
Oxidative Stress Induced by Lipotoxicity and Renal Hypoxia in Diabetic Kidney Disease and Possible Therapeutic Interventions: Targeting the Lipid Metabolism and Hypoxia
Abstract
:1. Introduction
2. Lipotoxicity and DKD
2.1. Lipid Metabolism—Physiologic Versus DKD
2.1.1. Fatty Acid (FA) (Figure 1)
FA Uptake—Normal and DKD Patients
FA Synthesis
FA Oxidation (FAO)
FA-Induced Post-Translational Modifications (PTMs)

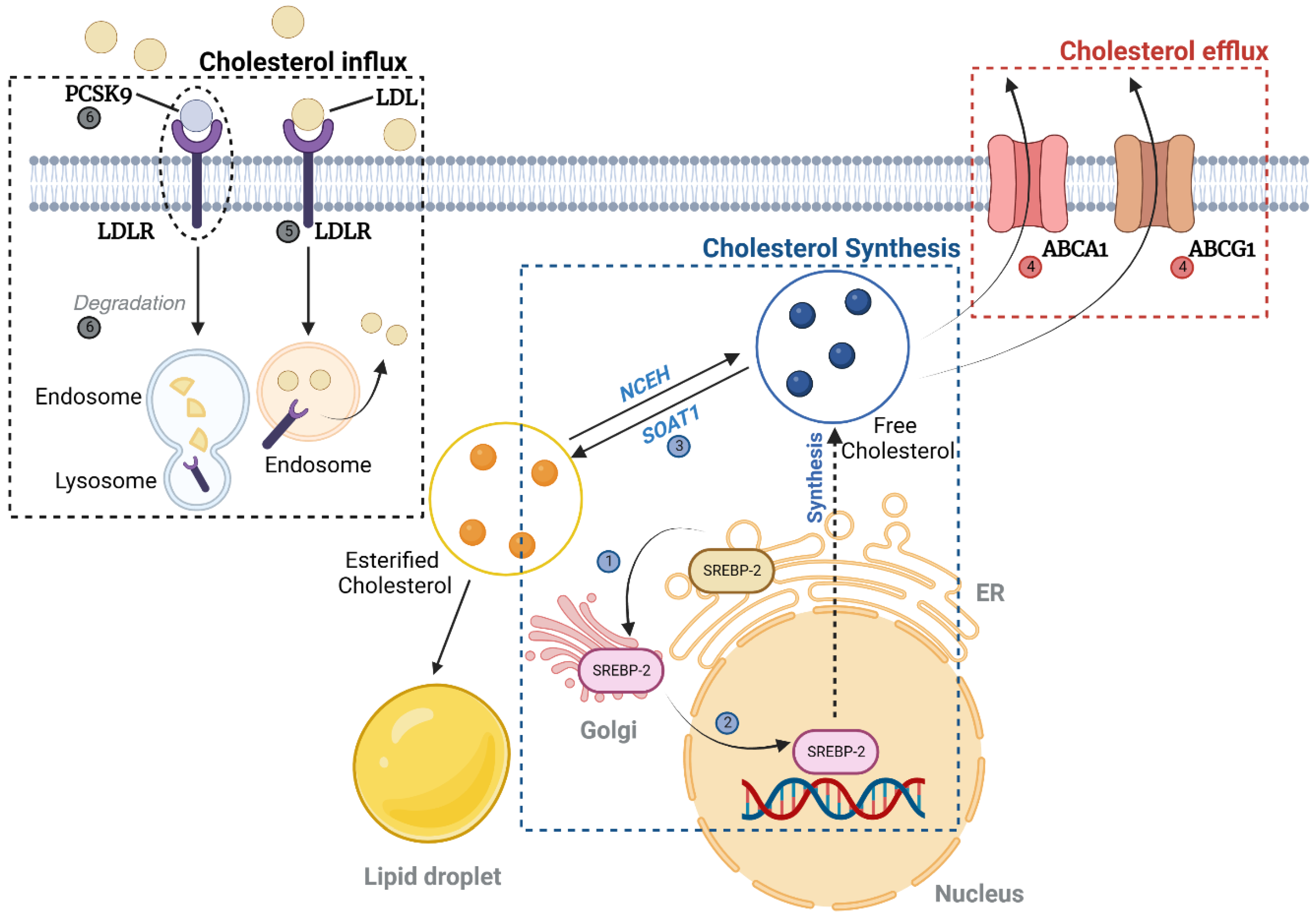
2.1.2. Cholesterol (Figure 2)
Cholesterol Uptake, Synthesis, and Efflux

2.2. Lipid-Induced Oxidative Stress in Lipid-Rich Kidney Disease, Including DKD
3. Hypoxia and DKD
3.1. Increased Risk of Hypoxic Injury in Kidney
3.2. Renal Hypoxia and Oxidative Stress in DKD
3.3. HIF-1 Activation in Hypoxia: A Double-Edged Sword
4. Oxidative Stress and DKD
4.1. The Role of ROS and Oxidative Stress in DKD
4.2. ROS, Oxidative Stress, and Intracellular Signaling Pathways
4.2.1. Keap1-Nrf2 Pathway
4.2.2. Forkhead Box O (FoxO) Proteins
4.2.3. Nuclear Factor (NF)-κB Pathway
4.3. ROS, Oxidative Stress, and Cellular Organelles in the Context of DKD
4.3.1. Mitochondria
4.3.2. ER and Peroxisome
4.3.3. Mitochondria-Associated ER Membrane (MAM)
5. Therapeutic Approaches to Modify DKD
5.1. Antilipidemic Drug
5.1.1. Statins/Fenofibrate
5.1.2. Ezetimibe
5.1.3. PCSK9 Inhibitors
5.1.4. ABCA1 Inducer
5.2. SGLT2 Inhibitor/GLP-1 Agonist
5.2.1. SGLT2 Inhibitor
5.2.2. GLP-1 Agonist
5.3. NRF2 Activators
5.3.1. Bardoxolone Methyl
5.3.2. Curcumin
5.3.3. Sulforaphane
5.4. Resveratrol
5.5. Vitamin D
5.6. Adiponectin Receptor Activator
5.7. HIF-1 Stabilizer (Prolyl Hydroxylase Inhibitor)
5.8. Potential Concerns in Applying Anti-Oxidants for Treating DKD
6. Conclusions and Perspectives


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Model | Advantages | Limitations | Reference Numbers in This Review |
|---|---|---|---|---|
| Type 1 DM | STZ |
|
| [49] |
| Unilateral nephrectomy + STZ |
|
| [45] | |
| Alloxan |
|
| [76] | |
| Type 1 DM (Auto-immune) | NOD |
|
| [50] |
| Type 2 DM | db/db |
|
| [19,23,46,49,57,63,120,128,129,157,193,200] |
| ob/ob |
|
| [49,164] | |
| HFD |
|
| [55,61,156,158,159,186] | |
| HFD + STZ |
|
| [64] | |
| HFD + ApoE−/− |
|
| [169] | |
| ZDF |
|
| [75] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, Y.; Hirose, H.; Ohneda, M.; Johnson, J.; McGarry, J.D.; Unger, R.H. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: Impairment in adipocyte-beta-cell relationships. Proc. Natl. Acad. Sci. USA 1994, 91, 10878–10882. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032. [Google Scholar] [CrossRef] [PubMed]
- Schelling, J.R. The contribution of lipotoxicity to diabetic kidney disease. Cells 2022, 11, 3236. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M. Mitochondria–power players in kidney function? Trends Endocrinol. Metab. 2016, 27, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.G.; Goddard, D.; Eppel, G.A.; O’Connor, P.M. Factors that render the kidney susceptible to tissue hypoxia in hypoxemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R931–R940. [Google Scholar] [CrossRef]
- Nourbakhsh, N.; Singh, P. Role of renal oxygenation and mitochondrial function in the pathophysiology of acute kidney injury. Nephron Clin. Pract. 2014, 127, 149–152. [Google Scholar] [CrossRef]
- Fu, Q.; Colgan, S.P.; Shelley, C.S. Hypoxia: The force that drives chronic kidney disease. Clin. Med. Res. 2016, 14, 15–39. [Google Scholar] [CrossRef]
- Chen, Y.; Dai, Y.; Song, K.; Huang, Y.; Zhang, L.; Zhang, C.; Yan, Q.; Gao, H. Pre-emptive pharmacological inhibition of fatty acid–binding protein 4 attenuates kidney fibrosis by reprogramming tubular lipid metabolism. Cell Death Dis. 2021, 12, 572. [Google Scholar] [CrossRef]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta Mol. Cell Biol. 2012, 1821, 852–857. [Google Scholar] [CrossRef]
- Khan, S.; Cabral, P.D.; Schilling, W.P.; Schmidt, Z.W.; Uddin, A.N.; Gingras, A.; Madhavan, S.M.; Garvin, J.L.; Schelling, J.R. Kidney proximal tubule lipoapoptosis is regulated by fatty acid transporter-2 (FATP2). J. Am. Soc. Nephrol. 2018, 29, 81. [Google Scholar] [CrossRef]
- Roche, C.M.; Blanch, H.W.; Clark, D.S.; Glass, N.L. Physiological role of acyl coenzyme A synthetase homologs in lipid metabolism in Neurospora crassa. Eukaryot. Cell 2013, 12, 1244–1257. [Google Scholar] [CrossRef]
- Castelblanco, E.; Sanjurjo, L.; Falguera, M.; Hernández, M.; Fernandez-Real, J.-M.; Sarrias, M.-R.; Alonso, N.; Mauricio, D. Circulating soluble CD36 is similar in type 1 and type 2 diabetes mellitus versus non-diabetic subjects. J. Clin. Med. 2019, 8, 710. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Moon, J.S.; Park, I.R.; Kim, J.H.; Yoon, J.S.; Won, K.C.; Lee, H.W. A novel index using soluble CD36 is associated with the prevalence of type 2 diabetes mellitus: Comparison study with triglyceride-glucose index. Endocrinol. Metab. 2017, 32, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Shiju, T.M.; Mohan, V.; Balasubramanyam, M.; Viswanathan, P. Soluble CD36 in plasma and urine: A plausible prognostic marker for diabetic nephropathy. J. Diabetes Complicat. 2015, 29, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, T.; Geng, J.; Wu, Z.; Xu, L.; Liu, J.; Tian, J.; Zhou, Z.; Nie, J.; Bai, X. Advanced oxidation protein products promote lipotoxicity and tubulointerstitial fibrosis via CD36/β-catenin pathway in diabetic nephropathy. Antioxid. Redox Signal. 2019, 31, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.-T.; Wu, C.-C.; Hung, W.-C.; Lee, T.-L.; Hsuan, C.-F.; Wei, C.-T.; Lu, Y.-C.; Yu, T.-H.; Chung, F.-M.; Lee, Y.-J.; et al. FABP1 and FABP2 as markers of diabetic nephropathy. Int. J. Med. Sci. 2020, 17, 2338. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Li, Z.; Ehara, T.; Yang, L.; Wang, D.; Feng, L.; Zhang, Y.; Wang, K.; Shi, Y.; Duan, H.; et al. Fatty acid-binding protein 4 mediates apoptosis via endoplasmic reticulum stress in mesangial cells of diabetic nephropathy. Mol. Cell. Endocrinol. 2015, 411, 232–242. [Google Scholar] [CrossRef]
