
Effects of Sequential Enzymolysis and Glycosylation on the Structural Properties and Antioxidant Activity of Soybean Protein Isolate


Abstract
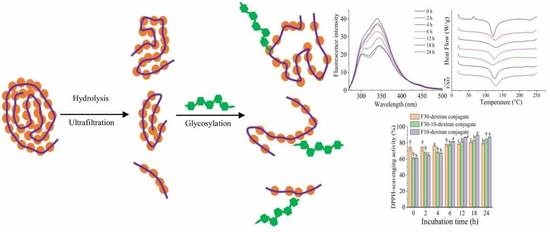
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Preparation of SPI Hydrolysates (SPIH)
2.3. Ultrafiltration of SPIH
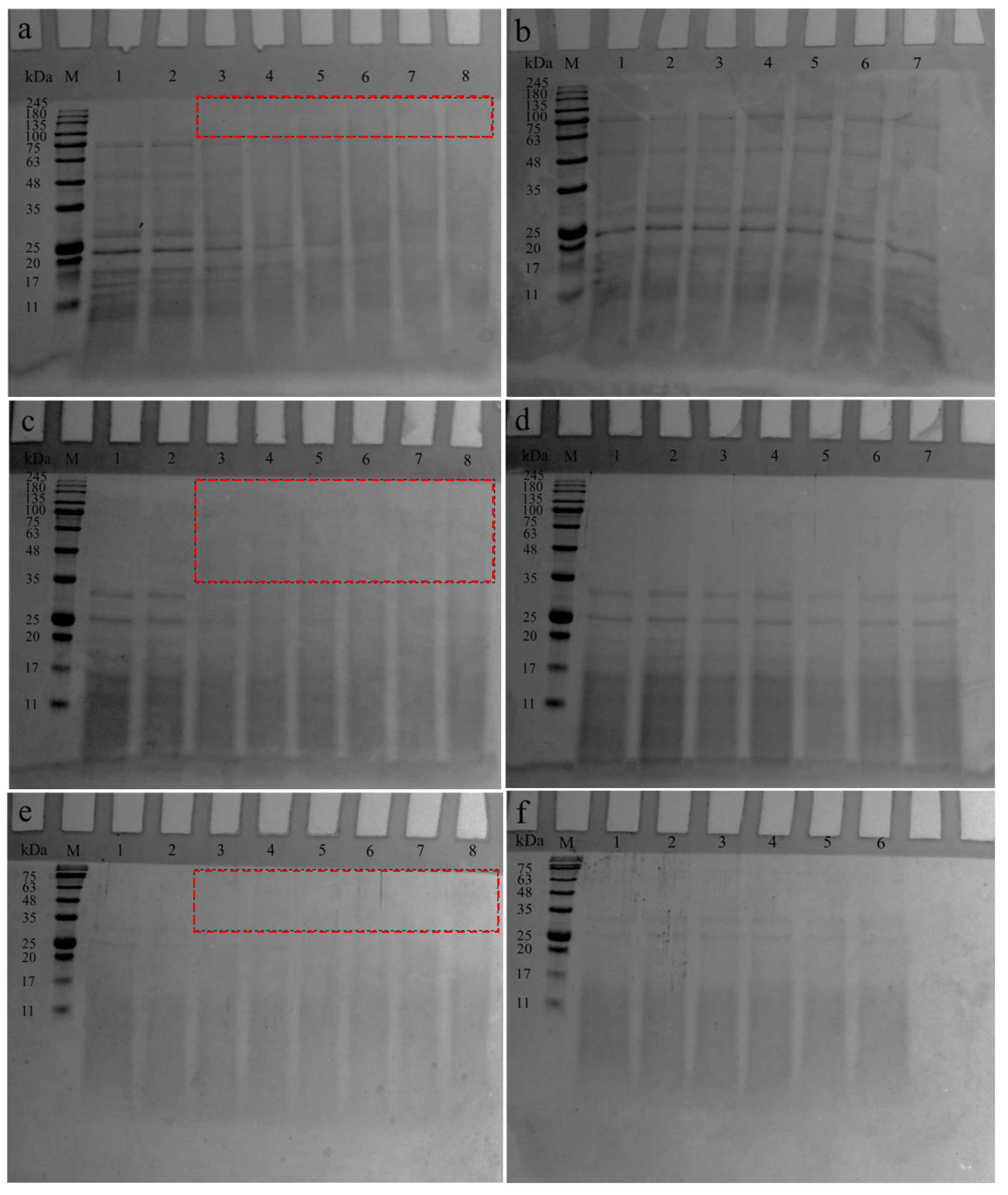
2.4. Determination of MW Distribution and Protein Composition of SPIH Fractions
2.5. Preparation of SPIH-Dextran Conjugates
2.6. Confirmation of F30, F30-10, or F10-Dextran Conjugates
2.7. Structural Analysis of F30, F30-10, or F10-Dextran Conjugates
2.7.1. Ultraviolet (UV)-Visible Spectroscopy
2.7.2. Intrinsic Fluorescence Emission Spectroscopy
2.7.3. Fourier Transform Infrared (FTIR) Spectroscopy
2.7.4. Differential Scanning Calorimetry (DSC)
2.8. Measurements of Antioxidant Activity
2.8.1. Determination of DPPH Radical Scavenging Activity
2.8.2. Determination of •OH Scavenging Activity
2.8.3. Determination of Ferrous Reducing Power
2.9. Statistical Analysis
3. Results and Discussion
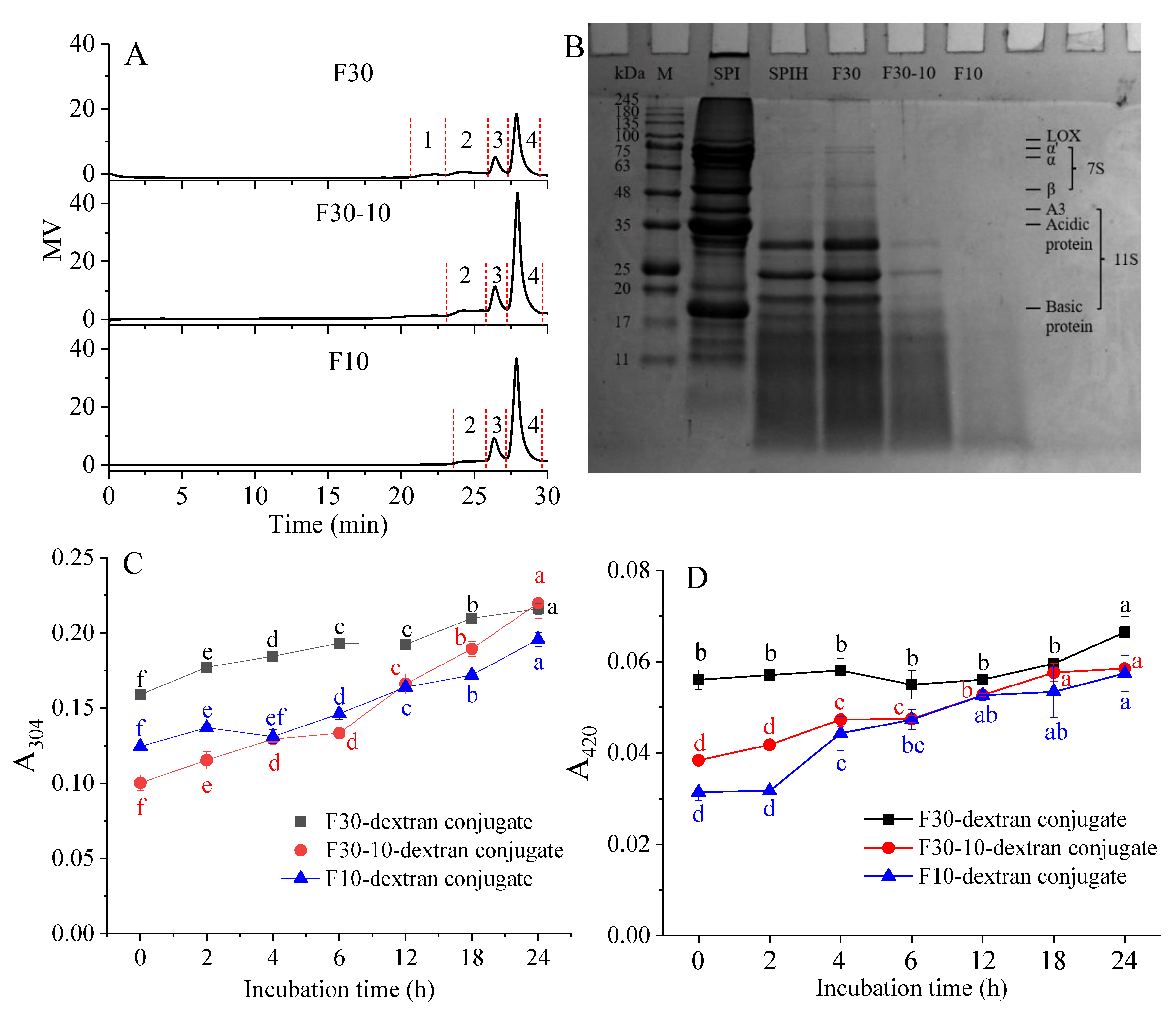
3.1. MW Distribution of F30, F30-10, and F10
3.2. Characterization of F30, F30-10, or F10-Dextran Conjugates
3.3. Structural Properties of F30, F30-10, or F10-Dextran Conjugates
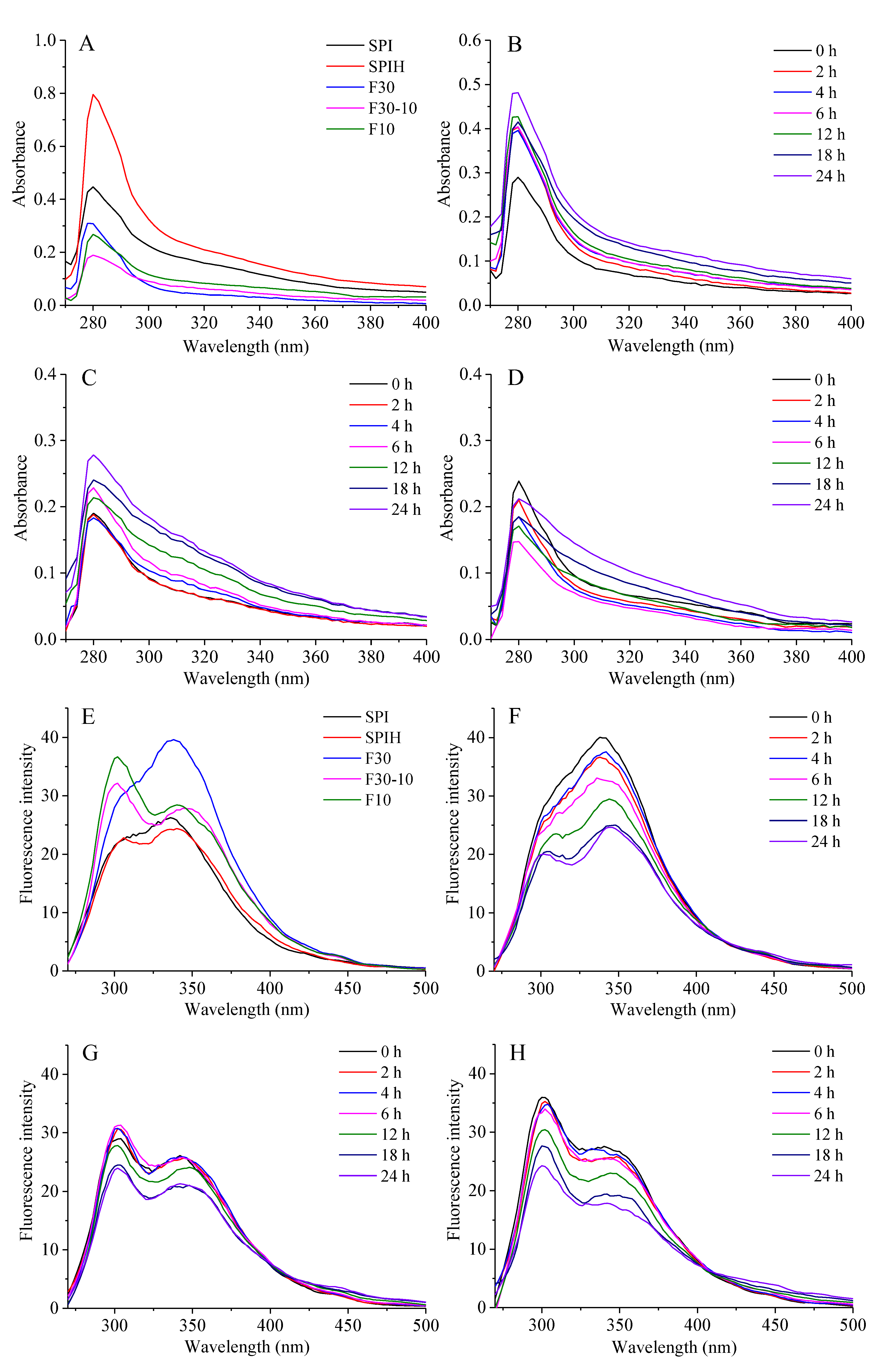
3.3.1. UV Absorption Intensity
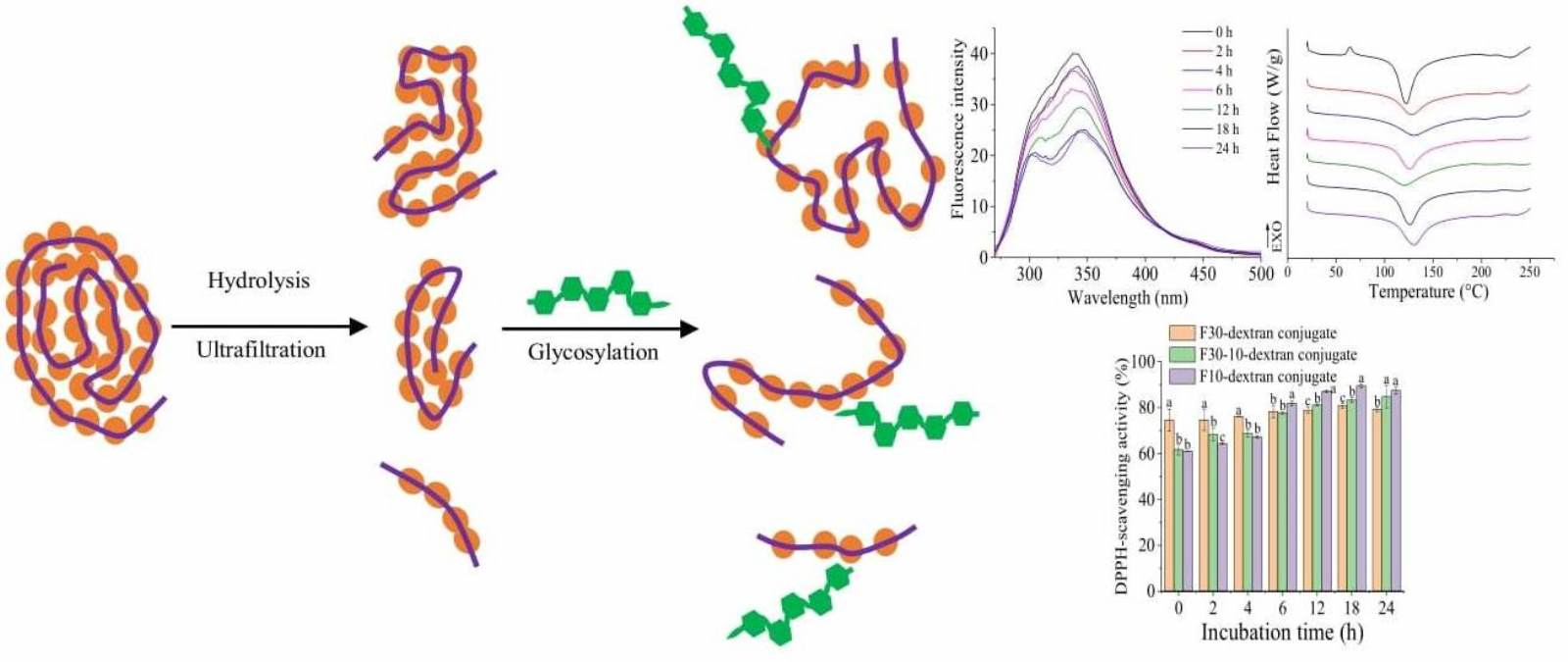
3.3.2. Tertiary Structure
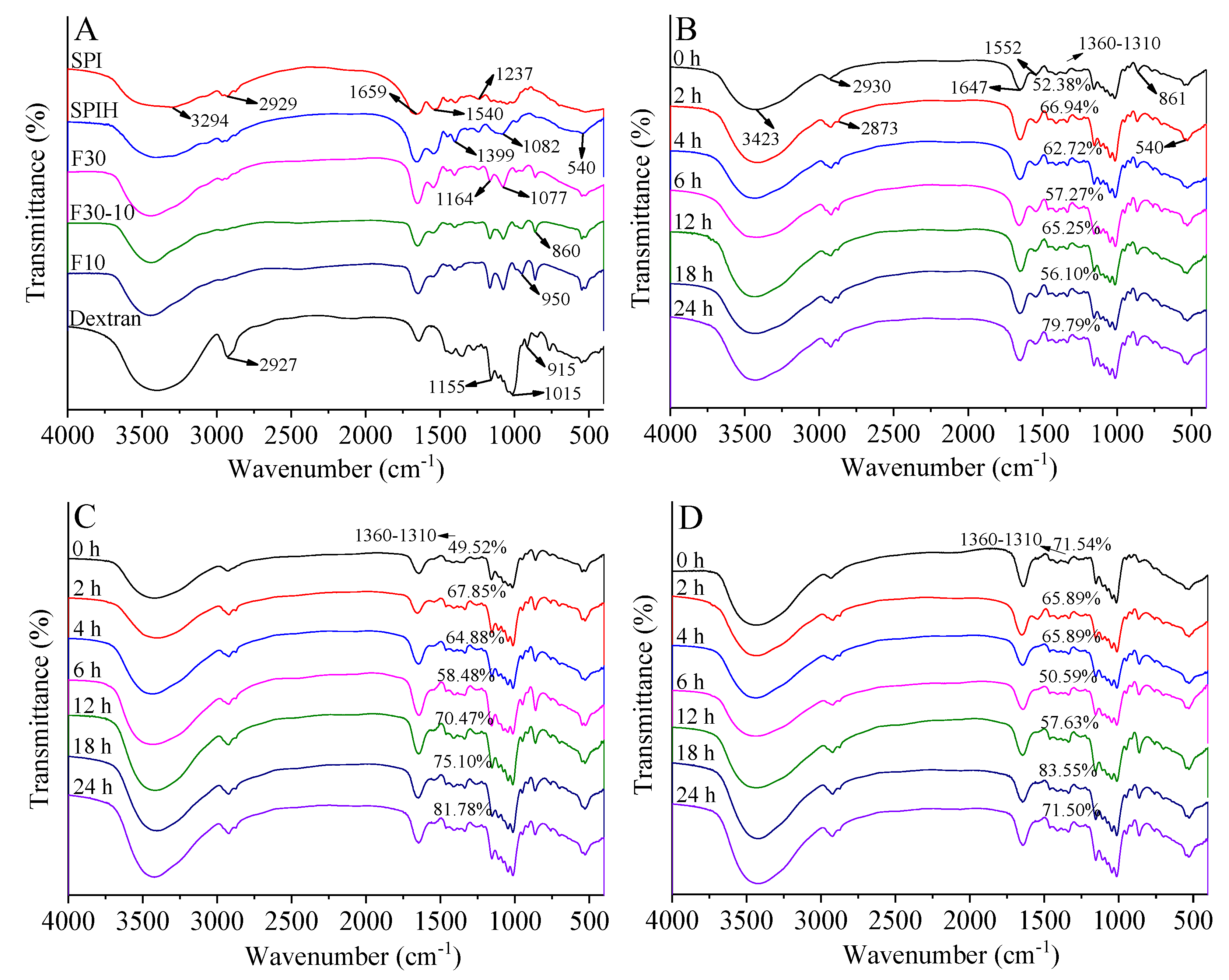
3.3.3. FTIR Spectroscopy
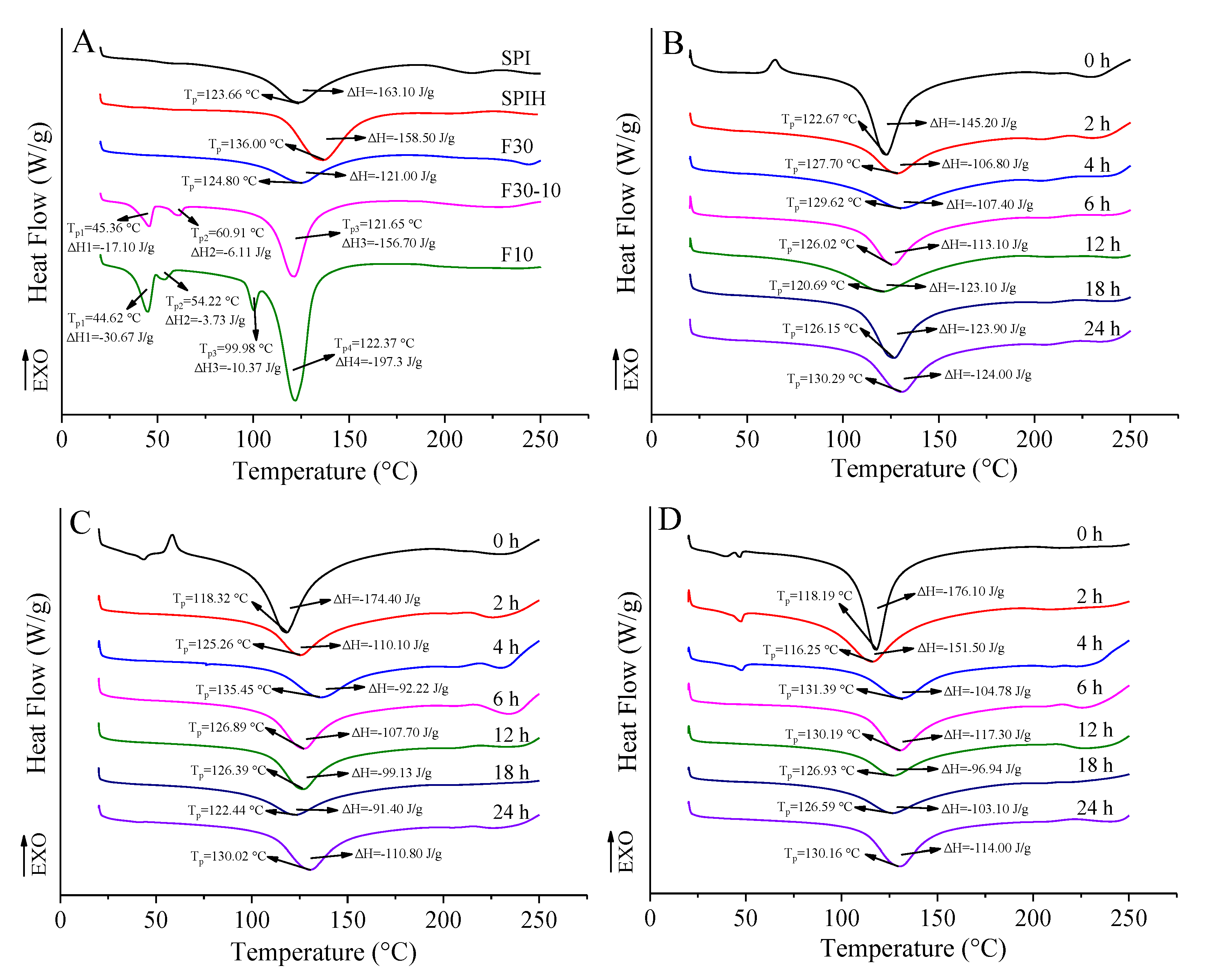
3.3.4. Thermal Stability
3.4. Antioxidant Activities
3.4.1. DPPH Radical Scavenging Activity
3.4.2. •OH Scavenging Activity
3.4.3. Ferrous Reducing Power
