.R.; Bioulac, B.; Brotchie, J.M.; Gross, C.E. Relationship between the Appearance of Symptoms and the Level of Nigrostriatal Degeneration in a Progressive 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Lesioned Macaque Model of Parkinson’s Disease. J. Neurosci. 2001, 21, 6853–6861. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-K.; Lee, M.J.; Yoo, H.S.; Lee, J.H.; Ryu, Y.H.; Lyoo, C.H. Temporal Trajectory Model for Dopaminergic Input to the Striatal Subregions in Parkinson’s Disease. Park. Relat. Disord. 2022, 103, 42–49. [Google Scholar] [CrossRef]
- Brennan-Minnella, A.M.; Shen, Y.; El-Benna, J.; Swanson, R.A. Phosphoinositide 3-Kinase Couples NMDA Receptors to Superoxide Release in Excitotoxic Neuronal Death. Cell Death Dis. 2013, 4, e580. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.X.; Lertvorachon, J.; Hou, S.T.; Konishi, Y.; Webster, J.; Mealing, G.; Brunette, E.; Tauskela, J.; Preston, E. Chlortetracycline and Demeclocycline Inhibit Calpains and Protect Mouse Neurons against Glutamate Toxicity and Cerebral Ischemia. J. Biol. Chem. 2005, 280, 33811–33818. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Yang, Y.; Chen, W.; Du, N.; Du, Y.; Gu, H.; Liu, Q. Minocycline, but Not Doxycycline Attenuates NMDA-Induced [Ca2+]i and Excitotoxicity. NeuroReport 2021, 32, 38–43. [Google Scholar] [CrossRef]
- Tang, Z.; Zhao, P.; Wang, H.; Liu, Y.; Bu, W. Biomedicine Meets Fenton Chemistry. Chem. Rev. 2021, 121, 1981–2019. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Mahoney-Sánchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.C. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog. NeuroBiol. 2021, 196, 101890. [Google Scholar] [CrossRef]
- Faller, B.; Nick, H. Kinetics and Mechanism of Iron(III) Removal from Citrate by Desferrioxamine B and 3-Hydroxy-1,2-Dimethyl-4-Pyridone. J. Am. Chem. Soc. 1994, 116, 3860–3865. [Google Scholar] [CrossRef]
- Schmaier, A.H. Transferrin: A blood coagulation modifier. Cell Res. 2020, 30, 101–102. [Google Scholar] [CrossRef]
- Beckman, J.S.; Minor, R.L.; White, C.W.; Repine, J.E.; Rosen, G.M.; Freeman, B.A. Superoxide Dismutase and Catalase Conjugated to Polyethylene Glycol Increases Endothelial Enzyme Activity and Oxidant Resistance. J. Biol. Chem. 1988, 263, 6884–6892. [Google Scholar] [CrossRef]
- Pandithavidana, D.R.; Jayawardana, S.B. Comparative Study of Antioxidant Potential of Selected Dietary Vitamins; Computational Insights. Molecules 2019, 24, 1646. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Sobreira, C.; Davidson, M.; King, M.P.; Miranda, A.F. Dihydrorhodamine 123 Identifies Impaired Mitochondrial Respiratory Chain Function in Cultured Cells Harboring Mitochondrial DNA Mutations. J. HistoChem. Cytochem. 1996, 44, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Grenier, D.; Huot, M.-P.; Mayrand, D. Iron-Chelating Activity of Tetracyclines and Its Impact on the Susceptibility of Actinobacillus Actinomycetemcomitans to These Antibiotics. Antimicrob. Agents Chemother. 2000, 44, 763–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markulin, I.; Matasin, M.; Turk, V.E.; Salković-Petrisic, M. Challenges of Repurposing Tetracyclines for the Treatment of Alzheimer’s and Parkinson’s Disease. J. Neural. Transm. 2022, 129, 773–804. [Google Scholar] [CrossRef] [PubMed]
- Faure, M.; Cilibrizzi, A.; Abbate, V.; Bruce, K.; Hider, R. Effect of Iron Chelation on Anti-Pseudomonal Activity of Doxycycline. Int. J. Antimicrob. Agents 2021, 58, 106438. [Google Scholar] [CrossRef] [PubMed]