- Falkevall, A.; Mehlem, A.; Palombo, I.; Sahlgren, B.H.; Ebarasi, L.; He, L.; Ytterberg, A.J.; Olauson, H.; Axelsson, J.; Sundelin, B.; et al. Reducing VEGF-B signaling ameliorates renal lipotoxicity and protects against diabetic kidney disease. Cell Metab. 2017, 25, 713–726. [Google Scholar] [CrossRef]
- Khan, S.; Gaivin, R.; Abramovich, C.; Boylan, M.; Calles, J.; Schelling, J.R. Fatty acid transport protein-2 regulates glycemic control and diabetic kidney disease progression. JCI Insight 2020, 5, e136845. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Dihingia, A.; Bordoloi, J.; Dutta, P.; Kalita, J.; Manna, P. Hexane-Isopropanolic Extract of Tungrymbai, a North-East Indian fermented soybean food prevents hepatic steatosis via regulating AMPK-mediated SREBP/FAS/ACC/HMGCR and PPARα/CPT1A/UCP2 pathways. Sci. Rep. 2018, 8, 10021. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, T.; Li, J.; Proctor, G.; McManaman, J.L.; Lucia, S.; Chua, S.; Levi, M. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVB db/db mice with type 2 diabetes. Diabetes 2005, 54, 2328–2335. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Halaihel, N.; Zhang, W.; Rogers, T.; Levi, M. Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J. Biol. Chem. 2002, 277, 18919–18927. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Mu, L.; Yang, Z.; Du, C.; Wu, M.; Song, S.; Yuan, C.; Shi, Y. Carbohydrate response element-binding protein regulates lipid metabolism via mTOR complex1 in diabetic nephropathy. J. Cell. Physiol. 2021, 236, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Park, C.W.; Kim, H.W.; Ko, S.H.; Chung, H.W.; Lim, S.W.; Yang, C.W.; Chang, Y.S.; Sugawara, A.; Guan, Y.; Breyer, M.D. Accelerated diabetic nephropathy in mice lacking the peroxisome proliferator–activated receptor α. Diabetes 2006, 55, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, C.; Yang, H.; Liu, S.; Lu, Y.; Fu, P.; Liu, J. Metabolomics reveal mitochondrial and fatty acid metabolism disorders that contribute to the development of DKD in T2DM patients. Mol. Biosyst. 2017, 13, 2392–2400. [Google Scholar] [CrossRef]
- Afshinnia, F.; Nair, V.; Lin, J.; Rajendiran, T.M.; Soni, T.; Byun, J.; Sharma, K.; Fort, P.E.; Gardner, T.W.; Looker, H.C.; et al. Increased lipogenesis and impaired β-oxidation predict type 2 diabetic kidney disease progression in American Indians. JCI Insight 2019, 4, e130317. [Google Scholar] [CrossRef]
- Vamecq, J.; Cherkaoui-Malki, M.; Andreoletti, P.; Latruffe, N. The human peroxisome in health and disease: The story of an oddity becoming a vital organelle. Biochimie 2014, 98, 4–15. [Google Scholar] [CrossRef]
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.-S.; Ha, H. Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes 2012, 61, 728–738. [Google Scholar] [CrossRef]
- Tserga, A.; Pouloudi, D.; Saulnier-Blache, J.S.; Stroggilos, R.; Theochari, I.; Gakiopoulou, H.; Mischak, H.; Zoidakis, J.; Schanstra, J.P.; Vlahou, A.; et al. Proteomic analysis of mouse kidney tissue associates peroxisomal dysfunction with early diabetic kidney disease. Biomedicines 2022, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Galdieri, L.; Chang, J.; Mehrotra, S.; Vancura, A. Yeast phospholipase C is required for normal acetyl-CoA homeostasis and global histone acetylation. J. Biol. Chem. 2013, 288, 27986–27998. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lin, S.-H.; Ren, F.; Li, J.-T.; Chen, J.-J.; Yao, C.-B.; Yang, H.-B.; Jiang, S.-X.; Yan, G.-Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef] [PubMed]
- Felix, J.B.; Cox, A.R.; Hartig, S.M. Acetyl-CoA and metabolite fluxes regulate white adipose tissue expansion. Trends Endocrinol. Metab. 2021, 32, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-L.; Doyle, H.A.; Clarke, S.G.; Herold, K.C.; Mamula, M.J. Oxidative modifications in tissue pathology and autoimmune disease. Antioxid. Redox Signal. 2018, 29, 1415–1431. [Google Scholar] [CrossRef]
- Hu, A.; Zou, H.; Chen, B.; Zhong, J. Posttranslational modifications in diabetes: Mechanisms and functions. Rev Endocr. Metab. Disord. 2022, 23, 1011–1033. [Google Scholar] [CrossRef]
- Ali, F.; Dar, J.S.; Magray, A.R.; Ganai, B.A.; Chishti, M. Posttranslational modifications of proteins and their role in biological processes and associated diseases. In Protein Modificomics; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–35. [Google Scholar]
- Zhao, L.; Zhang, C.; Luo, X.; Wang, P.; Zhou, W.; Zhong, S.; Xie, Y.; Jiang, Y.; Yang, P.; Tang, R.; et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 2018, 69, 705–717. [Google Scholar] [CrossRef]
- Ren, W.; Jhala, U.S.; Du, K. Proteomic analysis of protein palmitoylation in adipocytes. Adipocyte 2013, 2, 17–27. [Google Scholar] [CrossRef]
- Ravid, M.; Brosh, D.; Ravid-Safran, D.; Levy, Z.; Rachmani, R. Main risk factors for nephropathy in type 2 diabetes mellitus are plasma cholesterol levels, mean blood pressure, and hyperglycemia. Arch. Intern. Med. 1998, 158, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Park, A.S.D.; Mohtat, D.; Thomas, D.B.; Pullman, J.M.; Susztak, K. Transcriptome analysis of human diabetic kidney disease. Diabetes 2011, 60, 2354–2369. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Van Krieken, R.; Marway, M.; Parthasarathy, P.; Mehta, N.; Ingram, A.J.; Gao, B.; Krepinsky, J.C. Inhibition of SREBP with fatostatin does not attenuate early diabetic nephropathy in male mice. Endocrinology 2018, 159, 1479–1495. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ducasa, G.M.; Mallela, S.K.; Kim, J.-J.; Molina, J.; Mitrofanova, A.; Wilbon, S.S.; Ge, M.; Fontanella, A.; Pedigo, C.; et al. Sterol-O-acyltransferase-1 has a role in kidney disease associated with diabetes and Alport syndrome. Kidney Int. 2020, 98, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Jatem, E.; Lima, J.; Montoro, B.; Torres-Bondia, F.; Segarra, A. Efficacy and safety of PCSK9 inhibitors in hypercholesterolemia associated with refractory nephrotic syndrome. Kidney Int. Rep. 2021, 6, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Mitrofanova, A.; Burke, G.; Merscher, S.; Fornoni, A. New insights into renal lipid dysmetabolism in diabetic kidney disease. World J. Diabetes 2021, 12, 524. [Google Scholar] [CrossRef]
