
IGF-1 Controls Metabolic Homeostasis and Survival in HEI-OC1 Auditory Cells through AKT and mTOR Signaling
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Immunofluorescence
2.3. RNA Isolation and RT-qPCR
2.4. Protein Extraction and Western Blotting
2.5. Cell Viability
2.6. TUNEL Assay
2.7. Annexin V-FITC and Propidium Iodine Dual Staining
2.8. DNA Oxidative Damage Detection
2.9. Protein Carbonylation
2.10. Transfection with mCherry-GFP-Tagged LC3 and Live-Cell Microscopy
2.11. Statistical Analysis
3. Results
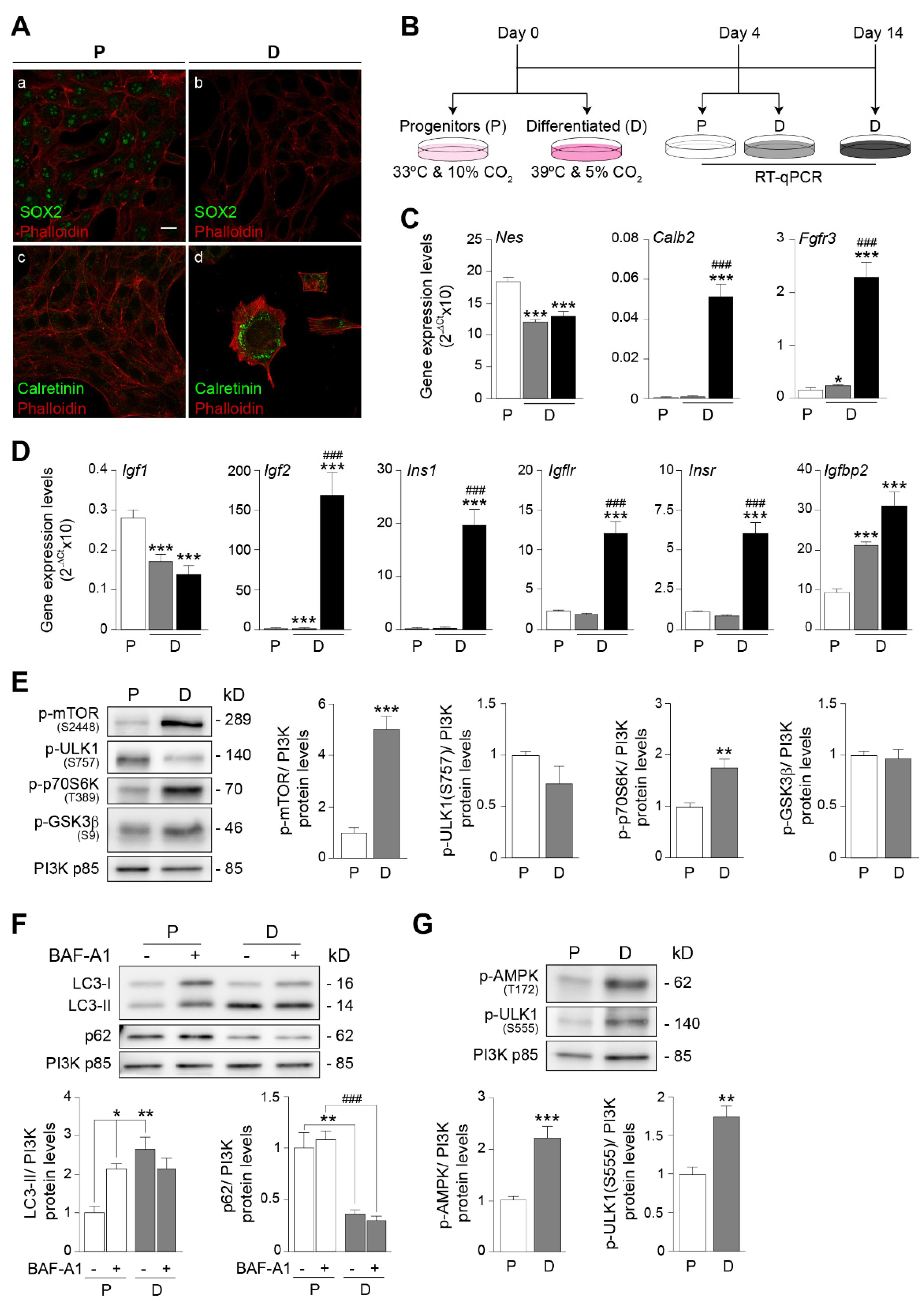
3.1. HEI-OC1 Cells Display Characteristic Phenotypes of Progenitor and Differentiated Auditory Cells
3.2. IGF-1 Stimulation of IGF1R Activates Both AKT and ERK1/2 Signaling in Progenitor and Differentiated HEI-OC1 Auditory Cells
3.3. IGF-1 Promotes Constructive Metabolism in Both Progenitor and Differentiated Auditory Cells but Differentially Modulates Autophagic Flux
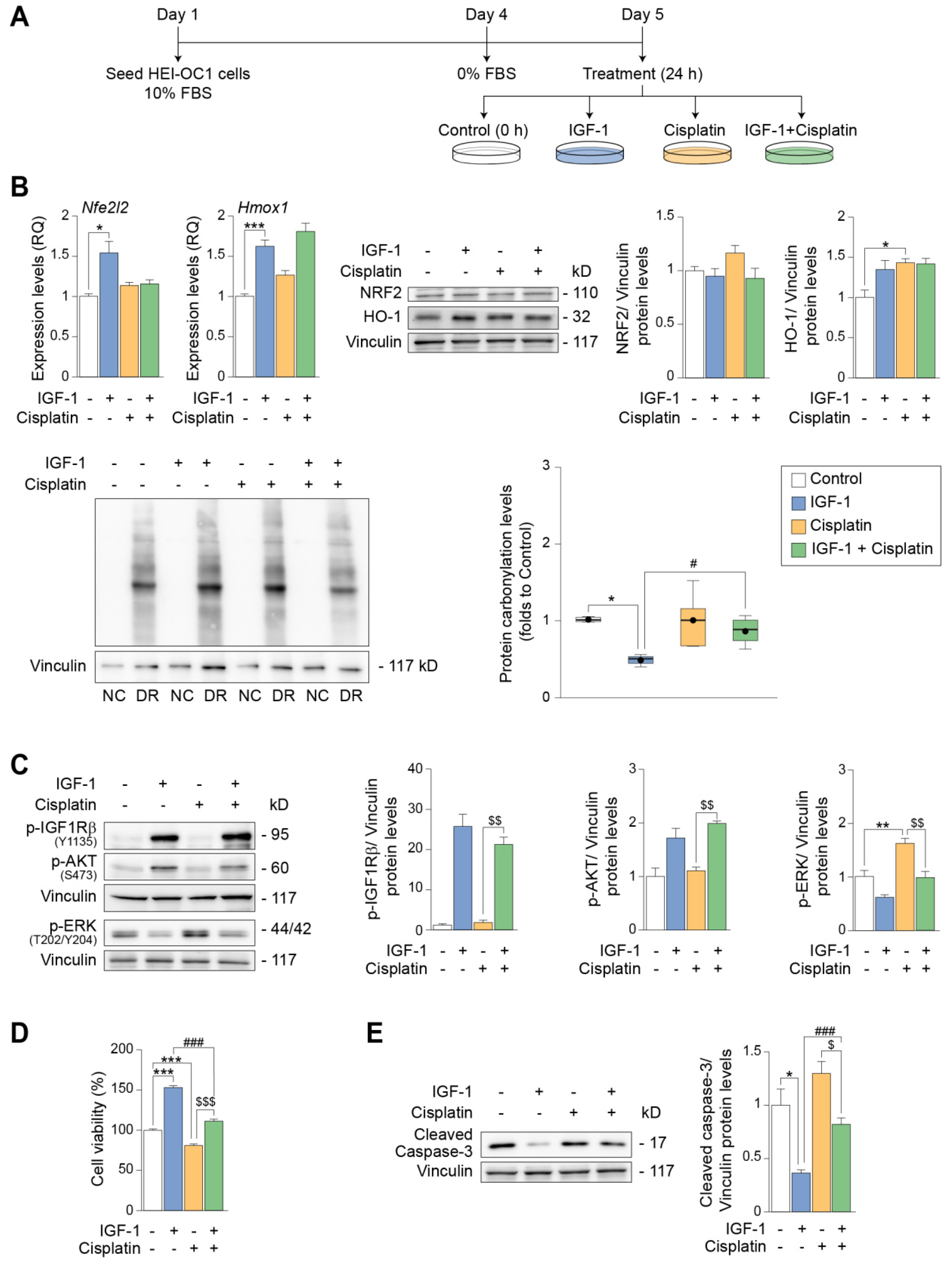
3.4. IGF-1 Stimulation of IGF1R Suppresses DNA Oxidative Damage and Apoptosis in HEI-OC1 Auditory Cells
3.5. IGF-1 Partially Protects against the Early- and Long-Term Ototoxic Effects of Cisplatin in HEI-OC1 Progenitor Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernandez, A.M.; Torres-Alemán, I. The Many Faces of Insulin-like Peptide Signalling in the Brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Hernandez, E.; Guerrero-Gomez, D.; Miranda-Vizuete, A.; Aleman, I.T. A Network of Insulin Peptides Regulate Glucose Uptake by Astrocytes: Potential New Druggable Targets for Brain Hypometabolism. Neuropharmacology 2018, 136, 216–222. [Google Scholar] [CrossRef]
- Nieto-Estévez, V.; Oueslati-Morales, C.O.; Li, L.; Pickel, J.; Morales, A.V.; Vicario-Abejón, C. Brain Insulin-Like Growth Factor-I Directs the Transition from Stem Cells to Mature Neurons During Postnatal/Adult Hippocampal Neurogenesis. Stem Cells 2016, 34, 2194–2209. [Google Scholar] [CrossRef] [Green Version]
- Rajala, A.; Teel, K.; Bhat, M.A.; Batushansky, A.; Griffin, T.M.; Purcell, L.; Rajala, R.V.S. Insulin-like Growth Factor 1 Receptor Mediates Photoreceptor Neuroprotection. Cell Death Dis. 2022, 13, 613. [Google Scholar] [CrossRef]
- Arroba, A.I.; Rodríguez-de la Rosa, L.; Murillo-Cuesta, S.; Vaquero-Villanueva, L.; Hurlé, J.M.; Varela-Nieto, I.; Valverde, Á.M. Autophagy Resolves Early Retinal Inflammation in Igf1-Deficient Mice. Dis. Model. Mech. 2016, 9, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Woods, K.A.; Camacho-Hübner, C.; Savage, M.O.; Clark, A.J. Intrauterine Growth Retardation and Postnatal Growth Failure Associated with Deletion of the Insulin-like Growth Factor I Gene. N. Engl. J. Med. 1996, 335, 1363–1367. [Google Scholar] [CrossRef]
- García-Mato, Á.; Cervantes, B.; Murillo-Cuesta, S.; Rodríguez-de la Rosa, L.; Varela-Nieto, I. Insulin-like Growth Factor 1 Signaling in Mammalian Hearing. Genes 2021, 12, 1553. [Google Scholar] [CrossRef]
