
Mitochondrial Stress Links Environmental Triggers with Pro-Inflammatory Signaling in Crohn’s Disease
 , , , ,
, , , ,  ,
,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Cell Culture and Drugs
2.3. Mitochondrial Function Assays
2.4. Gene Expression
2.5. Allelic Discrimination Assay
2.6. Immunoblotting
2.7. Assessment of mtDAMPs
2.8. Statistical Analysis
3. Results
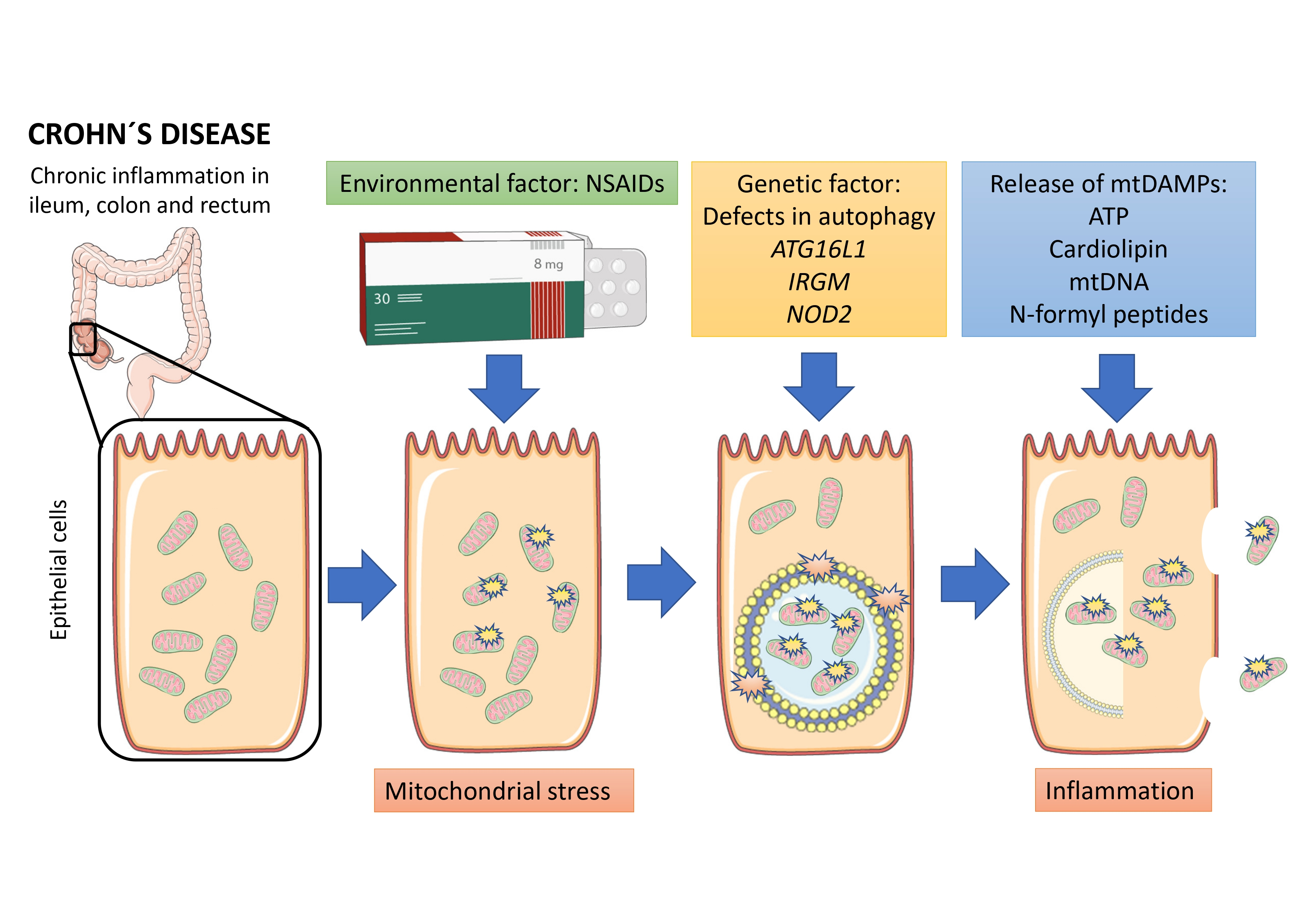
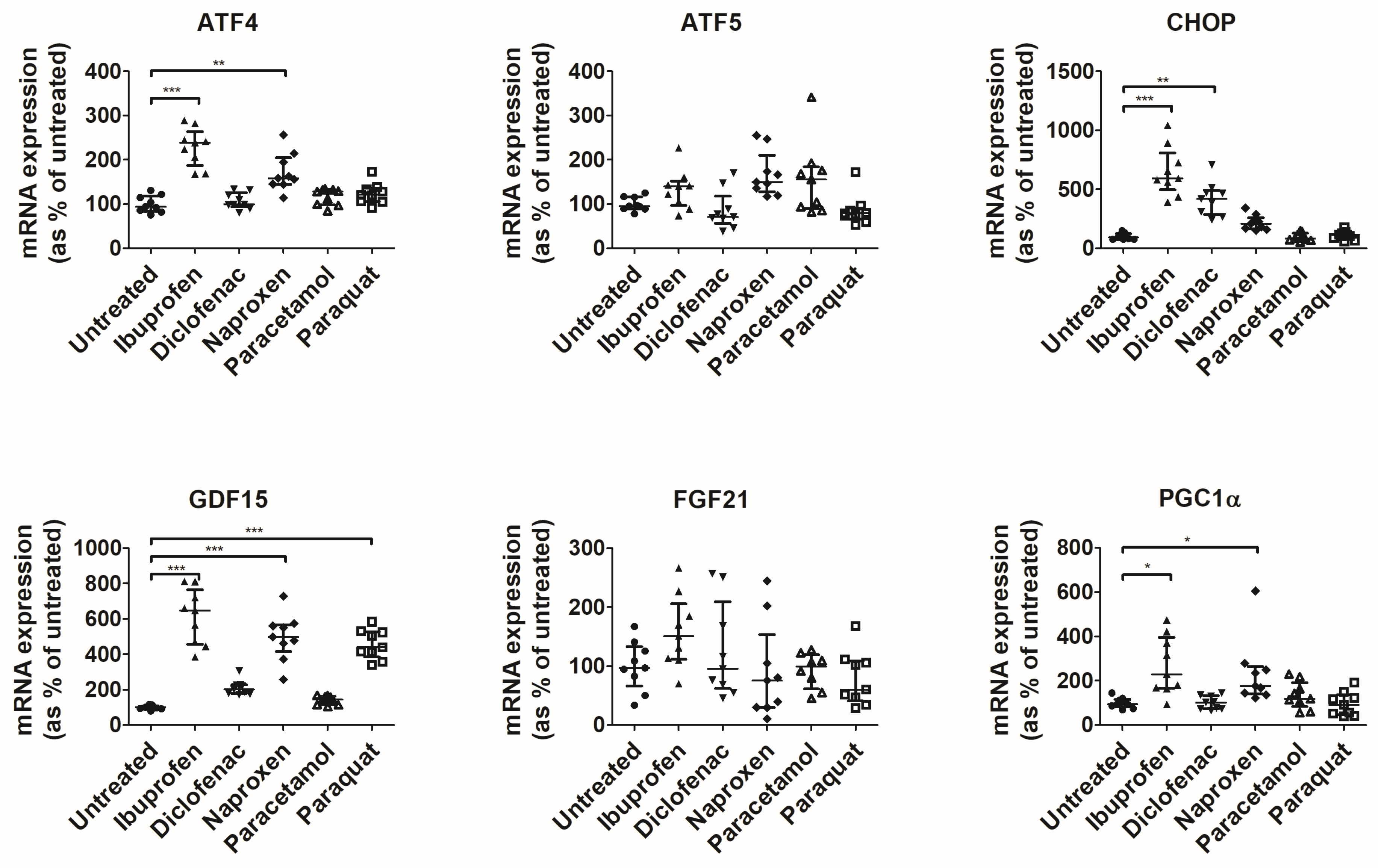
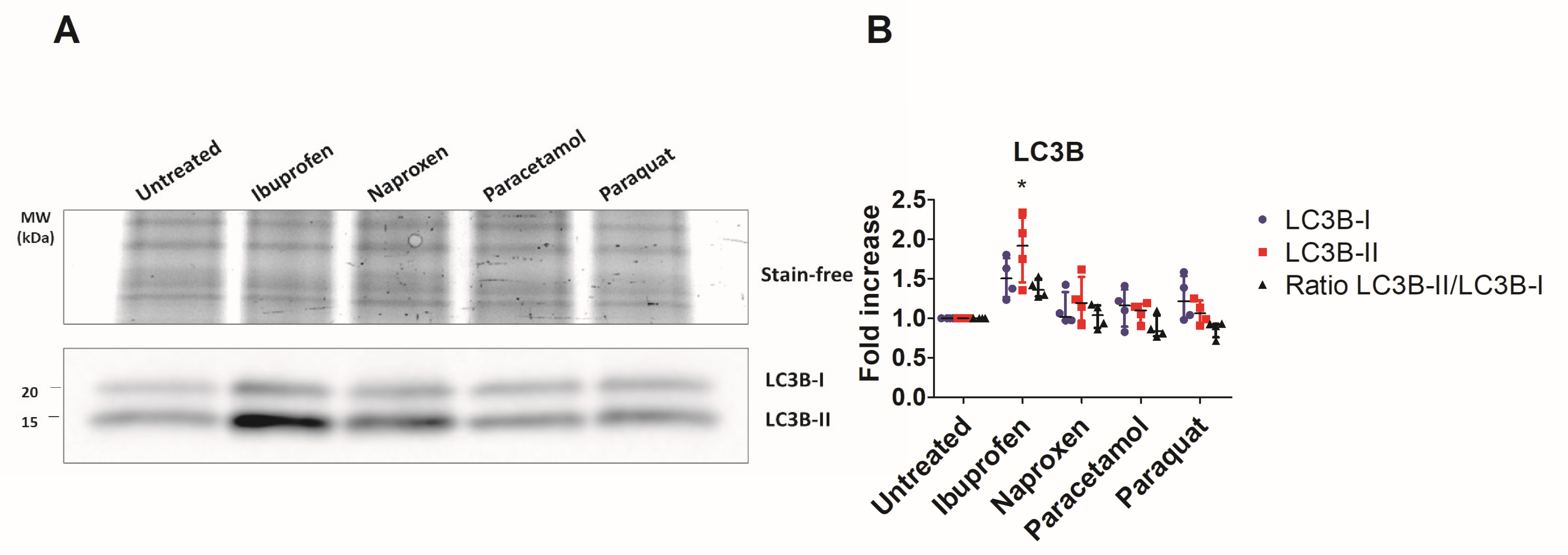
3.1. NSAIDs Induce Mitochondrial Stress in an In Vitro Model of Intestinal Epithelium
3.2. Under Impaired Mitophagy, Mitochondrial Stress Leads to Release of mtDAMPs
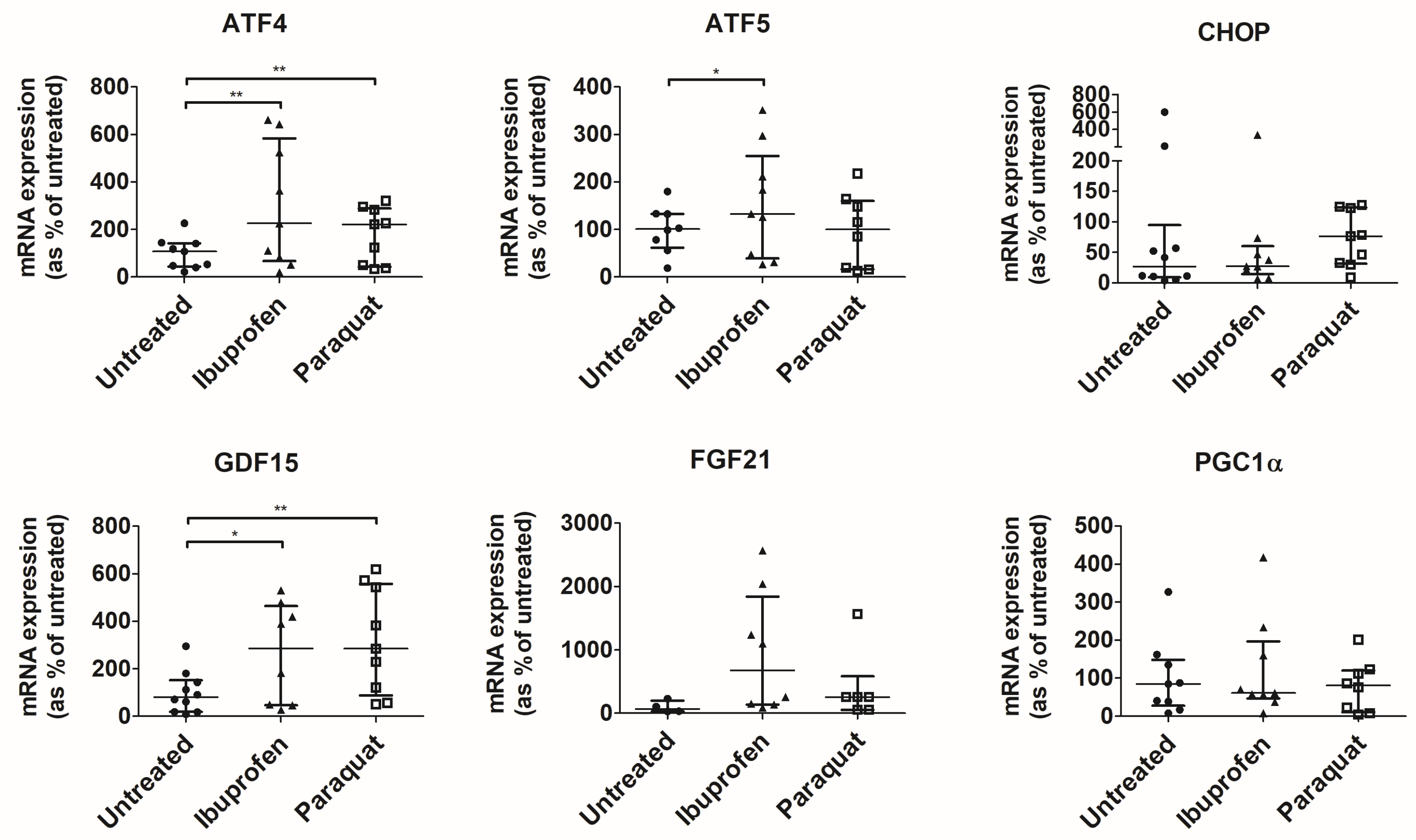
3.3. Ibuprofen Activates UPRmt in Colonic Organoids
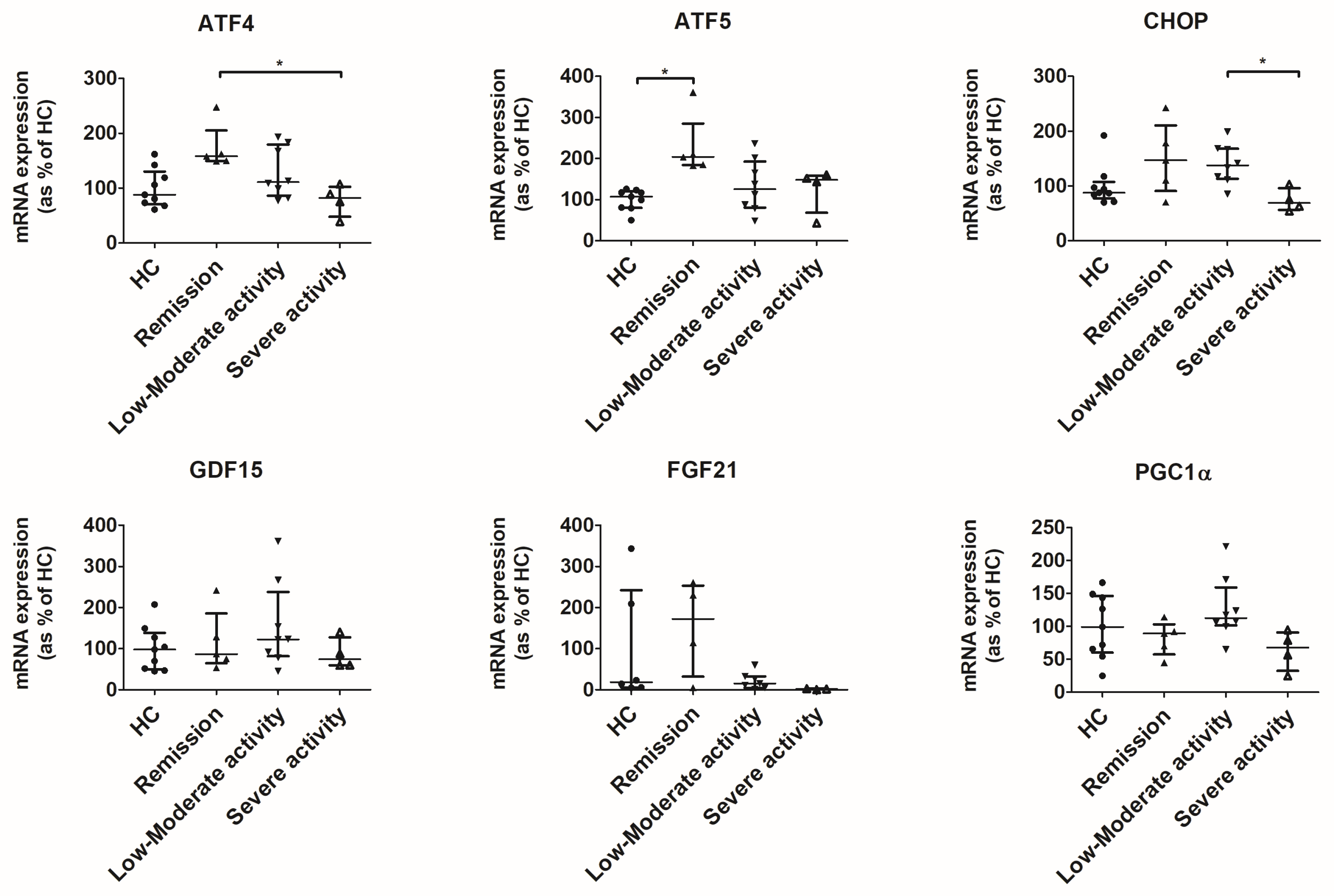
3.4. UPRmt Is Activated in the Colon of Patients in Remission and Low-to-Moderate Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uniken Venema, W.T.C.; Voskuil, M.D.; Dijkstra, G.; Weersma, R.K.; Festen, E.A.M. The Genetic Background of Inflammatory Bowel Disease: From Correlation to Causality. J. Pathol. 2017, 241, 146–158. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental Triggers in IBD: A Review of Progress and Evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Higuchi, L.M.; Huang, E.S.; Khalili, H.; Richter, J.M.; Fuchs, C.S.; Chan, A.T. Aspirin, Nonsteroidal Anti-Inflammatory Drug Use, and Risk for Crohn Disease and Ulcerative Colitis. Ann. Intern. Med. 