- Clemens, D.; Duryee, M.; Sarmiento, C.; Chiou, A.; McGowan, J.; Hunter, C.; Schlichte, S.; Tian, J.; Klassen, L.; O’Dell, J.; et al. Novel Antioxidant Properties of Doxycycline. IJMS 2018, 19, 4078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jové, M.; Mota-Martorell, N.; Pradas, I.; Martín-Gari, M.; Ayala, V.; Pamplona, R. The Advanced Lipoxidation End-Product Malondialdehyde-Lysine in Aging and Longevity. Antioxidants 2020, 9, 1132. [Google Scholar] [CrossRef]
- Long, J.; Liu, C.; Sun, L.; Gao, H.; Liu, J. Neuronal mitochondrial toxicity of malondialdehyde: Inhibitory effects on respiratory function and enzyme activities in rat brain mitochondria. Neurochem. Res. 2009, 34, 786–794. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial Membrane Potential Probes and the Proton Gradient: A Practical Usage Guide. BioTechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolaños, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on Experimental Methods to Assess Mitochondrial Dysfunction in Cellular Models of Neurodegenerative Diseases. Cell Death Differ. 2018, 25, 542–572. [Google Scholar] [CrossRef] [Green Version]
- Mottis, A.; Li, T.Y.; El Alam, G.; Rapin, A.; Katsyuba, E.; Liaskos, D.; D’Amico, D.; Harris, N.L.; Grier, M.C.; Mouchiroud, L.; et al. Tetracycline-Induced Mitohormesis Mediates Disease Tolerance against Influenza. J. Clin. Investig. 2022, 132, e151540. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free. Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Uncoupling is without an effect on the production of reactive oxygen species by in situ synaptic mitochondria. J. Neurochem. 2007, 103, 1864–1871. [Google Scholar] [CrossRef]
- Pozzi, D.; Ban, J.; Iseppon, F.; Torre, V. An Improved Method for Growing Neurons: Comparison with Standard Protocols. J. NeuroSci. Methods 2017, 280, 1–10. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCs | P. aeruginosa PAO1 | E. coli ATCC 25922 | S. aureus ATCC 25923 |
|---|---|---|---|
| MIC (µM) | |||
| DDMC | 200 | 200 | 12.5 |
| DDOX | >200 | >200 | >200 |
| DMC | 6.25 | 3.125 | 0.4 |
| CT | 6.25 | 6.25 | 1.6 |
| DOX | 12.5 | 3.125 | 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tourville, A.; Viguier, S.; González-Lizárraga, F.; Tomas-Grau, R.H.; Ramirez, P.; Brunel, J.-M.; Dos Santos Pereira, M.; Del-Bel, E.; Chehin, R.; Ferrié, L.; et al. Rescue of Dopamine Neurons from Iron-Dependent Ferroptosis by Doxycycline and Demeclocycline and Their Non-Antibiotic Derivatives. Antioxidants 2023, 12, 575. https://doi.org/10.3390/antiox12030575
Tourville A, Viguier S, González-Lizárraga F, Tomas-Grau RH, Ramirez P, Brunel J-M, Dos Santos Pereira M, Del-Bel E, Chehin R, Ferrié L, et al. Rescue of Dopamine Neurons from Iron-Dependent Ferroptosis by Doxycycline and Demeclocycline and Their Non-Antibiotic Derivatives. Antioxidants. 2023; 12(3):575. https://doi.org/10.3390/antiox12030575
Chicago/Turabian StyleTourville, Aurore, Sarah Viguier, Florencia González-Lizárraga, Rodrigo Hernán Tomas-Grau, Paola Ramirez, Jean-Michel Brunel, Mauricio Dos Santos Pereira, Elaine Del-Bel, Rosana Chehin, Laurent Ferrié, and et al. 2023. "Rescue of Dopamine Neurons from Iron-Dependent Ferroptosis by Doxycycline and Demeclocycline and Their Non-Antibiotic Derivatives" Antioxidants 12, no. 3: 575. https://doi.org/10.3390/antiox12030575