- Camarero, G.; Avendano, C.; Fernandez-Moreno, C.; Villar, A.; Contreras, J.; de Pablo, F.; Pichel, J.G.; Varela-Nieto, I. Delayed Inner Ear Maturation and Neuronal Loss in Postnatal Igf-1-Deficient Mice. J. Neurosci. 2001, 21, 7630–7641. [Google Scholar]
- Camarero, G.; Villar, M.A.; Contreras, J.; Fernández-Moreno, C.; Pichel, J.G.; Avendaño, C.; Varela-Nieto, I. Cochlear Abnormalities in Insulin-like Growth Factor-1 Mouse Mutants. Hear. Res. 2002, 170, 2–11. [Google Scholar] [CrossRef]
- Cediel, R.; Riquelme, R.; Contreras, J.; Díaz, A.; Varela-Nieto, I. Sensorineural Hearing Loss in Insulin-like Growth Factor I-Null Mice: A New Model of Human Deafness. Eur. J. Neurosci. 2006, 23, 587–590. [Google Scholar] [CrossRef]
- Rodríguez-de la Rosa, L.; Lassaletta, L.; Calvino, M.; Murillo-Cuesta, S.; Varela-Nieto, I. The Role of Insulin-Like Growth Factor 1 in the Progression of Age-Related Hearing Loss. Front. Aging Neurosci. 2017, 9, 411. [Google Scholar] [CrossRef]
- Celaya, A.M.; Rodríguez-de la Rosa, L.; Bermúdez-Muñoz, J.M.; Zubeldia, J.M.; Romá-Mateo, C.; Avendaño, C.; Pallardó, F.V.; Varela-Nieto, I. IGF-1 Haploinsufficiency Causes Age-Related Chronic Cochlear Inflammation and Increases Noise-Induced Hearing Loss. Cells 2021, 10, 1686. [Google Scholar] [CrossRef]
- LeRoith, D.; Holly, J.M.P.; Forbes, B.E. Insulin-like Growth Factors: Ligands, Binding Proteins, and Receptors. Mol. Metab. 2021, 52, 101245. [Google Scholar] [CrossRef]
- Sanchez-Calderon, H.; Rodriguez-de la Rosa, L.; Milo, M.; Pichel, J.G.; Holley, M.; Varela-Nieto, I. RNA Microarray Analysis in Prenatal Mouse Cochlea Reveals Novel IGF-I Target Genes: Implication of MEF2 and FOXM1 Transcription Factors. PLoS ONE 2010, 5, e8699. [Google Scholar] [CrossRef] [Green Version]
- Okano, T.; Xuan, S.; Kelley, M.W. Insulin-like Growth Factor Signaling Regulates the Timing of Sensory Cell Differentiation in the Mouse Cochlea. J. Neurosci. 2011, 31, 18104–18118. [Google Scholar] [CrossRef] [Green Version]
- Okano, T.; Kelley, M.W. Expression of Insulin-like Growth Factor Binding Proteins during Mouse Cochlear Development. Dev. Dyn. 2013, 242, 1210–1221. [Google Scholar] [CrossRef]
- Gross, J.; Machulik, A.; Moller, R.; Fuchs, J.; Amarjargal, N.; Ungethüm, U.; Kuban, R.-J.; Szczepek, A.J.; Haupt, H.; Mazurek, B. MRNA Expression of Members of the IGF System in the Organ of Corti, the Modiolus and the Stria Vascularis of Newborn Rats. Growth Factors 2008, 26, 180–191. [Google Scholar] [CrossRef]
- Liu, H.; Chen, L.; Giffen, K.P.; Stringham, S.T.; Li, Y.; Judge, P.D.; Beisel, K.W.; He, D.Z.Z. Cell-Specific Transcriptome Analysis Shows That Adult Pillar and Deiters’ Cells Express Genes Encoding Machinery for Specializations of Cochlear Hair Cells. Front. Mol. Neurosci. 2018, 11, 356. [Google Scholar] [CrossRef]