2012, 156, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Bonner, G.F.; Fakhri, A.; Vennamaneni, S.R. A Long-Term Cohort Study of Nonsteroidal Anti-Inflammatory Drug Use and Disease Activity in Outpatients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2004, 10, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Li, Y.; Peng, I.; Reichelt, M.; Katakam, A.K.; Noubade, R.; Roose-Girma, M.; Devoss, J.; Diehl, L.; Graham, R.R.; et al. A Crohn’s Disease Variant in Atg16l1 Enhances Its Degradation by Caspase 3. Nature 2014, 506, 456–462. [Google Scholar] [CrossRef]
- Söderholm, J.D.; Olaison, G.; Peterson, K.H.; Franzén, L.E.; Lindmark, T.; Wirén, M.; Tagesson, C.; Sjódahl, R. Augmented Increase in Tight Junction Permeability by Luminal Stimuli in the Non-Inflamed Ileum of Crohn’s Disease. Gut 2002, 50, 307–313. [Google Scholar] [CrossRef]
- Sifroni, K.G.; Damiani, C.R.; Stoffel, C.; Cardoso, M.R.; Ferreira, G.K.; Jeremias, I.C.; Rezin, G.T.; Scaini, G.; Schuck, P.F.; Dal-Pizzol, F.; et al. Mitochondrial Respiratory Chain in the Colonic Mucosal of Patients with Ulcerative Colitis. Mol. Cell. Biochem. 2010, 342, 111–115. [Google Scholar] [CrossRef]
- Rath, E.; Berger, E.; Messlik, A.; Nunes, T.; Liu, B.; Kim, S.C.; Hoogenraad, N.; Sans, M.; Sartor, R.B.; Haller, D. Induction of DsRNA-Activated Protein Kinase Links Mitochondrial Unfolded Protein Response to the Pathogenesis of Intestinal Inflammation. Gut 2012, 61, 1269–1278. [Google Scholar] [CrossRef]
