

Blue Light Damage and p53: Unravelling the Role of p53 in Oxidative-Stress-Induced Retinal Apoptosis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Organ Cultures
Isolation and Cultivation of Retinal Explants
2.2. Light Sources and Exposure Set Ups
Blue and White Light Exposure
2.3. Methods
2.3.1. Ex Vivo Optical Coherence Tomography (OCT)
2.3.2. Quantification of the Retinal Sections (OCT Measurements)
2.3.3. Cell Viability, ROS Assay, and Caspase 3/7 Activity Assay
2.3.4. Immunostainings
2.3.5. Western Blot
2.3.6. Inhibition of p53
2.3.7. Quantitative Real-Time PCR
2.3.8. TUNEL (TdT-Mediated dUTP-Biotin Nick end Labeling) Analysis
2.3.9. Statistical Analysis
2.3.10. Image Generation
3. Results
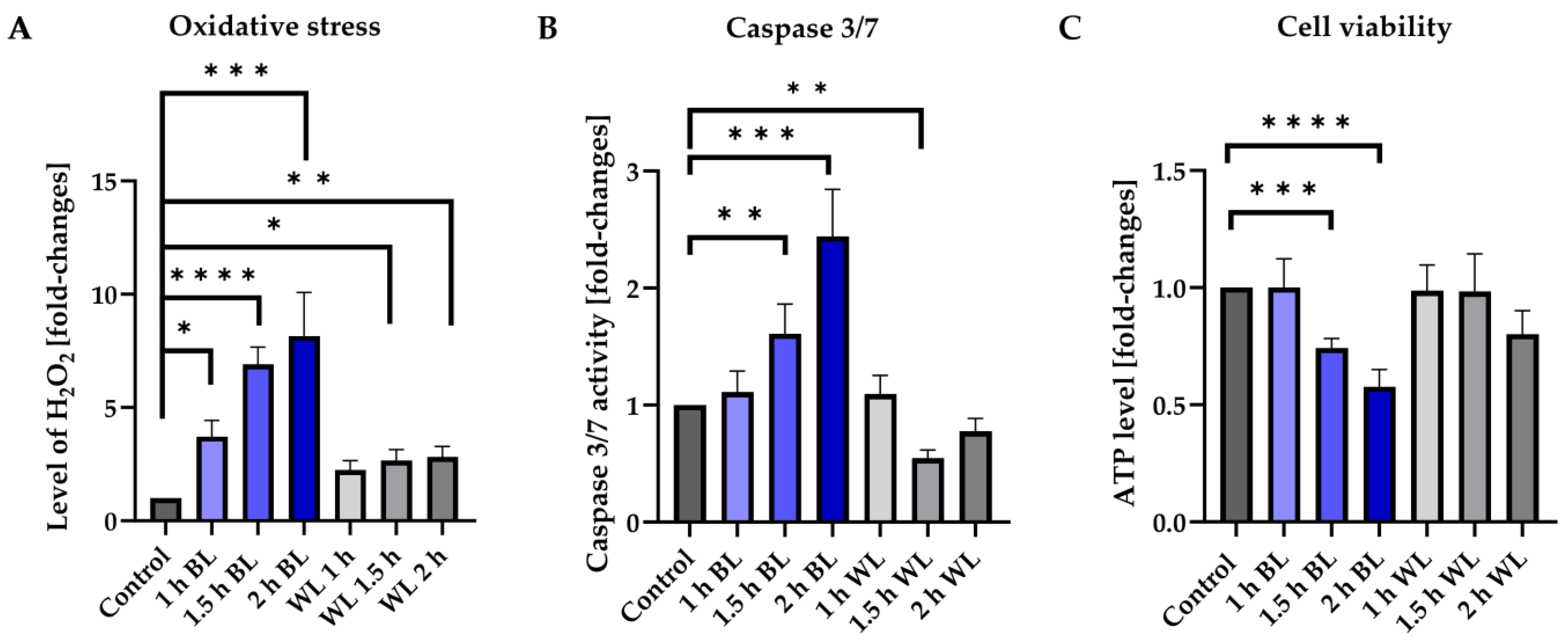
3.1. BL Is the Damaging Component of WL
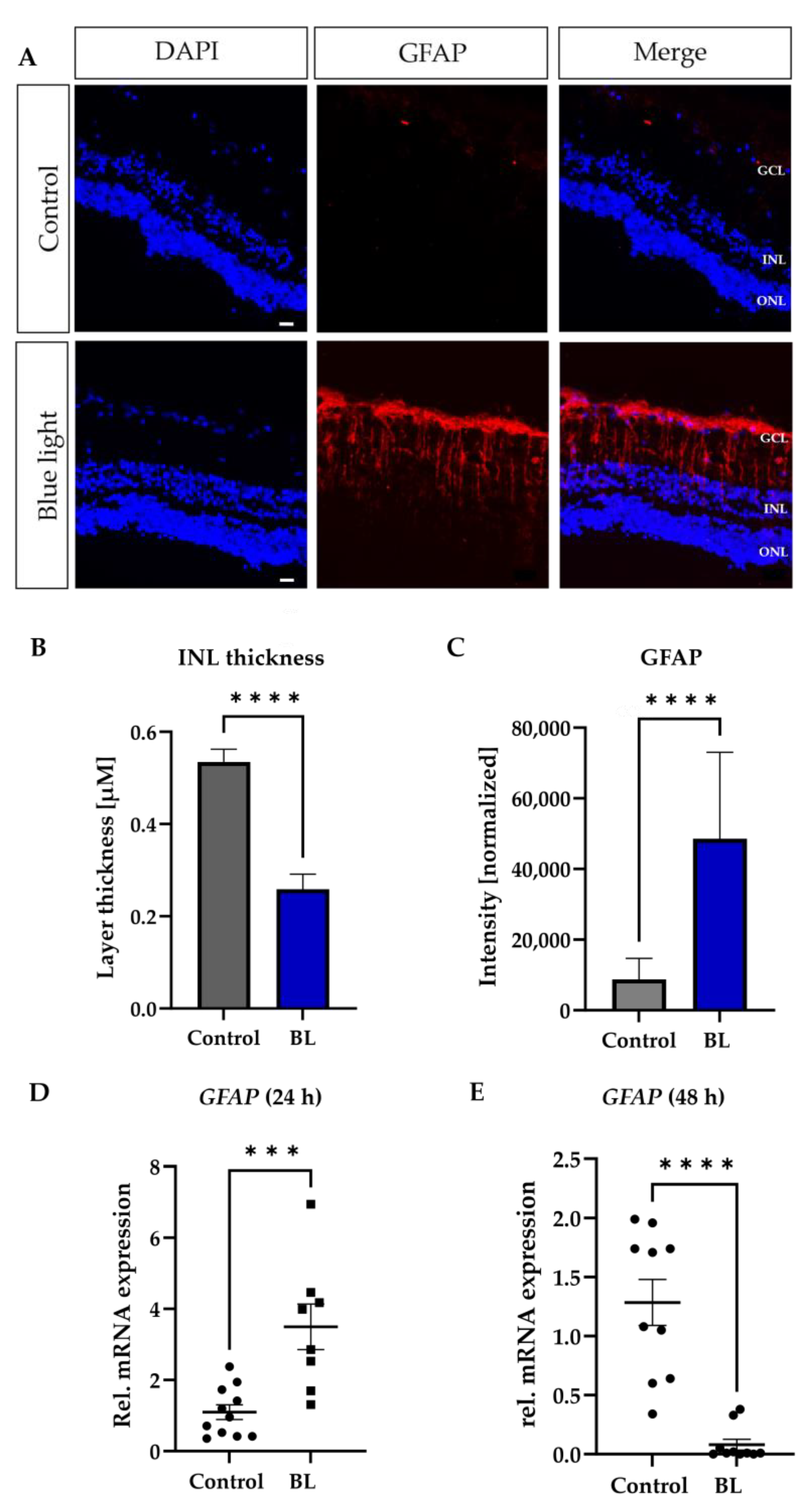
3.2. Müller Cells React to BL Exposure
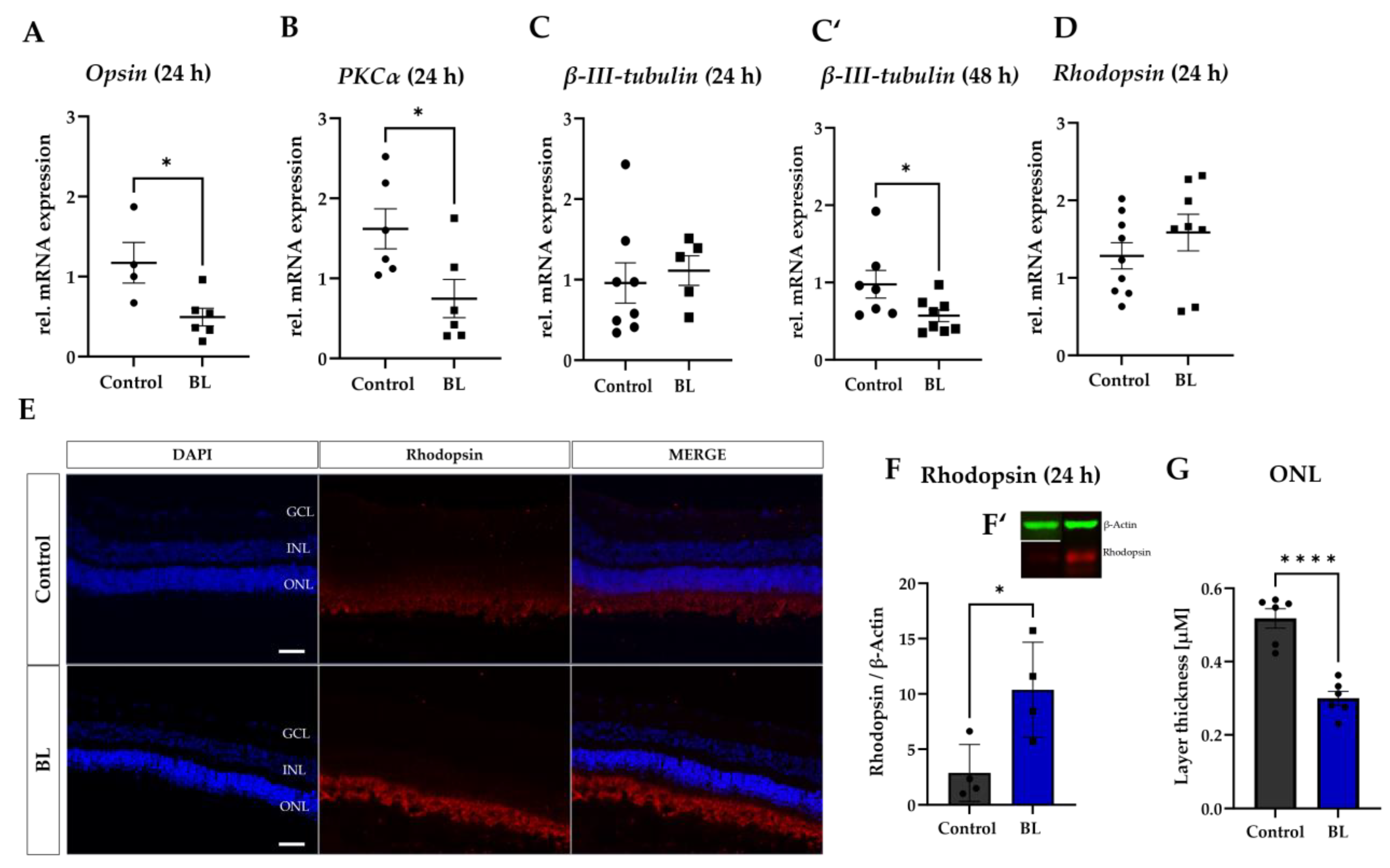
3.3. BL Damages Retinal Cells and Leads to Accumulation of Rhodopsin