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Day, L.; Cakebread, J.A.; Loveday, S.M. Food proteins from animals and plants: Differences in the nutritional and functional properties. Trends Food Sci. Technol. 2022, 119, 428–442. [Google Scholar] [CrossRef]
- Mirmoghtadaie, L.; Shojaee Aliabadi, S.; Hosseini, S.M. Recent approaches in physical modification of protein functionality. Food Chem. 2016, 199, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.C.; Kumar, M.; Molla, R.; Rai, V. Chemical methods for modification of proteins. Org. Biomol. Chem. 2020, 18, 4669–4691. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dai, L.; Gao, Y.X. Progress in enzymatic modification of proteins. Food Sci. 2018, 39, 233–239. (In Chinese) [Google Scholar] [CrossRef]
- Li, M.; Yu, R.; Fu, R.X.; He, Y.T.; Zhao, P.P.; Jiang, Z.M.; Hou, J.C. Limited hydrolysis of glycosylated whey protein isolate ameliorates the oxidative and physical stabilities of conjugated linoleic acid oil-in-water emulsions. Food Chem. 2021, 362, 130212. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, L.; Lan, Q.Y.; Li, M.L.; Wu, D.T.; Chen, H.; Liu, Y.W.; Lin, D.R.; Qin, W.; Zhang, Z.Q.; et al. Protein glycosylation: A promising way to modify the functional properties and extend the application in food system. Crit. Rev. Food Sci. Nutr. 2019, 59, 2506–2533. [Google Scholar] [CrossRef]
- Jiang, Z.M.; Li, M.; Zhao, J.J.; Wang, X.D.; Yu, P.P.; Qayum, A.; Li, A.L.; Hou, J.C. Effects of ultrafiltration and hydrolysis on antioxidant activities of Maillard reaction products derived from whey protein isolate and galactose. LWT-Food Sci. Technol. 2019, 113, 108313. [Google Scholar] [CrossRef]