- Kolla, L.; Kelly, M.C.; Mann, Z.F.; Anaya-Rocha, A.; Ellis, K.; Lemons, A.; Palermo, A.T.; So, K.S.; Mays, J.C.; Orvis, J.; et al. Characterization of the Development of the Mouse Cochlear Epithelium at the Single Cell Level. Nat. Commun. 2020, 11, 2389. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S.-I. IGF1 Receptor Signaling Pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Sha, S.-H.; Schacht, J. Kanamycin Alters Cytoplasmic and Nuclear Phosphoinositide Signaling in the Organ of Corti in Vivo. J. Neurochem. 2006, 99, 269–276. [Google Scholar] [CrossRef]
- Hayashi, Y.; Yamamoto, N.; Nakagawa, T.; Ito, J. Insulin-like Growth Factor 1 Inhibits Hair Cell Apoptosis and Promotes the Cell Cycle of Supporting Cells by Activating Different Downstream Cascades after Pharmacological Hair Cell Injury in Neonatal Mice. Mol. Cell. Neurosci. 2013, 56, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Yamamoto, N.; Nakagawa, T.; Ito, J. Insulin-like Growth Factor 1 Induces the Transcription of Gap43 and Ntn1 during Hair Cell Protection in the Neonatal Murine Cochlea. Neurosci. Lett. 2014, 560, 7–11. [Google Scholar] [CrossRef]
- Brand, Y.; Levano, S.; Radojevic, V.; Naldi, A.M.; Setz, C.; Ryan, A.F.; Pak, K.; Hemmings, B.A.; Bodmer, D. All Akt Isoforms (Akt1, Akt2, Akt3) Are Involved in Normal Hearing, but Only Akt2 and Akt3 Are Involved in Auditory Hair Cell Survival in the Mammalian Inner Ear. PLoS ONE 2015, 10, e0121599. [Google Scholar] [CrossRef]
- Lai, R.; Li, W.; Hu, P.; Xie, D.; Wen, J. Role of Hsp90/Akt Pathway in the Pathogenesis of Gentamicin-Induced Hearing Loss. Int. J. Clin. Exp. Pathol. 2018, 11, 4431–4438. [Google Scholar]
- Sha, S.-H.; Chen, F.-Q.; Schacht, J. PTEN Attenuates PIP3/Akt Signaling in the Cochlea of the Aging CBA/J Mouse. Hear. Res. 2010, 264, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Camarero, G.; Leon, Y.; Gorospe, I.; De Pablo, F.; Alsina, B.; Giraldez, F.; Varela-Nieto, I. Insulin-like Growth Factor 1 Is Required for Survival of Transit-Amplifying Neuroblasts and Differentiation of Otic Neurons. Dev. Biol. 2003, 262, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Magariños, M.; Aburto, M.R.; Sánchez-Calderón, H.; Muñoz-Agudo, C.; Rapp, U.R.; Varela-Nieto, I. RAF Kinase Activity Regulates Neuroepithelial Cell Proliferation and Neuronal Progenitor Cell Differentiation during Early Inner Ear Development. PLoS ONE 2010, 5, e14435. [Google Scholar] [CrossRef] [Green Version]
- Aburto, M.R.; Magariños, M.; Leon, Y.; Varela-Nieto, I.; Sanchez-Calderon, H. AKT Signaling Mediates IGF-I Survival Actions on Otic Neural Progenitors. PLoS ONE 2012, 7, e30790. [Google Scholar] [CrossRef] [Green Version]
- León, Y.; Magariños, M.; Varela-Nieto, I. Ceramide Kinase Inhibition Blocks IGF-1-Mediated Survival of Otic Neurosensory Progenitors by Impairing AKT Phosphorylation. Front. Cell Dev. Biol. 2021, 9, 678760. [Google Scholar] [CrossRef]