- Boyapati, R.K.; Dorward, D.A.; Tamborska, A.; Kalla, R.; Ventham, N.T.; Doherty, M.K.; Whitfield, P.D.; Gray, M.; Loane, J.; Rossi, A.G.; et al. Mitochondrial DNA Is a Pro-Inflammatory Damage-Associated Molecular Pattern Released during Active IBD. Inflamm. Bowel Dis. 2018, 24, 2113–2122. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Alula, K.M.; Dowdell, A.S.; LeBere, B.; Lee, J.S.; Levens, C.L.; Kuhn, K.A.; Kaipparettu, B.A.; Thompson, W.E.; Blumberg, R.S.; Colgan, S.P.; et al. Interplay of Gut Microbiota and Host Epithelial Mitochondrial Dysfunction Is Necessary for the Development of Spontaneous Intestinal Inflammation in Mice. Microbiome 2023, 11, 256. [Google Scholar] [CrossRef]
- Thai, P.N.; Ren, L.; Xu, W.; Overton, J.; Timofeyev, V.; Nader, C.E.; Haddad, M.; Yang, J.; Gomes, A.V.; Hammock, B.D.; et al. Chronic Diclofenac Exposure Increases Mitochondrial Oxidative Stress, Inflammatory Mediators, and Cardiac Dysfunction. Cardiovasc. Drugs Ther. 2023, 37, 25–37. [Google Scholar] [CrossRef]
- Alnahdi, A.; John, A.; Raza, H. Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in HepG2 Cells by Palmitic Acid. Nutrients 2019, 11, 1979. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Devriese, S.; van den Bossche, L.; van Welden, S.; Holvoet, T.; Pinheiro, I.; Hindryckx, P.; de Vos, M.; Laukens, D. T84 Monolayers Are Superior to Caco-2 as a Model System of Colonocytes. Histochem. Cell Biol. 2017, 148, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Bramlage, P.; Goldis, A. Bioequivalence Study of Three Ibuprofen Formulations after Single Dose Administration in Healthy Volunteers. BMC Pharmacol. 