- Song, C.L.; Ren, J.; Chen, J.P.; Sun, X.H.; Kopparapu, N.K.; Xue, Y.G. Effect of glycosylation and limited hydrolysis on structural and functional properties of soybean protein isolate. J. Food Meas. Charact. 2018, 12, 2946–2954. [Google Scholar] [CrossRef]
- Nooshkam, M.; Madadlou, A. Microwave-assisted isomerisation of lactose to lactulose and Maillard conjugation of lactulose and lactose with whey proteins and peptides. Food Chem. 2016, 200, 1–9. [Google Scholar] [CrossRef]
- Xiao, Q.; Woo, M.W.; Hu, J.W.; Xiong, H.; Zhao, Q. The role of heating time on the characteristics, functional properties and antioxidant activity of enzyme-hydrolyzed rice proteins-glucose Maillard reaction products. Food Biosci. 2021, 43, 101225. [Google Scholar] [CrossRef]
- Li, L.; He, H.; Wu, D.Z.; Lin, D.R.; Qin, W.; Meng, D.M.; Yang, R.; Zhang, Q. Rheological and textural properties of acid-induced soybean protein isolate gel in the presence of soybean protein isolate hydrolysates or their glycosylated products. Food Chem. 2021, 360, 129991. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ismail, B. Effect of Maillard-induced glycosylation on the nutritional quality, solubility, thermal stability and molecular configuration of whey protein. Int. Dairy J. 2012, 25, 112–122. [Google Scholar] [CrossRef]
- Zhu, D.; Damodaran, S.; Lucey, J.A. Physicochemical and emulsifying properties of whey protein isolate (WPI)-dextran conjugates produced in aqueous solution. J. Agric. Food Chem. 2010, 58, 2988–2994. [Google Scholar] [CrossRef] [PubMed]
- Creusot, N.; Gruppen, H.; van Koningsveld, G.A.; de Kruif, C.G.; Voragen, A.G.J. Peptide–peptide and protein–peptide interactions in mixtures of whey protein isolate and whey protein isolate hydrolysates. Int. Dairy J. 2006, 16, 840–849. [Google Scholar] [CrossRef]