- Murillo-Cuesta, S.; Camarero, G.; González-Rodríguez, A.; De La Rosa, L.R.; Burks, D.J.; Avendaño, C.; Valverde, A.M.; Varela-Nieto, I. Insulin Receptor Substrate 2 (IRS2)-Deficient Mice Show Sensorineural Hearing Loss That Is Delayed by Concomitant Protein Tyrosine Phosphatase 1B (PTP1B) Loss of Function. Mol. Med. Camb. Mass 2012, 18, 260–269. [Google Scholar] [CrossRef] [Green Version]
- de Iriarte Rodríguez, R.; Magariños, M.; Pfeiffer, V.; Rapp, U.R.; Varela-Nieto, I. C-Raf Deficiency Leads to Hearing Loss and Increased Noise Susceptibility. Cell. Mol. Life Sci. 2015, 72, 3983–3998. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMPK and Autophagy Get Connected. EMBO J. 2011, 30, 634–635. [Google Scholar] [CrossRef] [Green Version]
- Ebnoether, E.; Ramseier, A.; Cortada, M.; Bodmer, D.; Levano-Huaman, S. Sesn2 Gene Ablation Enhances Susceptibility to Gentamicin-Induced Hair Cell Death via Modulation of AMPK/MTOR Signaling. Cell Death Discov. 2017, 3, 17024. [Google Scholar] [CrossRef] [Green Version]
- Bodmer, D.; Levano-Huaman, S. Sesn2/AMPK/MTOR Signaling Mediates Balance between Survival and Apoptosis in Sensory Hair Cells under Stress. Cell Death Dis. 2017, 8, e3068. [Google Scholar] [CrossRef]
- Gentilin, E.; Simoni, E.; Candito, M.; Cazzador, D.; Astolfi, L. Cisplatin-Induced Ototoxicity: Updates on Molecular Targets. Trends Mol. Med. 2019, 25, 1123–1132. [Google Scholar] [CrossRef]
- Murillo-Cuesta, S.; Celaya, A.M.; Cervantes, B.; Bermúdez-Muñoz, J.M.; Rodríguez-de la Rosa, L.; Contreras, J.; Sánchez-Pérez, I.; Varela-Nieto, I. Therapeutic Efficiency of the APAF-1 Antagonist LPT99 in a Rat Model of Cisplatin-Induced Hearing Loss. Clin. Transl. Med. 2021, 11, e363. [Google Scholar] [CrossRef]
- Langer, T.; am Zehnhoff-Dinnesen, A.; Radtke, S.; Meitert, J.; Zolk, O. Understanding Platinum-Induced Ototoxicity. Trends Pharmacol. Sci. 2013, 34, 458–469. [Google Scholar] [CrossRef]
- Hayashi, Y.; Yamamoto, N.; Nakagawa, T.; Omori, K.; Ito, J. Activation of IGF1 Signaling in the Cochlea Induces the Transcription of Its Mediators During the Protection of Cochlear Hair Cells Against Aminoglycoside. Otol. Neurotol. 2017, 38, 278–282. [Google Scholar] [CrossRef]
- Yamahara, K.; Nakagawa, T.; Ito, J.; Kinoshita, K.; Omori, K.; Yamamoto, N. Netrin 1 Mediates Protective Effects Exerted by Insulin-like Growth Factor 1 on Cochlear Hair Cells. Neuropharmacology 2017, 119, 26–39. [Google Scholar] [CrossRef]
- Kalinec, G.M.; Webster, P.; Lim, D.J.; Kalinec, F. A Cochlear Cell Line as an in Vitro System for Drug Ototoxicity Screening. Audiol. Neurootol. 2003, 8, 177–189. [Google Scholar] [CrossRef]
- Kalinec, G.; Thein, P.; Park, C.; Kalinec, F. HEI-OC1 Cells as a Model for Investigating Drug Cytotoxicity. Hear. Res. 2016, 335, 105–117. [Google Scholar] [CrossRef]