- Ducasa, G.M.; Mitrofanova, A.; Mallela, S.K.; Liu, X.; Molina, J.; Sloan, A.; Pedigo, C.E.; Ge, M.; Santos, J.V.; Hernandez, Y.; et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Investig. 2019, 129, 3387–3400. [Google Scholar] [CrossRef]
- Tang, C.; Kanter, J.E.; Bornfeldt, K.E.; Leboeuf, R.C.; Oram, J.F. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys 1. J. Lipid Res. 2010, 51, 1719–1728. [Google Scholar] [CrossRef]
- Tsun, J.G.; Yung, S.; Chau, M.K.; Shiu, S.W.; Chan, T.M.; Tan, K.C. Cellular cholesterol transport proteins in diabetic nephropathy. PLoS ONE 2014, 9, e105787. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Anton, M.I.; Floria, M.; Seritean Isac, P.N.; Hurjui, L.L.; Tarniceriu, C.C.; Costea, C.F.; Ciocoiu, M.; Rezus, C. Oxidative stress and NRF2/KEAP1/ARE pathway in diabetic kidney disease (DKD): New perspectives. Biomolecules 2022, 12, 1227. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Kume, S.; Koya, D.; Araki, S.-i.; Isshiki, K.; Chin-Kanasaki, M.; Sugimoto, T.; Haneda, M.; Sugaya, T.; Kashiwagi, A.; et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic. Biol. Med. 2011, 51, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Su, Y.; Ju, Y.; Ma, K.; Li, W.; Li, W. Astragalosides IV protected the renal tubular epithelial cells from free fatty acids-induced injury by reducing oxidative stress and apoptosis. Biomed. Pharmacother. 2018, 108, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Kang, J.S.; Kim, H.M.; Kim, S.J.; Kim, N.; Lee, J.O.; Kim, H.S.; Lee, E.Y.; Chung, C.H. Dehydrozingerone inhibits renal lipotoxicity in high-fat diet–induced obese mice. J. Cell. Mol. Med. 2021, 25, 8725–8733. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.W.; Lim, J.H.; Kim, M.Y.; Shin, S.J.; Chung, S.; Choi, B.S.; Kim, H.W.; Kim, Y.-S.; Park, C.W.; Chang, Y.S. High-fat diet-induced renal cell apoptosis and oxidative stress in spontaneously hypertensive rat are ameliorated by fenofibrate through the PPARα–FoxO3a–PGC-1α pathway. Nephrol. Dial. Transplant. 2012, 27, 2213–2225. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, T.W.; Kim, Y.; Yang, K.S.; Park, H.S.; Choi, S.R.; Chung, S.; Kim, H.W.; et al. Fenofibrate improves renal lipotoxicity through activation of AMPK-PGC-1α in db/db mice. PLoS ONE 2014, 9, e96147. [Google Scholar] [CrossRef] [PubMed]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: Implications for prevention and therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Ren, L.; Cui, H.; Wang, Y.; Ju, F.; Cai, Y.; Gang, X.; Wang, G. The role of lipotoxicity in kidney disease: From molecular mechanisms to therapeutic prospects. Biomed. Pharmacother. 2023, 161, 114465. [Google Scholar] [CrossRef]
- Park, M.-J.; Han, H.J.; Kim, D.-i. Lipotoxicity-induced PRMT1 exacerbates mesangial cell apoptosis via endoplasmic reticulum stress. Int. J. Mol. Sci. 2017, 18, 1421. [Google Scholar] [CrossRef]
- Li, B.; Leung, J.C.; Chan, L.Y.; Yiu, W.H.; Li, Y.; Lok, S.W.; Liu, W.H.; Chan, K.W.; Tse, H.F.; Lai, K.N.; et al. Amelioration of endoplasmic reticulum stress by mesenchymal stem cells via hepatocyte growth factor/c-Met signaling in obesity-associated kidney injury. Stem Cells Transl. Med. 2019, 8, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-C.; Tang, S.-Q.; Liu, Y.-T.; Li, A.-M.; Zhan, M.; Yang, M.; Song, N.; Zhang, W.; Wu, X.-Q.; Peng, C.-H.; et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 2021, 12, 925. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.; Kurtcuoglu, V. Renal arteriovenous oxygen shunting. Curr. Opin. Nephrol. Hypertens. 2017, 26, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J.; Baumgärtl, H.; Lübbers, D.; Wilcox, C.S. Nephron pO2 and renal oxygen usage in the hypertensive rat kidney. Kidney Int. 2001, 59, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Leong, C.-L.; Anderson, W.P.; O’Connor, P.M.; Evans, R.G. Evidence that renal arterial-venous oxygen shunting contributes to dynamic regulation of renal oxygenation. Am. J. Physiol. Renal Physiol. 2007, 292, F1726–F1733. [Google Scholar] [CrossRef]
- Van Bommel, J.; Siegemund, M.; Henny, C.P.; Ince, C. Heart, kidney, and intestine have different tolerances for anemia. Transl. Res. 2008, 151, 110–117. [Google Scholar] [CrossRef]
- Evans, R.G.; Eppel, G.A.; Anderson, W.P.; Denton, K.M. Mechanisms underlying the differential control of blood flow in the renal medulla and cortex. J. Hypertens. 2004, 22, 1439–1451. [Google Scholar] [CrossRef]
- Hansell, P.; Welch, W.J.; Blantz, R.C.; Palm, F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 2013, 40, 123–137. [Google Scholar] [CrossRef]
- Hesp, A.C.; Schaub, J.A.; Prasad, P.V.; Vallon, V.; Laverman, G.D.; Bjornstad, P.; Van Raalte, D.H. The role of renal hypoxia in the pathogenesis of diabetic kidney disease: A promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int. 2020, 98, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Körner, A.; Eklöf, A.-C.; Celsi, G.; Aperia, A. Increased renal metabolism in diabetes: Mechanism and functional implications. Diabetes 1994, 43, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Palm, F.; Cederberg, J.; Hansell, P.; Liss, P.; Carlsson, P.-O. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia 2003, 46, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Vrbjar, N.; Jasenovec, T.; Kollarova, M.; Snurikova, D.; Chomova, M.; Radosinska, D.; Shawkatova, I.; Tothova, L.; Radosinska, J. Na, K-ATPase Kinetics and Oxidative Stress in Kidneys of Zucker Diabetic Fatty (Fa/Fa) Rats Depending on the Diabetes Severity—Comparison with Lean (Fa/+) and Wistar Rats. Biology 2022, 11, 1519. [Google Scholar] [CrossRef] [PubMed]