2008, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Sugár, D.; Francombe, D.; da Silva, T.; Adams, R.; Hutchings, S. Bioequivalence of 2 Naproxen Sodium Tablet Formulations in Healthy Male and Female Volunteers. Curr. Ther. Res. Clin. Exp. 2019, 90, 33–38. [Google Scholar] [CrossRef]
- Xu, J.J.; Henstock, P.V.; Dunn, M.C.; Smith, A.R.; Chabot, J.R.; de Graaf, D. Cellular Imaging Predictions of Clinical Drug-Induced Liver Injury. Toxicol. Sci. 2008, 105, 97–105. [Google Scholar] [CrossRef]
- Rana, P.; Aleo, M.D.; Gosink, M.; Will, Y. Evaluation of in Vitro Mitochondrial Toxicity Assays and Physicochemical Properties for Prediction of Organ Toxicity Using 228 Pharmaceutical Drugs. Chem. Res. Toxicol. 2019, 32, 156–167. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-Wide Association Study Identifies New Susceptibility Loci for Crohn Disease and Implicates Autophagy in Disease Pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.C.; Heath, R.J.; et al. Atg16L1 T300A Variant Decreases Selective Autophagy Resulting in Altered Cytokine Signaling and Decreased Antibacterial Defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef] [PubMed]