- Lv, Y.; Guo, S.T.; Yang, B.C. Aggregation of hydrophobic soybean protein hydrolysates: Changes in molecular weight distribution during storage. LWT-Food Sci. Technol. 2009, 42, 914–917. [Google Scholar] [CrossRef]
- Li, L.; Wang, C.Z.; Li, K.X.; Qin, W.; Wu, D.T.; Hu, B.; Yang, W.Y.; Dong, H.M.; Zhang, Q. Influence of soybean protein isolate-dextran conjugates on the characteristics of glucono-δ-lactone-induced tofu. LWT-Food Sci. Technol. 2021, 139, 110588. [Google Scholar] [CrossRef]
- Ahmadifard, N.; Murueta, J.H.; Abedian-Kenari, A.; Motamedzadegan, A.; Jamali, H. Comparison the effect of three commercial enzymes for enzymatic hydrolysis of two substrates (rice bran protein concentrate and soy-been protein) with SDS-PAGE. J. Food Sci. Technol. 2016, 53, 1279–1284. [Google Scholar] [CrossRef]
- Li, Y.; Zhong, F.; Ji, W.; Yokoyama, W.; Shoemaker, C.F.; Zhu, S.; Xia, W.S. Functional properties of Maillard reaction products of rice protein hydrolysates with mono-, oligo- and polysaccharides. Food Hydrocolloid. 2013, 30, 53–60. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Tan, C.; Zhang, X.M.; Xia, S.Q.; Jia, C.S.; Eric, K.; Abbas, S.; Feng, B.; Zhong, F. Effects of maltodextrin glycosylation following limited enzymatic hydrolysis on the functional and conformational properties of soybean protein isolate. Eur. Res. Technol. 2014, 238, 957–968. [Google Scholar] [CrossRef]
- Wang, C.Z.; Li, L.; Zhang, Q.; Raheem, D.; Qin, W.; Wu, D.T.; Dong, H.M.; Vasanthan, T.; Zhang, Q. Incorporation of high-speed shearing in the fabrication of whole soybean curd: Effects on aggregation behaviors and microstructures. Food Bioprocess Technol. 2020, 13, 611–624. [Google Scholar] [CrossRef]