- Franzén, S.; Pihl, L.; Khan, N.; Gustafsson, H.; Palm, F. Pronounced kidney hypoxia precedes albuminuria in type 1 diabetic mice. Am. J. Physiol. Renal Physiol. 2016, 310, F807–F809. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, E.A.; Li, L.-P.; Ji, L.; Prasad, P.V. Early changes with diabetes in renal medullary hemodynamics as evaluated by fiberoptic probes and BOLD magnetic resonance imaging. Investig. Radiol. 2007, 42, 157. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Basseau, F.; Tyndal, B.; Jones, R.; Deminière, C.; Catargi, B.; Combe, C.; Moonen, C.W.; Grenier, N. Renal diffusion and BOLD MRI in experimental diabetic nephropathy. J. Magn. Reson. Imaging 2003, 17, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.-J.; Liu, F.; Li, X.-M.; Yang, L.; Zhao, S.; Huang, Z.-X.; Huang, Y.-Q.; Liu, R.-B. Noninvasive evaluation of renal oxygenation in diabetic nephropathy by BOLD-MRI. Eur. J. Radiol. 2012, 81, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Nangaku, M. The suffocating kidney: Tubulointerstitial hypoxia in end-stage renal disease. Nat. Rev. Nephrol. 2010, 6, 667–678. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef]
- Lovshin, J.A.; Boulet, G.; Lytvyn, Y.; Lovblom, L.E.; Bjornstad, P.; Farooqi, M.A.; Lai, V.; Cham, L.; Tse, J.; Orszag, A.; et al. Renin-angiotensin-aldosterone system activation in long-standing type 1 diabetes. JCI Insight 2018, 3, e96968. [Google Scholar] [CrossRef] [PubMed]
- Layton, A.T.; Vallon, V.; Edwards, A. Predicted consequences of diabetes and SGLT inhibition on transport and oxygen consumption along a rat nephron. Am. J. Physiol. Renal Physiol. 2016, 310, F1269–F1283. [Google Scholar] [CrossRef] [PubMed]
- Ooi, Q.L.; Tow, F.K.N.-F.H.; Deva, R.; Alias, M.A.; Kawasaki, R.; Wong, T.Y.; Mohamad, N.; Colville, D.; Hutchinson, A.; Savige, J. The microvasculature in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 1872. [Google Scholar] [CrossRef] [PubMed]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Murea, M.; Freedman, B.I.; Parks, J.S.; Antinozzi, P.A.; Elbein, S.C.; Ma, L. Lipotoxicity in diabetic nephropathy: The potential role of fatty acid oxidation. Clin. J. Am. Soc. Nephrol. 2010, 5, 2373–2379. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Satriano, J.; Thomas, J.L.; Miyamoto, S.; Sharma, K.; Pastor-Soler, N.M.; Hallows, K.R.; Singh, P. Interactions between HIF-1α and AMPK in the regulation of cellular hypoxia adaptation in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2015, 309, F414–F428. [Google Scholar] [CrossRef] [PubMed]
- Lindblom, R.; Higgins, G.; Coughlan, M.; de Haan, J.B. Targeting mitochondria and reactive oxygen species-driven pathogenesis in diabetic nephropathy. Rev. Diabet. Stud. 2015, 12, 134. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and kidney disease: Role of oxidative stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 8416743. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Adam-Vizi, V. Production of reactive oxygen species in brain mitochondria: Contribution by electron transport chain and non–electron transport chain sources. Antioxid. Redox Signal. 2005, 7, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Palm, F.; Nangaku, M.; Fasching, A.; Tanaka, T.; Nordquist, L.; Hansell, P.; Kawakami, T.; Nishijima, F.; Fujita, T. Uremia induces abnormal oxygen consumption in tubules and aggravates chronic hypoxia of the kidney via oxidative stress. Am. J. Physiol. Renal Physiol. 2010, 299, F380–F386. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.; Kim, K.-W. Hypoxia-inducible factor (HIF-1) α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Gunaratnam, L.; Bonventre, J.V. HIF in kidney disease and development. J. Am. Soc. Nephrol. 2009, 20, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nangaku, M. Angiogenesis and hypoxia in the kidney. Nat. Rev. Nephrol. 2013, 9, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Hypoxic regulation of erythropoiesis and iron metabolism. Am. J. Physiol. Renal Physiol. 2010, 299, F1–F13. [Google Scholar] [CrossRef] [PubMed]
- Foresto-Neto, O.; da Silva, A.R.P.A.; Cipelli, M.; Santana-Novelli, F.P.R.; Camara, N.O.S. The impact of hypoxia-inducible factors in the pathogenesis of kidney diseases: A link through cell metabolism. Kidney Res. Clin. Pract. 2023, 42, 561–578. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef]
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta-Rev. Cancer. 2018, 1870, 15–22. [Google Scholar] [CrossRef]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.-w.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.-Y.; Wang, X.; Jiang, C. HIF1α-induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediat. Inflamm. 2017, 2017, 9029327. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.-H.; Barbi, J.; Pan, F. Hypoxia-inducible factors in T lymphocyte differentiation and function. A review in the theme: Cellular responses to hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C580–C589. [Google Scholar] [CrossRef] [PubMed]
- Liikanen, I.; Lauhan, C.; Quon, S.; Omilusik, K.; Phan, A.T.; Bartrolí, L.B.; Ferry, A.; Goulding, J.; Chen, J.; Scott-Browne, J.P. Hypoxia-inducible factor activity promotes antitumor effector function and tissue residency by CD8+ T cells. J. Clin. Investig. 2021, 131, e143729. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A. Molecular interactions between reactive oxygen species and autophagy in kidney disease. Int. J. Mol. Sci. 2019, 20, 3791. [Google Scholar] [CrossRef]