- Johann, K.; Kleinert, M.; Klaus, S. The Role of Gdf15 as a Myomitokine. Cells 2021, 10, 2990. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Ito, M.; Kojima, T.; Yatsuga, S.; Koga, Y.; Tanaka, M. GDF15 Is a Novel Biomarker to Evaluate Efficacy of Pyruvate Therapy for Mitochondrial Diseases. Mitochondrion 2015, 20, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.H.; Tao, L.L.; Su, D.Q.; Guo, C.J.; Liu, H. Diagnostic Utility of GDF15 in Neurodegenerative Diseases: A Systematic Review and Meta-Analysis. Brain Behav. 2022, 12, e2502. [Google Scholar] [CrossRef] [PubMed]
- Keranov, S.; Widmann, L.; Jafari, L.; Liebetrau, C.; Keller, T.; Troidl, C.; Kriechbaum, S.; Voss, S.; Bauer, P.; Richter, M.J.; et al. GDF-15 and Soluble ST2 as Biomarkers of Right Ventricular Dysfunction in Pulmonary Hypertension. Biomark Med. 2023, 16, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, J.A.; Pothuraju, R.; Khan, P.; Sharma, G.; Muniyan, S.; Seshacharyulu, P.; Jain, M.; Nasser, M.W.; Batra, S.K. Pathophysiological Role of Growth Differentiation Factor 15 (GDF15) in Obesity, Cancer, and Cachexia. Cytokine Growth Factor Rev. 2022, 64, 71–83. [Google Scholar] [CrossRef]
- Liu, H.; Huang, Y.; Lyu, Y.; Dai, W.; Tong, Y.; Li, Y. GDF15 as a Biomarker of Ageing. Exp. Gerontol. 2021, 146, 111228. [Google Scholar] [CrossRef]
- Townsend, L.K.; Medak, K.; Weber, A.J.; Dibe, H.; Shamshoum, H.; Wright, D.C. CHOP Is Dispensable for Exercise-Induced Increases in GDF15. J. Appl. Physiol. 2022, 132, 413–422. [Google Scholar] [CrossRef]