- Liu, L.L.; Li, Y.; Prakash, S.; Dai, X.N.; Meng, Y.Y. Enzymolysis and glycosylation synergistic modified ovalbumin: Functional and structural characteristics. Int. J. Food Prop. 2018, 21, 395–406. [Google Scholar] [CrossRef]
- de Oliveira, F.C.; Coimbra, J.S.; de Oliveira, E.B.; Zuniga, A.D.; Rojas, E.E. Food protein-polysaccharide conjugates obtained via the Maillard reaction: A review. Crit. Rev. Food Sci. Nutr. 2016, 56, 1108–1125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Zhang, A.Q.; Wang, X.B.; Xu, N.; Jiang, L.Z. The radiation assisted-Maillard reaction comprehensively improves the freeze-thaw stability of soy protein-stabilized oil-in-water emulsions. Food Hydrocolloid. 2020, 103, 105684. [Google Scholar] [CrossRef]
- Spotti, M.J.; Martinez, M.J.; Pilosof, A.M.R.; Candioti, M.; Rubiolo, A.C.; Carrara, C.R. Influence of Maillard conjugation on structural characteristics and rheological properties of whey protein/dextran systems. Food Hydrocolloid. 2014, 39, 223–230. [Google Scholar] [CrossRef]
- Zhao, C.B.; Yin, H.H.; Yan, J.N.; Niu, X.; Qi, B.K.; Liu, J.S. Structure and acid-induced gelation properties of soy protein isolate–maltodextrin glycation conjugates with ultrasonic pretreatment. Food Hydrocolloid. 2021, 112, 106278. [Google Scholar] [CrossRef]
- Wan, Y.L.; Liu, J.Y.; Guo, S.T. Effects of succinylation on the structure and thermal aggregation of soy protein isolate. Food Chem. 2018, 245, 542–550. [Google Scholar] [CrossRef]
- Su, J.F.; Huang, Z.; Yuan, X.Y.; Wang, X.Y.; Li, M. Structure and properties of carboxymethyl cellulose soy protein isolate blend edible films crosslinked by Maillard reactions. Carbohydr. Polym. 2010, 79, 145–153. [Google Scholar] [CrossRef]