- Satoh, M.; Fujimoto, S.; Haruna, Y.; Arakawa, S.; Horike, H.; Komai, N.; Sasaki, T.; Tsujioka, K.; Makino, H.; Kashihara, N. NAD (P) H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2005, 288, F1144–F1152. [Google Scholar] [CrossRef]
- Kunsch, C.; Medford, R.M. Oxidative stress as a regulator of gene expression in the vasculature. Circ. Res. 1999, 85, 753–766. [Google Scholar] [CrossRef]
- Satoh, M.; Fujimoto, S.; Arakawa, S.; Yada, T.; Namikoshi, T.; Haruna, Y.; Horike, H.; Sasaki, T.; Kashihara, N. Angiotensin II type 1 receptor blocker ameliorates uncoupled endothelial nitric oxide synthase in rats with experimental diabetic nephropathy. Nephrol. Dial. Transplant. 2008, 23, 3806–3813. [Google Scholar] [CrossRef]
- Sakashita, M.; Tanaka, T.; Inagi, R. Metabolic changes and oxidative stress in diabetic kidney disease. Antioxidants 2021, 10, 1143. [Google Scholar] [CrossRef] [PubMed]
- Nezu, M.; Suzuki, N. Roles of Nrf2 in protecting the kidney from oxidative damage. Int. J. Mol. Sci. 2020, 21, 2951. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Nezu, M.; Suzuki, N.; Yamamoto, M. Targeting the KEAP1-NRF2 system to prevent kidney disease progression. Am. J. Nephrol. 2017, 45, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Daitoku, H.; Fukamizu, A. FOXO transcription factors in the regulatory networks of longevity. J. Biochem. 2007, 141, 769–774. [Google Scholar] [CrossRef]
- Putker, M.; Madl, T.; Vos, H.R.; de Ruiter, H.; Visscher, M.; van den Berg, M.C.; Kaplan, M.; Korswagen, H.C.; Boelens, R.; Vermeulen, M.; et al. Redox-dependent control of FOXO/DAF-16 by transportin-1. Mol. Cell 2013, 49, 730–742. [Google Scholar] [CrossRef]
- Kim, M.; Lim, J.; Youn, H.; Hong, Y.; Yang, K.; Park, H.; Chung, S.; Koh, S.; Shin, S.; Choi, B.; et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK–SIRT1–PGC1α axis in db/db mice. Diabetologia 2013, 56, 204–217. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Y.; Ma, X.; Zhang, N.; Qin, G. The effect of resveratrol on FoxO1 expression in kidneys of diabetic nephropathy rats. Mol. Biol. Rep. 2012, 39, 9085–9093. [Google Scholar] [CrossRef]
- Wang, Y.; He, W. Improving the dysregulation of FoxO1 activity is a potential therapy for alleviating diabetic kidney disease. Front. Pharmacol. 2021, 12, 630617. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.-S.; Yu, S.; Kong, A.-N. Activation of Nrf2-antioxidant signaling attenuates NFκB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Cai, J.; Yin, X.-M.; Weinberg, J.M.; Venkatachalam, M.A.; Dong, Z. Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhu, X.; Yang, S.; Liu, F.; Zhou, Z.; Zhan, M.; Xie, P.; Zhang, D.; Li, J.; Song, P. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes 2014, 63, 1366–1380. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304. [Google Scholar] [CrossRef] [PubMed]
- Ayanga, B.A.; Badal, S.S.; Wang, Y.; Galvan, D.L.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Dynamin–related protein 1 deficiency improves mitochondrial fitness and protects against progression of diabetic nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2733. [Google Scholar] [CrossRef]
- Qin, X.; Zhao, Y.; Gong, J.; Huang, W.; Su, H.; Yuan, F.; Fang, K.; Wang, D.; Li, J.; Zou, X. Berberine protects glomerular podocytes via inhibiting Drp1-mediated mitochondrial fission and dysfunction. Theranostics 2019, 9, 1698. [Google Scholar] [CrossRef]
- Chen, K.; Dai, H.; Yuan, J.; Chen, J.; Lin, L.; Zhang, W.; Wang, L.; Zhang, J.; Li, K.; He, Y. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 2018, 9, 105. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Liu, G.; Sun, Y.; Li, Z.; Song, T.; Wang, H.; Zhang, Y.; Ge, Z. Apoptosis induced by endoplasmic reticulum stress involved in diabetic kidney disease. Biochem. Biophys. Res. 2008, 370, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Plaisance, V.; Brajkovic, S.; Tenenbaum, M.; Favre, D.; Ezanno, H.; Bonnefond, A.; Bonner, C.; Gmyr, V.; Kerr-Conte, J.; Gauthier, B.R. Endoplasmic reticulum stress links oxidative stress to impaired pancreatic beta-cell function caused by human oxidized LDL. PLoS ONE 2016, 11, e0163046. [Google Scholar] [CrossRef] [PubMed]
- van der Vlies, D.; Makkinje, M.; Jansens, A.; Braakman, I.; Verkleij, A.J.; Wirtz, K.W.; Post, J.A. Oxidation of ER resident proteins upon oxidative stress: Effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid. Redox Signal. 2003, 5, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Hasanain, M.; Bhattacharjee, A.; Pandey, P.; Ashraf, R.; Singh, N.; Sharma, S.; Vishwakarma, A.; Datta, D.; Mitra, K.; Sarkar, J. α-Solanine induces ROS-mediated autophagy through activation of endoplasmic reticulum stress and inhibition of Akt/mTOR pathway. Cell Death Dis. 2015, 6, e1860. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Kishi, S.; Nagasu, H.; Kidokoro, K.; Kashihara, N. Oxidative stress and the role of redox signalling in chronic kidney disease. Nat. Rev. Nephrol. 2023, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Tamura, S.; Honsho, M.; Yada, H.; Yagita, Y.; Kosako, H.; Fujiki, Y. Mitotic phosphorylation of Pex14p regulates peroxisomal import machinery. J. Cell Biol. 2020, 219, e202001003. [Google Scholar] [CrossRef]
- van Vliet, A.R.; Agostinis, P. Mitochondria-associated membranes and ER stress. In Coordinating Organismal Physiology Through the Unfolded Protein Response; Springer: Berlin/Heidelberg, Germany, 2018; Volume 414, pp. 73–102. [Google Scholar]
- Janikiewicz, J.; Szymański, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszyński, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef]