- Langan, T.J.; Rodgers, K.R.; Chou, R.C. Synchronization of Mammalian Cell Cultures by Serum Deprivation. Methods Mol. Biol. 2017, 1524, 97–105. [Google Scholar] [CrossRef]
- Tang, Q.; Wang, X.; Jin, H.; Mi, Y.; Liu, L.; Dong, M.; Chen, Y.; Zou, Z. Cisplatin-Induced Ototoxicity: Updates on Molecular Mechanisms and Otoprotective Strategies. Eur. J. Pharm. Biopharm. 2021, 163, 60–71. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bernis, M.E.; Oksdath, M.; Dupraz, S.; Nieto Guil, A.; Fernández, M.M.; Malchiodi, E.L.; Rosso, S.B.; Quiroga, S. Wingless-Type Family Member 3A Triggers Neuronal Polarization via Cross-Activation of the Insulin-like Growth Factor-1 Receptor Pathway. Front. Cell. Neurosci. 2013, 7, 194. [Google Scholar] [CrossRef] [Green Version]
- Cervantes, B.; Arana, L.; Murillo-Cuesta, S.; Bruno, M.; Alkorta, I.; Varela-Nieto, I. Solid Lipid Nanoparticles Loaded with Glucocorticoids Protect Auditory Cells from Cisplatin-Induced Ototoxicity. J. Clin. Med. 2019, 8, 1464. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- de Chaumont, F.; Dallongeville, S.; Chenouard, N.; Hervé, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; Lecomte, T.; Le Montagner, Y.; et al. Icy: An Open Bioimage Informatics Platform for Extended Reproducible Research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Halevy, O.; Cantley, L.C. Differential Regulation of the Phosphoinositide 3-Kinase and MAP Kinase Pathways by Hepatocyte Growth Factor vs. Insulin-like Growth Factor-I in Myogenic Cells. Exp. Cell Res. 2004, 297, 224–234. [Google Scholar] [CrossRef]
- Zheng, W.-H.; Quirion, R. Insulin-like Growth Factor-1 (IGF-1) Induces the Activation/Phosphorylation of Akt Kinase and CAMP Response Element-Binding Protein (CREB) by Activating Different Signaling Pathways in PC12 Cells. BMC Neurosci. 2006, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- García-Echeverría, C.; Pearson, M.A.; Marti, A.; Meyer, T.; Mestan, J.; Zimmermann, J.; Gao, J.; Brueggen, J.; Capraro, H.-G.; Cozens, R.; et al. In Vivo Antitumor Activity of NVP-AEW541-A Novel, Potent, and Selective Inhibitor of the IGF-IR Kinase. Cancer Cell 2004, 5, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Higashi, Y.; Sukhanov, S.; Anwar, A.; Shai, S.-Y.; Delafontaine, P. IGF-1, Oxidative Stress and Atheroprotection. Trends Endocrinol. Metab. 2010, 21, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- So, H.; Kim, H.; Lee, J.-H.; Park, C.; Kim, Y.; Kim, E.; Kim, J.-K.; Yun, K.-J.; Lee, K.-M.; Lee, H.-Y.; et al. Cisplatin Cytotoxicity of Auditory Cells Requires Secretions of Proinflammatory Cytokines via Activation of ERK and NF-KappaB. J. Assoc. Res. Otolaryngol. 2007, 8, 338–355. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Shi, S.; Ren, T.; Zhang, Y.; Guo, P.; Wang, J.; Wang, W. U0126 Pretreatment Inhibits Cisplatin-Induced Apoptosis and Autophagy in HEI-OC1 Cells and Cochlear Hair Cells. Toxicol. Appl. Pharmacol. 2021, 415, 115447. [Google Scholar] [CrossRef]