- Zheng, L.; Zhao, Y.J.; Xiao, C.Q.; Sun-Waterhouse, D.X.; Zhao, M.M.; Su, G.W. Mechanism of the discrepancy in the enzymatic hydrolysis efficiency between defatted peanut flour and peanut protein isolate by Flavorzyme. Food Chem. 2015, 168, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Krilov, D.; Balarin, M.; Kosović, M.; Gamulin, O.; Brnjas-Kraljević, J. FT-IR spectroscopy of lipoproteins-A comparative study. Spectrochim Acta A Mol. Biomol. Spectrosc. 2009, 73, 701–706. [Google Scholar] [CrossRef]
- Boostani, S.; Aminlari, M.; Moosavi-Nasab, M.; Niakosari, M.; Mesbahi, G. Fabrication and characterisation of soy protein isolate-grafted dextran biopolymer: A novel ingredient in spray-dried soy beverage formulation. Int. J. Biol. Macromol. 2017, 102, 297–307. [Google Scholar] [CrossRef]
- Hu, Q.H.; Wu, Y.L.; Zhong, L.; Ma, N.; Zhao, L.Y.; Ma, G.X.; Cheng, N.H.; Nakata, P.A.; Xu, J. In vitro digestion and cellular antioxidant activity of β-carotene-loaded emulsion stabilized by soy protein isolate-Pleurotus eryngii polysaccharide conjugates. Food Hydrocolloid. 2021, 112, 106340. [Google Scholar] [CrossRef]
- Zhao, C.B.; Zhang, H.; Xu, X.Y.; Zheng, M.Z.; Cao, Y.; Xiu, L.; Cai, D.; Liu, J.S. Effect of dextran with different molecular masses on structure and functional properties of zein glycosylation products. Food Sci. 2018, 39, 68–73. (In Chinese) [Google Scholar] [CrossRef]
- Wang, K.Q.; Luo, S.Z.; Cai, J.; Sun, Q.Q.; Zhao, Y.Y.; Zhong, X.Y.; Jiang, S.T.; Zheng, Z. Effects of partial hydrolysis and subsequent cross-linking on wheat gluten physicochemical properties and structure. Food Chem. 2016, 197, 168–174. [Google Scholar] [CrossRef]