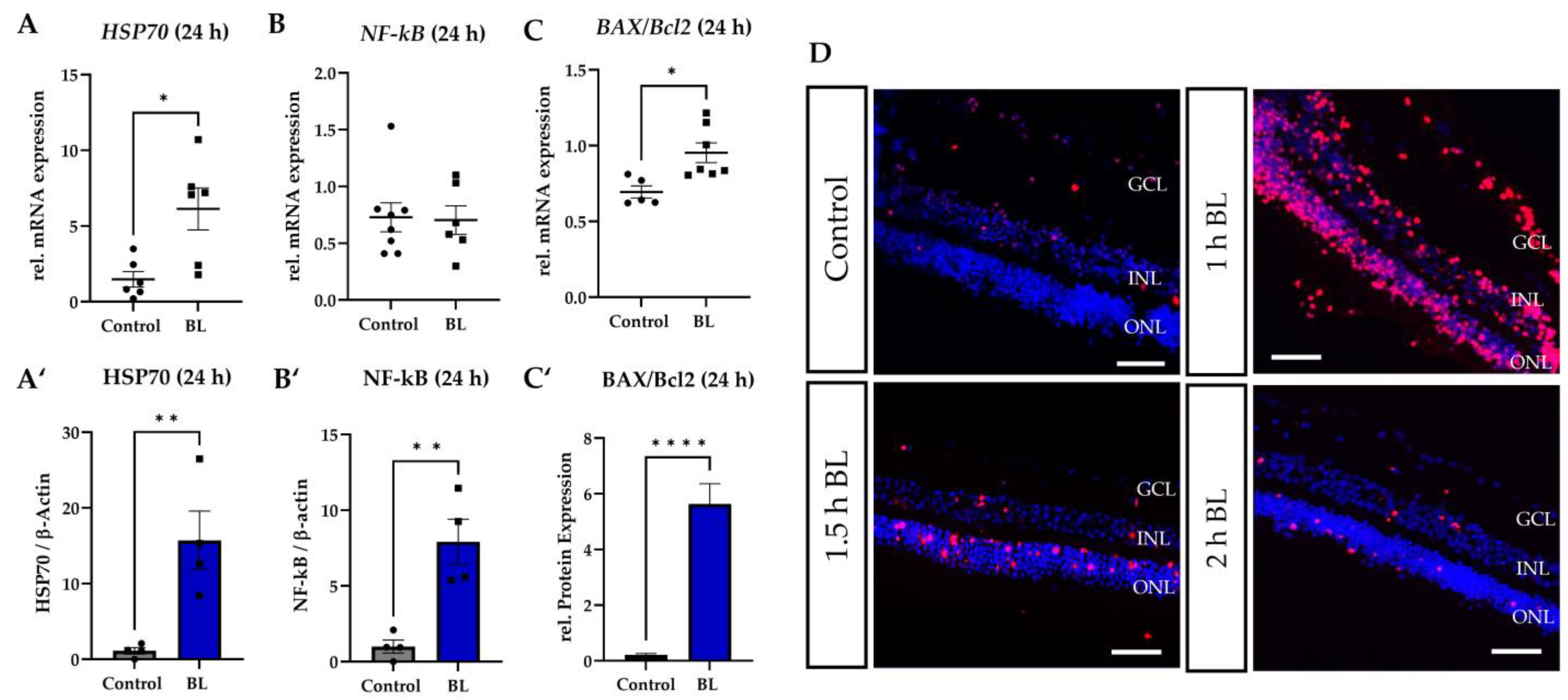
3.4. Apoptosis Induced by Blue Light Depends on the Exposure Duration
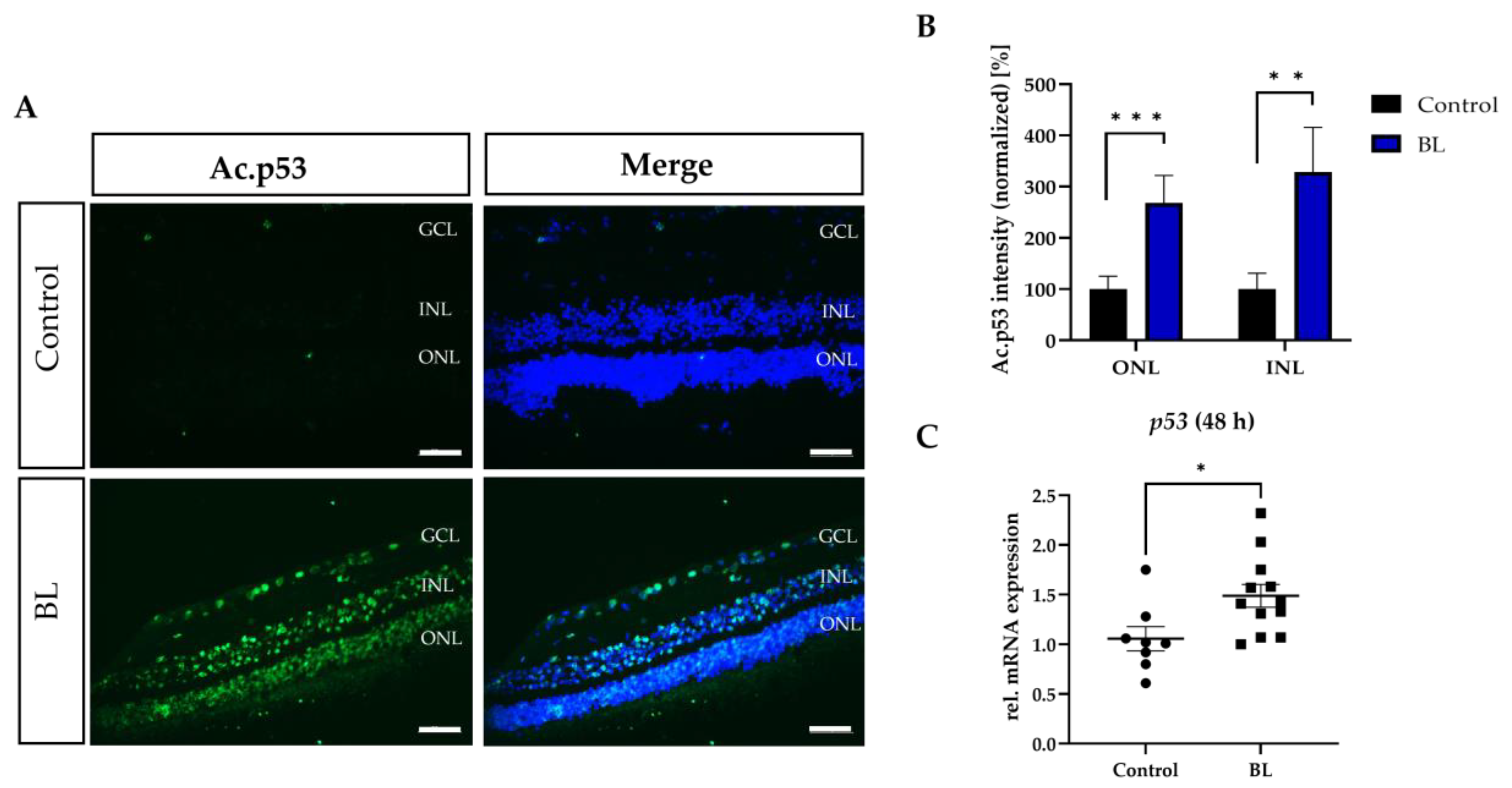
3.5. BL Induced the Expression of Modified p53
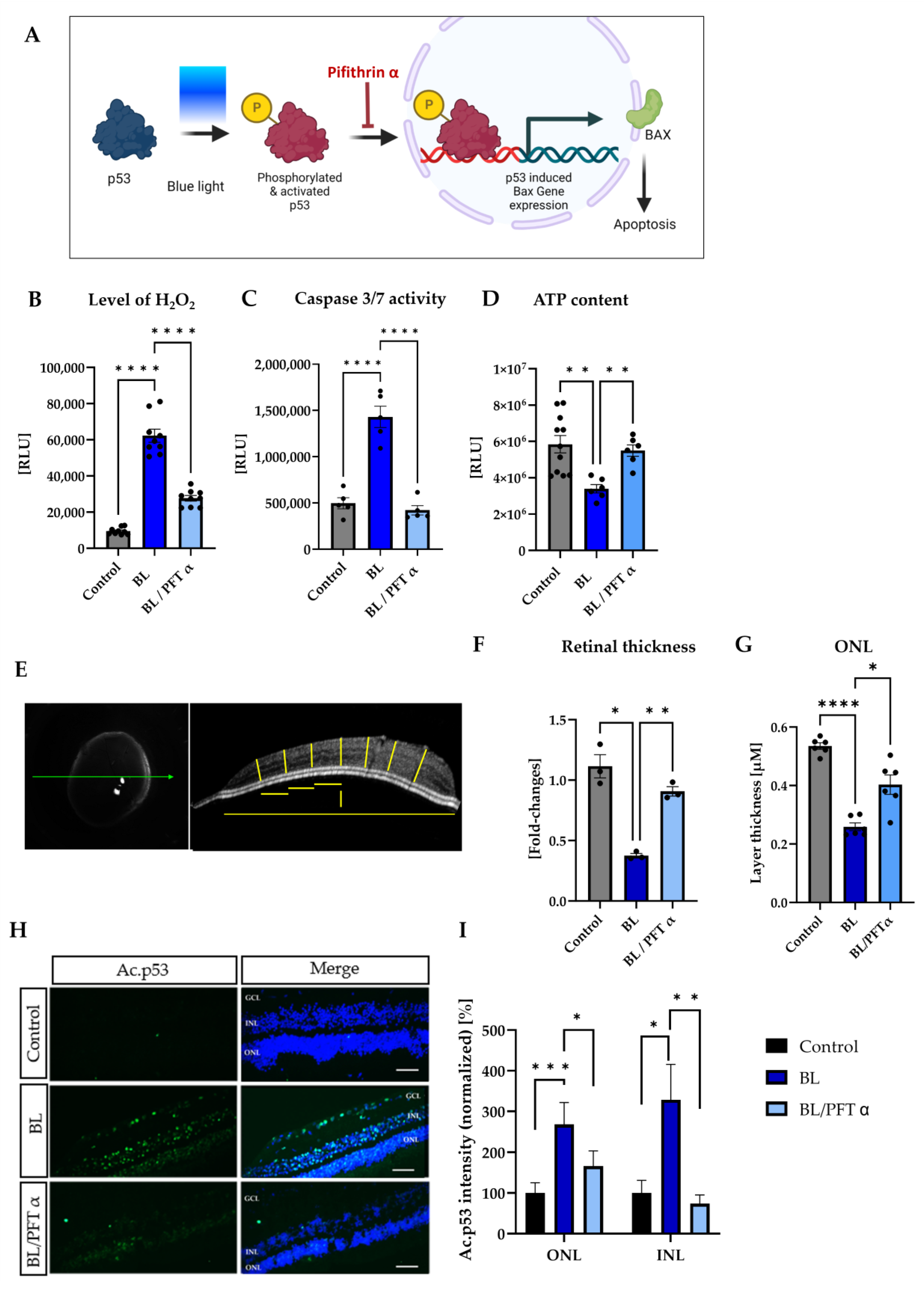
3.6. BL-Induced Retinal Damage Can Be Reduced by p53 Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsubota, K.; Inaba, T.; Dogru, M.; Matsumoto, Y.; Ishida, R.; Kaido, M.; Kojima, T.; Uchino, M.; Uchino, Y.; Ogawa, Y.; et al. The antiaging approach for the treatment of dry eye. Cornea 2012, 31, S3–S8. [Google Scholar] [CrossRef]
- Li, J.; O, W.; Li, W.; Jiang, Z.-G.; Ghanbari, H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4412. [Google Scholar] [CrossRef] [PubMed]
- Spector, A. Oxidative stress-induced cataract: Mechanism of action. FASEB J. 1995, 9, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 3, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Gritz, D.C.; Montes, C.; Atalla, L.R.; Wu, G.S.; Sevanian, A.; Rao, N.A. Histochemical