- Yang, M.; Zhao, L.; Gao, P.; Zhu, X.; Han, Y.; Chen, X.; Li, L.; Xiao, Y.; Wei, L.; Li, C. DsbA-L ameliorates high glucose induced tubular damage through maintaining MAM integrity. EBioMedicine 2019, 43, 607–619. [Google Scholar] [CrossRef]
- Wanner, C.; Tonelli, M. Kidney Disease: Improving Global Outcomes Lipid Guideline Development Work Group Members. KDIGO clinical practice guideline for lipid management in CKD: Summary of recommendation statements and clinical approach to the patient. Kidney Int. 2014, 85, 1303–1309. [Google Scholar] [CrossRef]
- Sandhu, S.; Wiebe, N.; Fried, L.F.; Tonelli, M. Statins for improving renal outcomes: A meta-analysis. J. Am. Soc. Nephrol. 2006, 17, 2006–2016. [Google Scholar] [CrossRef] [PubMed]
- Robins, S.J.; Collins, D.; Wittes, J.T.; Papademetriou, V.; Deedwania, P.C.; Schaefer, E.J.; McNamara, J.R.; Kashyap, M.L.; Hershman, J.M.; Wexler, L.F. Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: A randomized controlled trial. JAMA 2001, 285, 1585–1591. [Google Scholar] [CrossRef] [PubMed]
- Imai, E.; Imai, A. Effect of Pemafibrate on Serum Creatinine in Patients with Chronic Kidney Disease. JMA J. 2022, 5, 328–333. [Google Scholar] [PubMed]
- Hadjivasilis, A.; Kouis, P.; Kousios, A.; Panayiotou, A. the effect of fibrates on kidney function and chronic kidney disease progression: A systematic review and meta-analysis of randomised studies. J. Clin. Med. 2022, 11, 768. [Google Scholar] [CrossRef] [PubMed]
- Aomura, D.; Harada, M.; Yamada, Y.; Nakajima, T.; Hashimoto, K.; Tanaka, N.; Kamijo, Y. Pemafibrate protects against fatty acid-induced nephropathy by maintaining renal fatty acid metabolism. Metabolites 2021, 11, 372. [Google Scholar] [CrossRef] [PubMed]
- Schlackow, I.; Kent, S.; Herrington, W.; Emberson, J.; Haynes, R.; Reith, C.; Collins, R.; Landray, M.J.; Gray, A.; Baigent, C.; et al. Cost-effectiveness of lipid lowering with statins and ezetimibe in chronic kidney disease. Kidney Int. 2019, 96, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, N.S.; Pedersen, R.P.; Vestergaard, M.B.; Lindberg, U.; Andersen, U.B.; Haddock, B.; Hansen, T.W.; Fornoni, A.; Larsson, H.B.W.; Rossing, P. Evaluation of the effects of ezetimibe on albuminuria and kidney fat in individuals with type 2 diabetes and chronic kidney disease. Diabetes Obes. Metab. 2023, 25, 2605–2615. [Google Scholar] [CrossRef]
- Toth, P.P.; Dwyer, J.P.; Cannon, C.P.; Colhoun, H.M.; Rader, D.J.; Upadhyay, A.; Louie, M.J.; Koren, A.; Letierce, A.; Mandel, J.; et al. Efficacy and safety of lipid lowering by alirocumab in chronic kidney disease. Kidney Int. 2018, 93, 1397–1408. [Google Scholar] [CrossRef]
- Charytan, D.M.; Sabatine, M.S.; Pedersen, T.R.; Im, K.; Park, J.-G.; Pineda, A.L.; Wasserman, S.M.; Deedwania, P.; Olsson, A.G.; Sever, P.S.; et al. Efficacy and safety of evolocumab in chronic kidney disease in the FOURIER trial. J. Am. Coll. Cardiol. 2019, 73, 2961–2970. [Google Scholar] [CrossRef]
- Byun, J.H.; Lebeau, P.F.; Platko, K.; Carlisle, R.E.; Faiyaz, M.; Chen, J.; MacDonald, M.E.; Makda, Y.; Yousof, T.; Lynn, E.G. Inhibitory antibodies against PCSK9 reduce surface CD36 and mitigate diet-induced renal lipotoxicity. Kidney360 2022, 3, 1394. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Y.; Zhang, J.; Zhang, R.; Wang, Y.; Liu, F. ABCA1 deficiency-mediated glomerular cholesterol accumulation exacerbates glomerular endothelial injury and dysfunction in diabetic kidney disease. Metabolism 2023, 139, 155377. [Google Scholar] [CrossRef] [PubMed]
- Pagtalunan, M.E.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Luo, Y.; Wang, X.; Orlicky, D.J.; Myakala, K.; Yang, P.; Levi, M. The sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents renal and liver disease in western diet induced obesity mice. Int. J. Mol. Sci. 2018, 19, 137. [Google Scholar] [CrossRef] [PubMed]
- Shibusawa, R.; Yamada, E.; Okada, S.; Nakajima, Y.; Bastie, C.C.; Maeshima, A.; Kaira, K.; Yamada, M. Dapagliflozin rescues endoplasmic reticulum stress-mediated cell death. Sci. Rep. 2019, 9, 9887. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Li, J.; Takagaki, Y.; Kitada, M.; Nitta, K.; Takasu, T.; Kanasaki, K.; Koya, D. Ipragliflozin improves mitochondrial abnormalities in renal tubules induced by a high-fat diet. J. Diabetes Investig. 2018, 9, 1025–1032. [Google Scholar] [CrossRef]
- Wei, D.; Liao, L.; Wang, H.; Zhang, W.; Wang, T.; Xu, Z. Canagliflozin ameliorates obesity by improving mitochondrial function and fatty acid oxidation via PPARα in vivo and in vitro. Life Sci. 2020, 247, 117414. [Google Scholar] [CrossRef]
- van Bommel, E.J.; Muskiet, M.H.; Tonneijck, L.; Kramer, M.H.; Nieuwdorp, M.; van Raalte, D.H. SGLT2 inhibition in the diabetic kidney—From mechanisms to clinical outcome. Clin. J. Am. Soc. Nephrol. 2017, 12, 700. [Google Scholar] [CrossRef]
- Vallon, V.; Richter, K.; Blantz, R.C.; Thomson, S.; Osswald, H. Glomerular hyperfiltration in experimental diabetes mellitus: Potential role of tubular reabsorption. J. Am. Soc. Nephrol. 1999, 10, 2569–2576. [Google Scholar] [CrossRef]
- Layton, A.T.; Vallon, V.; Edwards, A. Modeling oxygen consumption in the proximal tubule: Effects of NHE and SGLT2 inhibition. Am. J. Physiol. Renal Physiol. 2015, 308, F1343–F1357. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J.; Fasching, A.; Pihl, L.; Patinha, D.; Franzén, S.; Palm, F. Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol. Renal Physiol. 2015, 309, F227–F234. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Sugiura, Y.; Saito, H.; Sugahara, M.; Higashijima, Y.; Yamaguchi, J.; Inagi, R.; Suematsu, M.; Nangaku, M.; Tanaka, T. Sodium–glucose cotransporter 2 inhibition normalizes glucose metabolism and suppresses oxidative stress in the kidneys of diabetic mice. Kidney Int. 2018, 94, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Bessho, R.; Takiyama, Y.; Takiyama, T.; Kitsunai, H.; Takeda, Y.; Sakagami, H.; Ota, T. Hypoxia-inducible factor-1α is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci. Rep. 2019, 9, 14754. [Google Scholar] [CrossRef] [PubMed]