- Conway, K.L.; Kuballa, P.; Song, J.H.; Patel, K.K.; Castoreno, A.B.; Yilmaz, O.H.; Jijon, H.B.; Zhang, M.; Aldrich, L.N.; Villablanca, E.J.; et al. Atg16l1 Is Required for Autophagy in Intestinal Epithelial Cells and Protection of Mice from Salmonella Infection. Gastroenterology 2013, 145, 1347–1357. [Google Scholar] [CrossRef]
- Baqri, R.M.; Pietron, A.v; Gokhale, R.H.; Turner, B.A.; Kaguni, L.S.; Shingleton, A.W.; Kunes, S.; Miller, K.E. Mitochondrial Chaperone TRAP1 Activates the Mitochondrial UPR and Extends Healthspan in Drosophila. Mech. Ageing Dev. 2014, 141–142, 35–45. [Google Scholar] [CrossRef]
- Park, S.H.; Hong, M.; Lee, H.S.; Ye, B.D.; Hwang, S.W.; Jung, S.; Baek, J.; Moon, J.W.; Kim, B.M.; Oh, S.H.; et al. Association of TRAP1 with Infliximab-Induced Mucosal Healing in Crohn’s Disease. J. Gastroenterol. Hepatol. 2019, 34, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lu, J.; Yang, M.; Cai, J.; Fu, Q.; Ma, J.; Zhu, L. The Mitochondrial Unfolded Protein Response (UPRmt) Protects against Osteoarthritis. Exp. Mol. Med. 2022, 54, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Scorrano, L. Mitochondrial Morphology in Mitophagy and Macroautophagy. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 205–212. [Google Scholar] [CrossRef]
- Svaguša, T.; Martinić, M.; Martinić, M.; Kovačević, L.; Šepac, A.; Miličić, D.; Bulum, J.; Starčević, B.; Sirotković-Skerlev, M.; Seiwerth, F.; et al. Mitochondrial Unfolded Protein Response, Mitophagy and Other Mitochondrial Quality Control Mechanisms in Heart Disease and Aged Heart. Croat. Med. J. 2020, 61, 126–138. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Replicate | % of T | % of C |