localization of superoxide production in experimental autoimmune uveitis. Curr. Eye Res. 1991, 10, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Niesman, M.R.; Johnson, K.; Penn, J.S. Therapeutic effect of liposomal superoxide dismutase in an animal model of retinopathy of prematurity. Neurochem. Res. 1997, 22, 597–605. [Google Scholar] [CrossRef]
- Alio, J.L.; Ayala, M.; Mulet, M.E.; Artola, A.; Ruiz, J.M.; Bellot, J. Antioxidant therapy in the treatment of experimental acute corneal inflammation. Ophthalmic Res. 1995, 27, 136–143. [Google Scholar] [CrossRef]
- Jonas, J.B.; Songhomitra, P.-J. Updates on the epidemiology of age-related macular degeneration. Asia-Pac. J. Ophthalmol. 2017, 6, 493–497. [Google Scholar]
- Li, J.Q.; Welchowski, T.; Schmid, M.; Letow, J.; Wolpers, A.C.; Holz, F.G.; Finger, R.P. Retinal Diseases in Europe; EURETINA: Dublin, Ireland, 2017. [Google Scholar]
- Li, J.Q.; Welchowski, T.; Schmid, M.; Mauschitz, M.M.; Holz, F.G.; Finger, R.P. Prevalence and incidence of age-related macular degeneration in Europe: A systematic review and meta-analysis. Br. J. Ophthalmol. 2020, 104, 1077–1084. [Google Scholar] [CrossRef]
- Arnault, E.; Barrau, C.; Nanteau, C.; Gondouin, P.; Bigot, K.; Viénot, F.; Gutman, E.; Fontaine, V.; Villette, T.; Cohen-Tannoudji, D.; et al. Phototoxic action spectrum on a retinal pigment epithelium model of age-related macular degeneration exposed to sunlight normalized conditions. PLoS ONE 2013, 8, e71398. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Oveson, B.; Jo, Y.J.; Lauer, T.W.; Usui, S.; Komeima, K.; Xie, B.; Campochiaro, P.A. Increased expression of glutathione peroxidase 4 strongly protects retina from oxidative damage. Antioxid. Redox Signal. 2009, 11, 715–724. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Boulton, M. Consequences of oxidative stress in age-related macular degeneration. Mol. Aspects Med. 2012, 33, 399–417. [Google Scholar] [CrossRef]