- Rojano Toimil, A.; Ciudin, A. GLP-1 receptor agonists in diabetic kidney disease: From physiology to clinical outcomes. J. Clin. Med. 2021, 10, 3955. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Su, K.; Yi, B.; Yao, B.-q.; Xia, T.; Yang, Y.-f.; Zhang, Z.-h.; Chen, C. Liraglutide attenuates renal tubular ectopic lipid deposition in rats with diabetic nephropathy by inhibiting lipid synthesis and promoting lipolysis. Pharmacol. Res. 2020, 156, 104778. [Google Scholar] [CrossRef]
- Yin, Q.-H.; Zhang, R.; Li, L.; Wang, Y.-T.; Liu, J.-P.; Zhang, J.; Bai, L.; Cheng, J.-Q.; Fu, P.; Liu, F. Exendin-4 ameliorates lipotoxicity-induced glomerular endothelial cell injury by improving ABC transporter A1-mediated cholesterol efflux in diabetic apoE knockout mice. J. Biol. Chem. 2016, 291, 26487–26501. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T.; Yore, M.M.; Fu, L.; Lopchuk, J.M.; Gribble, G.W. New synthetic triterpenoids: Potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J. Nat. Prod. 2011, 74, 537–545. [Google Scholar] [CrossRef]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef]
- Pergola, P.E.; Krauth, M.; Huff, J.W.; Ferguson, D.A.; Ruiz, S.; Meyer, C.J.; Warnock, D.G. Effect of bardoxolone methyl on kidney function in patients with T2D and stage 3b–4 CKD. Am. J. Nephrol. 2011, 33, 469–476. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Reisman, S.A.; Bakris, G.L.; O’Grady, M.; Linde, P.G.; McCullough, P.A.; Packham, D.; Vaziri, N.D.; Ward, K.W.; Warnock, D.G.; et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 2014, 39, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Wrolstad, D.; Bakris, G.L.; Chertow, G.M.; de Zeeuw, D.; Goldsberry, A.; Linde, P.G.; McCullough, P.A.; McMurray, J.J.; Wittes, J.; et al. Risk factors for heart failure in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. J. Card. Fail. 2014, 20, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Kanda, H.; Takama, H.; Ichikawa, T.; Hase, H.; Akizawa, T. Randomized clinical trial on the effect of bardoxolone methyl on GFR in diabetic kidney disease patients (TSUBAKI study). Kidney Int. Rep. 2020, 5, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Yamawaki, K. Bardoxolone methyl: Drug development for diabetic kidney disease. Clin. Exp. Nephrol. 2020, 24, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Al-Waili, N.; Al-Waili, H.; Al-Waili, T.; Salom, K. Natural antioxidants in the treatment and prevention of diabetic nephropathy; a potential approach that warrants clinical trials. Redox Rep. 2017, 22, 99–118. [Google Scholar] [CrossRef]
- Jie, Z.; Chao, M.; Jun, A.; Wei, S.; LiFeng, M. Effect of curcumin on diabetic kidney disease: A systematic review and meta-analysis of randomized, double-blind, placebo-controlled clinical trials. Evid. Based Complement. Altern. Med. 2021, 2021, 6109406. [Google Scholar] [CrossRef]
- Park, J.-Y.; Sohn, H.-Y.; Koh, Y.H.; Jo, C. Curcumin activates Nrf2 through PKCδ-mediated p62 phosphorylation at Ser351. Sci. Rep. 2021, 11, 8430. [Google Scholar] [CrossRef]
- Rahban, M.; Habibi-Rezaei, M.; Mazaheri, M.; Saso, L.; Moosavi-Movahedi, A.A. Anti-viral potential and modulation of Nrf2 by curcumin: Pharmacological implications. Antioxidants 2020, 9, 1228. [Google Scholar] [CrossRef]
- Li, C.; Miao, X.; Wang, S.; Adhikari, B.K.; Wang, X.; Sun, J.; Liu, Q.; Tong, Q.; Wang, Y. Novel curcumin C66 that protects diabetes-induced aortic damage was associated with suppressing JNK2 and upregulating Nrf2 expression and function. Oxid. Med. Cell. Longev. 2018, 2018, 5783239. [Google Scholar] [CrossRef]
- Wu, H.; Kong, L.; Tan, Y.; Epstein, P.N.; Zeng, J.; Gu, J.; Liang, G.; Kong, M.; Chen, X.; Miao, L. C66 ameliorates diabetic nephropathy in mice by both upregulating NRF2 function via increase in miR-200a and inhibiting miR-21. Diabetologia 2016, 59, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Janczewski, Ł. Sulforaphane and its bifunctional analogs: Synthesis and biological activity. Molecules 2022, 27, 1750. [Google Scholar] [CrossRef]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and other nutrigenomic Nrf2 activators: Can the clinician’s expectation be matched by the reality? Oxid. Med. Cell. Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Kong, L.; Cheng, Y.; Zhang, Z.; Wang, Y.; Luo, M.; Tan, Y.; Chen, X.; Miao, L.; Cai, L. Metallothionein plays a prominent role in the prevention of diabetic nephropathy by sulforaphane via up-regulation of Nrf2. Free Radic. Biol. Med. 2015, 89, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Tang, X.; Gao, P.; Guo, F.; Liu, H.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N.; Li, H. Sulforaphane attenuation of experimental diabetic nephropathy involves GSK-3 beta/Fyn/Nrf2 signaling pathway. J. Nutr. Biochem. 2015, 26, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Liebman, S.E.; Le, T.H. Eat your broccoli: Oxidative stress, NRF2, and sulforaphane in chronic kidney disease. Nutrients 2021, 13, 266. [Google Scholar] [CrossRef] [PubMed]
- Nyambuya, T.M.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Mxinwa, V.; Mokgalaboni, K.; Orlando, P.; Silvestri, S.; Louw, J.; Tiano, L.; Dludla, P.V. A meta-analysis of the impact of resveratrol supplementation on markers of renal function and blood pressure in type 2 diabetic patients on hypoglycemic therapy. Molecules 2020, 25, 5645. [Google Scholar]
- Truong, V.L.; Jun, M.; Jeong, W.S. Role of resveratrol in regulation of cellular defense systems against oxidative stress. BioFactors. 2018, 44, 36–49. [Google Scholar] [CrossRef]
- Salami, M.; Salami, R.; Mafi, A.; Aarabi, M.-H.; Vakili, O.; Asemi, Z. Therapeutic potential of resveratrol in diabetic nephropathy according to molecular signaling. Curr. Mol. Pharmacol. 2022, 15, 716–735. [Google Scholar] [CrossRef]