- Riquelme, R.; Cediel, R.; Contreras, J.; la Rosa Lourdes, R.; Murillo-Cuesta, S.; Hernandez-Sanchez, C.; Zubeldia, J.M.; Cerdan, S.; Varela-Nieto, I. A Comparative Study of Age-Related Hearing Loss in Wild Type and Insulin-like Growth Factor I Deficient Mice. Front. Neuroanat. 2010, 4, 27. [Google Scholar] [CrossRef]
- Kempfle, J.S.; Turban, J.L.; Edge, A.S.B. Sox2 in the Differentiation of Cochlear Progenitor Cells. Sci. Rep. 2016, 6, 23293. [Google Scholar] [CrossRef] [Green Version]
- Chow, C.L.; Trivedi, P.; Pyle, M.P.; Matulle, J.T.; Fettiplace, R.; Gubbels, S.P. Evaluation of Nestin Expression in the Developing and Adult Mouse Inner Ear. Stem Cells Dev. 2016, 25, 1419–1432. [Google Scholar] [CrossRef] [Green Version]
- Dechesne, C.J.; Rabejac, D.; Desmadryl, G. Development of Calretinin Immunoreactivity in the Mouse Inner Ear. J. Comp. Neurol. 1994, 346, 517–529. [Google Scholar] [CrossRef]
- Sampath, P.; Pritchard, D.K.; Pabon, L.; Reinecke, H.; Schwartz, S.M.; Morris, D.R.; Murry, C.E. A Hierarchical Network Controls Protein Translation during Murine Embryonic Stem Cell Self-Renewal and Differentiation. Cell Stem Cell 2008, 2, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Easley, C.A.; Ben-Yehudah, A.; Redinger, C.J.; Oliver, S.L.; Varum, S.T.; Eisinger, V.M.; Carlisle, D.L.; Donovan, P.J.; Schatten, G.P. MTOR-Mediated Activation of P70 S6K Induces Differentiation of Pluripotent Human Embryonic Stem Cells. Cell. Reprogramming 2010, 12, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-S.; Liu, F.; Segil, N. A Morphogenetic Wave of P27Kip1 Transcription Directs Cell Cycle Exit during Organ of Corti Development. Development 2006, 133, 2817–2826. [Google Scholar] [CrossRef] [Green Version]
- Laine, H.; Doetzlhofer, A.; Mantela, J.; Ylikoski, J.; Laiho, M.; Roussel, M.F.; Segil, N.; Pirvola, U. P19(Ink4d) and P21(Cip1) Collaborate to Maintain the Postmitotic State of Auditory Hair Cells, Their Codeletion Leading to DNA Damage and P53-Mediated Apoptosis. J. Neurosci. 2007, 27, 1434–1444. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B. Autophagy in Mammalian Development and Differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z. Induction of Autophagy Is Essential for Monocyte-Macrophage Differentiation. Blood 2012, 119, 2895–2905. [Google Scholar] [CrossRef]
- Romanelli, D.; Casati, B.; Franzetti, E.; Tettamanti, G. A Molecular View of Autophagy in Lepidoptera. BioMed Res. Int. 2014, 2014, 902315. [Google Scholar] [CrossRef] [Green Version]
- de Iriarte Rodríguez, R.; Pulido, S.; Rodríguez-de la Rosa, L.; Magariños, M.; Varela-Nieto, I. Age-Regulated Function of Autophagy in the Mouse Inner Ear. Hear. Res. 2015, 330, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, C.; Iwasaki, S.; Urata, S.; Morishita, H.; Sakamaki, Y.; Fujioka, M.; Kondo, K.; Mizushima, N.; Yamasoba, T. Autophagy Is Essential for Hearing in Mice. Cell Death Dis. 2017, 8, e2780. [Google Scholar] [CrossRef] [Green Version]