- Masuda, T.; Shimazawa, M.; Hara, H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxid. Med. Cell Longev. 2017, 2017, 9208489. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Oliveira, C.R. Mitochondria as potential targets in antidiabetic therapy. In Handbook of Experimental Pharmacology; Michel, M.C., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 331–356. [Google Scholar]
- Sasaki, M.; Yuki, K.; Kurihara, T.; Miyake, S.; Noda, K.; Kobayashi, S.; Ishida, S.; Tsubota, K.; Ozawa, Y. Biological role of lutein in the light-induced retinal degeneration. J. Nutr. Biochem. 2012, 23, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y. Oxidative stress in the light-exposed retina and its implication in age-related macular degeneration. Redox Biol. 2020, 37, 101779. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy protects the retina from light-induced degeneration. J. Biol. Chem. 2013, 288, 7506–7518. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Ozawa, Y.; Osada, H.; Lin, J.B.; Toda, E.; Watanabe, M.; Yuki, K.; Kubota, S.; Apte, R.S.; Tsubota, K. Neuroprotective role of retinal SIRT3 against acute photo-stress. NPJ Aging Mech. Dis. 2017, 3, 19. [Google Scholar] [CrossRef]
- Pardhan, S. Eye Complications of Exposure to Blue-Violet Light; Points de Vue International Review of Ophthalmic Optics: Paris, France, 2016. [Google Scholar]
- Dunaief, J.L.; Dentchev, T.; Ying, G.S.; Milam, A.H. The role of apoptosis in age-related macular degeneration. Arch. Ophthalmol. 2002, 120, 1435–1442. [Google Scholar] [CrossRef]
- Loughman, J.; Davison, P.; Nolan, J.M.; Akkali, M.C.; Beatty, S. Macular pigment and its contribution to visual performance and experience. J. Optom. 2010, 3, 74–90. [Google Scholar] [CrossRef]
- Czepita, D.; Mojsa, A.; Ustianowska, M.; Czepita, M.; Lachowicz, E. Reading, writing, working on a computer or watching television, and myopia. Klin. Ocz. Acta Ophthalmol. Pol. 2010, 112, 293–295. [Google Scholar]