|---|---|---|
| 1 | 29.64 | 70.36 |
| 2 | 35.43 | 64.57 |
| 3 | 30.35 | 69.65 |
| 4 | 32.16 | 67.84 |
| 5 | 29.22 | 70.78 |
| 6 | 31.59 | 68.41 |
| 7 | 29.85 | 70.15 |
| 8 | 30.77 | 69.23 |
| 9 | 26.00 | 74.00 |
| 10 | 31.18 | 68.82 |
| 11 | 30.27 | 69.73 |
| Mean ± SD | 30.59 ± 2.28 | 69.41 ± 2.28 |
| ATF4 | ATF5 | CHOP | GDF15 | FGF21 | PGC1α | TRAP1 | LON | ||
| ATF4 | Pearson’s r | 0.745 | 0.539 | 0.71 | 0.798 | 0.63 | 0.208 | 0.73 | |
| p value | <0.001 | 0.004 | <0.001 | <0.001 | 0.001 | 0.297 | <0.001 | ||
| ATF5 | Pearson’s r | 0.745 | 0.636 | 0.613 | 0.811 | 0.779 | 0.285 | 0.579 | |
| p value | <0.001 | 0.001 | 0.001 | <0.001 | <0.001 | 0.167 | 0.002 | ||
| CHOP | Pearson’s r | 0.539 | 0.636 | 0.243 | 0.613 | 0.862 | 0.015 | 0.342 | |
| p value | 0.004 | 0.001 | 0.222 | 0.007 | <0.001 | 0.94 | 0.087 | ||
| GDF15 | Pearson’s r | 0.71 | 0.613 | 0.243 | 0.702 | 0.49 | 0.462 | 0.834 | |
| p value | <0.001 | 0.001 | 0.222 | 0.001 | 0.013 | 0.017 | <0.001 | ||
| FGF21 | Pearson’s r | 0.798 | 0.811 | 0.613 | 0.702 | 0.809 | 0.472 | 0.622 | |
| p value | <0.001 | <0.001 | 0.007 | 0.001 | <0.001 | 0.048 | 0.006 | ||
| PGC1α | Pearson’s r | 0.63 | 0.779 | 0.862 | 0.49 | 0.809 | 0.127 | 0.513 | |
| p value | 0.001 | <0.001 | <0.001 | 0.013 | <0.001 | 0.536 | 0.009 | ||
| TRAP1 | Pearson’s r | 0.208 | 0.285 | 0.015 | 0.462 | 0.472 | 0.127 | 0.606 | |