- He, Z.-H.; Li, M.; Fang, Q.-J.; Liao, F.-L.; Zou, S.-Y.; Wu, X.; Sun, H.-Y.; Zhao, X.-Y.; Hu, Y.-J.; Xu, X.-X.; et al. FOXG1 Promotes Aging Inner Ear Hair Cell Survival through Activation of the Autophagy Pathway. Autophagy 2021, 17, 4341–4362. [Google Scholar] [CrossRef]
- Varela-Nieto, I.; Palmero, I.; Magariños, M. Complementary and Distinct Roles of Autophagy, Apoptosis and Senescence during Early Inner Ear Development. Hear. Res. 2019, 376, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Iwai, K.; Nakagawa, T.; Endo, T.; Matsuoka, Y.; Kita, T.; Kim, T.-S.; Tabata, Y.; Ito, J. Cochlear Protection by Local Insulin-like Growth Factor-1 Application Using Biodegradable Hydrogel. Laryngoscope 2006, 116, 529–533. [Google Scholar] [CrossRef]
- Fujiwara, T.; Hato, N.; Nakagawa, T.; Tabata, Y.; Yoshida, T.; Komobuchi, H.; Takeda, S.; Hyodo, J.; Hakuba, N.; Gyo, K. Insulin-like Growth Factor 1 Treatment via Hydrogels Rescues Cochlear Hair Cells from Ischemic Injury. Neuroreport 2008, 19, 1585–1588. [Google Scholar] [CrossRef]
- Gao, L.; Kita, T.; Katsuno, T.; Yamamoto, N.; Omori, K.; Nakagawa, T. Insulin-Like Growth Factor 1 on the Maintenance of Ribbon Synapses in Mouse Cochlear Explant Cultures. Front. Cell. Neurosci. 2020, 14, 571155. [Google Scholar] [CrossRef]
- Bieniussa, L.; Kahraman, B.; Skornicka, J.; Schulte, A.; Voelker, J.; Jablonka, S.; Hagen, R.; Rak, K. Pegylated Insulin-Like Growth Factor 1 Attenuates Hair Cell Loss and Promotes Presynaptic Maintenance of Medial Olivocochlear Cholinergic Fibers in the Cochlea of the Progressive Motor Neuropathy Mouse. Front. Neurol. 2022, 13, 885026. [Google Scholar] [CrossRef]
- Nakagawa, T.; Yamamoto, M.; Kumakawa, K.; Usami, S.-I.; Hato, N.; Tabuchi, K.; Takahashi, M.; Fujiwara, K.; Sasaki, A.; Komune, S.; et al. Prognostic Impact of Salvage Treatment on Hearing Recovery in Patients with Sudden Sensorineural Hearing Loss Refractory to Systemic Corticosteroids: A Retrospective Observational Study. Auris Nasus Larynx 2016, 43, 489–494. [Google Scholar] [CrossRef]
- Mahran, Y.F. New Insights into the Protection of Growth Hormone in Cisplatin-Induced Nephrotoxicity: The Impact of IGF-1 on the Keap1-Nrf2/HO-1 Signaling. Life Sci. 2020, 253, 117581. [Google Scholar] [CrossRef]
- Bailey-Downs, L.C.; Mitschelen, M.; Sosnowska, D.; Toth, P.; Pinto, J.T.; Ballabh, P.; Valcarcel-Ares, M.N.; Farley, J.; Koller, A.; Henthorn, J.C.; et al. Liver-Specific Knockdown of IGF-1 Decreases Vascular Oxidative Stress Resistance by Impairing the Nrf2-Dependent Antioxidant Response: A Novel Model of Vascular Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 313–329. [Google Scholar] [CrossRef]