- Yin, S.W.; Tang, C.H.; Cao, J.S.; Hu, E.K.; Wen, Q.B.; Yang, X.Q. Effects of limited enzymatic hydrolysis with trypsin on the functional properties of hemp (Cannabis sativa L.) protein isolate. Food Chem. 2008, 106, 1004–1013. [Google Scholar] [CrossRef]
- Liu, J.H.; Luo, Y.H.; Gu, S.Q.; Xu, Q.H.; Zhang, J.J.; Zhao, P.C.; Ding, Y.T. Physicochemical, conformational and functional properties of silver carp myosin glycated with konjac oligo-glucomannan Implications for structure-function relationships. Food Hydrocolloid. 2017, 72, 136–144. [Google Scholar] [CrossRef]
- Wang, L.; Wu, M.; Liu, H.M. Emulsifying and physicochemical properties of soy hull hemicelluloses-soy protein isolate conjugates. Carbohydr. Polym. 2017, 163, 181–190. [Google Scholar] [CrossRef]
- Tang, C.H.; Sun, X.; Foegeding, E.A. Modulation of physicochemical and conformational properties of kidney bean vicilin (phaseolin) by glycation with glucose: Implications for structure-function relationships of legume vicilins. J. Agric. Food Chem. 2011, 59, 10114–10123. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, G.L.; Zhao, M.M.; Ren, J.Y.; Yang, B. Improvement of functional properties of peanut protein isolate by conjugation with dextran through Maillard reaction. Food Chem. 2012, 131, 901–906. [Google Scholar] [CrossRef]
- Yi, J.; Fan, Y.T.; Zhang, Y.Z.; Wen, Z.; Zhao, L.Q.; Lu, Y.J. Glycosylated α-lactalbumin-based nanocomplex for curcumin: Physicochemical stability and DPPH-scavenging activity. Food Hydrocolloid. 2016, 61, 369–377. [Google Scholar] [CrossRef]