| p value | 0.297 | 0.167 | 0.94 | 0.017 | 0.048 | 0.536 | 0.001 | ||
| LON | Pearson’s r | 0.73 | 0.579 | 0.342 | 0.834 | 0.622 | 0.513 | 0.606 | |
| p value | <0.001 | 0.002 | 0.087 | <0.001 | 0.006 | 0.009 | 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Reyes, F.; Bernal, M.; Rodríguez-Díaz, C.; Rodríguez-de los Reyes, D.; Ho-Plagaro, A.; Rodríguez-Pacheco, F.; Camacho-Martel, L.; Camargo-Camero, R.; Rodríguez-González, F.J.; Alcain-Martínez, G.; et al. Mitochondrial Stress Links Environmental Triggers with Pro-Inflammatory Signaling in Crohn’s Disease. Antioxidants 2023, 12, 2105. https://doi.org/10.3390/antiox12122105
Martín-Reyes F, Bernal M, Rodríguez-Díaz C, Rodríguez-de los Reyes D, Ho-Plagaro A, Rodríguez-Pacheco F, Camacho-Martel L, Camargo-Camero R, Rodríguez-González FJ, Alcain-Martínez G, et al. Mitochondrial Stress Links Environmental Triggers with Pro-Inflammatory Signaling in Crohn’s Disease. Antioxidants. 2023; 12(12):2105. https://doi.org/10.3390/antiox12122105
Chicago/Turabian StyleMartín-Reyes, Flores, Manuel Bernal, Cristina Rodríguez-Díaz, Damaris Rodríguez-de los Reyes, Ailec Ho-Plagaro, Francisca Rodríguez-Pacheco, Laura Camacho-Martel, Raquel Camargo-Camero, Francisco J. Rodríguez-González, Guillermo Alcain-Martínez, and et al. 2023. "Mitochondrial Stress Links Environmental Triggers with Pro-Inflammatory Signaling in Crohn’s Disease" Antioxidants 12, no. 12: 2105. https://doi.org/10.3390/antiox12122105