- Riis, S.; Murray, J.B.; O’Connor, R. IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β-Nrf2-BNIP3 Pathway. Cells 2020, 9, 147. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Oh, G.-S.; Shen, A.; Lee, S.-B.; Choe, S.-K.; Kwon, K.-B.; Lee, S.; Seo, K.-S.; Kwak, T.H.; Park, R.; et al. Augmentation of NAD(+) by NQO1 Attenuates Cisplatin-Mediated Hearing Impairment. Cell Death Dis. 2014, 5, e1292. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Fukushima, K.; Omichi, R.; Kariya, S.; Nishizaki, K. Time Courses of Changes in Phospho- and Total- MAP Kinases in the Cochlea after Intense Noise Exposure. PLoS ONE 2013, 8, e58775. [Google Scholar] [CrossRef] [Green Version]
- Kurioka, T.; Matsunobu, T.; Satoh, Y.; Niwa, K.; Endo, S.; Fujioka, M.; Shiotani, A. ERK2 Mediates Inner Hair Cell Survival and Decreases Susceptibility to Noise-Induced Hearing Loss. Sci. Rep. 2015, 5, 16839. [Google Scholar] [CrossRef] [Green Version]
- Herranen, A.; Ikäheimo, K.; Virkkala, J.; Pirvola, U. The Stress Response in the Non-Sensory Cells of the Cochlea Under Pathological Conditions-Possible Role in Mediating Noise Vulnerability. J. Assoc. Res. Otolaryngol. 2018, 19, 637–652. [Google Scholar] [CrossRef]
- Yu, X.; Fan, Z.; Han, Y.; Zhang, D.; Xu, L.; Wang, M.; Yang, Q.; Li, H.; Zhou, M.; Zhang, L.; et al. Paeoniflorin Reduces Neomycin-Induced Ototoxicity in Hair Cells by Suppression of Reactive Oxygen Species Generation and Extracellularly Regulated Kinase Signalization. Toxicol. Lett. 2018, 285, 9–19. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, J.-H.; Kim, S.-J.; Oh, G.S.; Moon, H.-D.; Kwon, K.-B.; Park, C.; Park, B.H.; Lee, H.-K.; Chung, S.-Y.; et al. Roles of NADPH Oxidases in Cisplatin-Induced Reactive Oxygen Species Generation and Ototoxicity. J. Neurosci. 2010, 30, 3933–3946. [Google Scholar] [CrossRef] [Green Version]
- Porter, A.G.; Jänicke, R.U. Emerging Roles of Caspase-3 in Apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX Phosphorylation: A Marker for DNA Damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Mato, Á.; Cervantes, B.; Rodríguez-de la Rosa, L.; Varela-Nieto, I. IGF-1 Controls Metabolic Homeostasis and Survival in HEI-OC1 Auditory Cells through AKT and mTOR Signaling. Antioxidants 2023, 12, 233. https://doi.org/10.3390/antiox12020233
García-Mato Á, Cervantes B, Rodríguez-de la Rosa L, Varela-Nieto I. IGF-1 Controls Metabolic Homeostasis and Survival in HEI-OC1 Auditory Cells through AKT and mTOR Signaling. Antioxidants. 2023; 12(2):233. https://doi.org/10.3390/antiox12020233
Chicago/Turabian StyleGarcía-Mato, Ángela, Blanca Cervantes, Lourdes Rodríguez-de la Rosa, and Isabel Varela-Nieto. 2023. "IGF-1 Controls Metabolic Homeostasis and Survival in HEI-OC1 Auditory Cells through AKT and mTOR Signaling" Antioxidants 12, no. 2: 233. https://doi.org/10.3390/antiox12020233