- Gu, W.; Wang, X.; Zhao, H.; Geng, J.; Li, X.; Zheng, K.; Guan, Y.; Hou, X.; Wang, C.; Song, G. Resveratrol ameliorates diabetic kidney injury by reducing lipotoxicity and modulates expression of components of the junctional adhesion molecule-like/sirtuin 1 lipid metabolism pathway. Eur. J. Pharmacol. 2022, 918, 174776. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Lim, J.H.; Kim, M.Y.; Kim, Y.; Hong, Y.A.; Choi, S.R.; Chung, S.; Kim, H.W.; Choi, B.S.; Kim, Y.S.; et al. Resveratrol increases AdipoR1 and AdipoR2 expression in type 2 diabetic nephropathy. J. Transl. Med. 2016, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Said, M.A. Vitamin D attenuates endothelial dysfunction in streptozotocin induced diabetic rats by reducing oxidative stress. Arch. Physiol. Biochem. 2022, 128, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lin, L.; Xu, C.; Chai, D.; Peng, F.; Lin, J. VDR agonist prevents diabetic endothelial dysfunction through inhibition of prolyl isomerase-1-mediated mitochondrial oxidative stress and inflammation. Oxid. Med. Cell. Longev. 2018, 2018, 1714896. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, E.M.; Saadulla, L.; Reeves, W.B.; Awad, A.S. Therapeutic modalities in diabetic nephropathy: Standard and emerging approaches. J. Gen. Intern. 2012, 27, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Lin, T.-W.; Hong, Z.-X.; Lim, L.-M. Vitamin D and diabetic kidney disease. Int. J. Mol. Sci. 2023, 24, 3751. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, S.; Zhou, Q.; Zhang, H.; Yi, B. Effects of vitamin D supplementation on renal function, inflammation and glycemic control in patients with diabetic nephropathy: A systematic review and meta-analysis. Kidney Blood Press. Res. 2019, 44, 72–87. [Google Scholar] [CrossRef]
- Nguyen, T.M.D. Adiponectin: Role in physiology and pathophysiology. Int. J. Prev. Med. 2020, 11, 136. [Google Scholar] [CrossRef]
- Kim, Y.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Yoon, H.E.; Shin, S.J.; Choi, B.S.; Kim, Y.-S.; Chang, Y.S.; Park, C.W. The adiponectin receptor agonist AdipoRon ameliorates diabetic nephropathy in a model of type 2 diabetes. J. Am. Soc. Nephrol. 2018, 29, 1108. [Google Scholar] [CrossRef]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.-I.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Eckardt, K.-U. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat. Rev. Nephrol. 2016, 12, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.R.; Smith, M.T.; Maroni, B.J.; Zuraw, Q.C.; deGoma, E.M. Clinical trial of vadadustat in patients with anemia secondary to stage 3 or 4 chronic kidney disease. Am. J. Nephrol. 2017, 45, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Spinowitz, B.S.; Hartman, C.S.; Maroni, B.J.; Haase, V.H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016, 90, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Akizawa, T.; Tsubakihara, Y.; Nangaku, M.; Endo, Y.; Nakajima, H.; Kohno, T.; Imai, Y.; Kawase, N.; Hara, K.; Lepore, J.; et al. Effects of daprodustat, a novel hypoxia-inducible factor prolyl hydroxylase inhibitor on anemia management in Japanese hemodialysis subjects. Am. J. Nephrol. 2017, 45, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Holdstock, L.; Meadowcroft, A.M.; Maier, R.; Johnson, B.M.; Jones, D.; Rastogi, A.; Zeig, S.; Lepore, J.J.; Cobitz, A.R. Four-week studies of oral hypoxia-inducible factor–prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J. Am. Soc. Nephrol. 2016, 27, 1234. [Google Scholar] [CrossRef] [PubMed]
- Na, K. New Oral Agent for Treatment of Anemia in Patient with Chronic Kidney Disease: Prolyl Hydroxylase Inhibitor. Korean J. Med. 2019, 94, 11–16. [Google Scholar] [CrossRef]
- Hasegawa, S.; Tanaka, T.; Saito, T.; Fukui, K.; Wakashima, T.; Susaki, E.A.; Ueda, H.R.; Nangaku, M. The oral hypoxia-inducible factor prolyl hydroxylase inhibitor enarodustat counteracts alterations in renal energy metabolism in the early stages of diabetic kidney disease. Kidney Int. 2020, 97, 934–950. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Cernaro, V.; Gembillo, G.; Baggetta, R.; Buemi, M.; D’Arrigo, G. Antioxidant agents for delaying diabetic kidney disease progression: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0178699. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, D.; Zhang, Z.; Zhang, Y.; Zhang, Y.; Kang, X.; Jiang, L.; Tong, X.; Lian, F. Effects of antioxidants on diabetic kidney diseases: Mechanistic interpretations and clinical assessment. Chin. Med. 2023, 18, 3. [Google Scholar] [CrossRef]
- Vrbjar, N.; Vlkovicova, J.; Snurikova, D.; Kalocayova, B.; Zorad, S.; Culafic, T.; Tepavcevic, S.; Tothova, L.; Radosinska, D.; Kollarova, M.; et al. Alterations in Oxidative Stress Markers and Na, K-ATPase Enzyme Properties in Kidney after Fructose Intake and Quercetin Intervention in Rats. Life 2023, 13, 931. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chae, S.Y.; Kim, Y.; Park, C.W. Oxidative Stress Induced by Lipotoxicity and Renal Hypoxia in Diabetic Kidney Disease and Possible Therapeutic Interventions: Targeting the Lipid Metabolism and Hypoxia. Antioxidants 2023, 12, 2083. https://doi.org/10.3390/antiox12122083
Chae SY, Kim Y, Park CW. Oxidative Stress Induced by Lipotoxicity and Renal Hypoxia in Diabetic Kidney Disease and Possible Therapeutic Interventions: Targeting the Lipid Metabolism and Hypoxia. Antioxidants. 2023; 12(12):2083. https://doi.org/10.3390/antiox12122083
Chicago/Turabian StyleChae, Seung Yun, Yaeni Kim, and Cheol Whee Park. 2023. "Oxidative Stress Induced by Lipotoxicity and Renal Hypoxia in Diabetic Kidney Disease and Possible Therapeutic Interventions: Targeting the Lipid Metabolism and Hypoxia" Antioxidants 12, no. 12: 2083. https://doi.org/10.3390/antiox12122083