- Bkhairia, I.; Ben Slama Ben Salem, R.; Nasri, R.; Jridi, M.; Ghorbel, S.; Nasri, M. In-vitro antioxidant and functional properties of protein hydrolysates from golden grey mullet prepared by commercial, microbial and visceral proteases. J. Food Sci. Technol. 2016, 53, 2902–2912. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.H.; Jiang, B.; Zhang, T.; Mu, W.M.; Liu, J. Antioxidant and free radical-scavenging activities of chickpea protein hydrolysate (CPH). Food Chem. 2008, 106, 444–450. [Google Scholar] [CrossRef]
- Hu, M.; Liu, G.N.; Du, X.Q.; Zhang, X.Y.; Qi, B.K.; Li, Y. Molecular crowding prevents the aggregation of protein-dextran conjugate by inducing structural changes, improves its functional properties, and stabilizes it in nanoemulsions. Int. J. Biol. Macromol. 2020, 164, 4183–4192. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tu, Z.C.; Zhang, L.; Luo, J. Effect of heat processing conditions on the antioxidant activity of bovine serum albumin-glucose glycosylation system. Food Sci. 2020, 41, 7–14. (In Chinese) [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Li, L.; Chen, L.; Liu, S.; Cui, Q.; Qin, W. Effects of Sequential Enzymolysis and Glycosylation on the Structural Properties and Antioxidant Activity of Soybean Protein Isolate. Antioxidants 2023, 12, 430. https://doi.org/10.3390/antiox12020430
Zhang Q, Li L, Chen L, Liu S, Cui Q, Qin W. Effects of Sequential Enzymolysis and Glycosylation on the Structural Properties and Antioxidant Activity of Soybean Protein Isolate. Antioxidants. 2023; 12(2):430. https://doi.org/10.3390/antiox12020430
Chicago/Turabian StyleZhang, Qing, Lin Li, Lan Chen, Shuxiang Liu, Qiang Cui, and Wen Qin. 2023. "Effects of Sequential Enzymolysis and Glycosylation on the Structural Properties and Antioxidant Activity of Soybean Protein Isolate" Antioxidants 12, no. 2: 430. https://doi.org/10.3390/antiox12020430