- Lou, L.; Arumugam, B.; Hung, L.-F.; Beach, K.M.; She, Z.; Smith, E.L.; Ostrin, L.A. Effects of narrowband light rearing on activity and the pupil in infant rhesus monkeys. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1338. [Google Scholar]
- Smick, K.; Villette, T.; Boulton, M.E.; Brainard, G.C.; Jones, W.; Karpecki, P.; Melton, R.; Thomas, R.; Sliney, D.H.; Shechtman, D.L. Blue Light Hazard: New Knowledge, New Approaches to Maintaining Ocular Health; Essilor of America: New York City, NY, USA, 2013. [Google Scholar]
- Nakamura, S.; Chichibu, S. Introduction to Nitride Semiconductor Blue Lasers and Light Emitting Diodes; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Peng, M.; Tsai, C.; Chien, C.; Hsiao, J.C.; Huang, S.; Lee, C.; Lin, H.; Wen, Y.; Tseng, K. The Influence of Low-powered Family LED Lighting on Eyes in Mice Experimental Model. Life Sci. J. 2012, 9, 477–482. [Google Scholar]
- Ham, W.T., Jr.; Mueller, H.A.; Sliney, D.H. Retinal sensitivity to damage from short wavelength light. Nature 1976, 260, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Klettner, A.; Roider, J. Comparison of bevacizumab, ranibizumab and pegaptanib in vitro: Efficiency and possible additional pathways. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4523–4527. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Nakanishi, K.; Parish, C.A. The Lipofuscin Fluorophore A2E Mediates Blue Light–Induced Damage to Retinal Pigmented Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1981–1989. [Google Scholar]
- Grimm, C.; Remé, C.E.; Rol, P.O.; Williams, T.P. Blue Light’s Effects on Rhodopsin: Photoreversal of Bleaching in Living Rat Eye. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3984–3990. [Google Scholar]
- Iandiev, I.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Grimm, C.; Remé, C.E.; Reichenbach, A.; Pannicke, T.; Bringmann, A. Müller Cell Response to Blue Light Injury of the Rat Retina. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3559–3567. [Google Scholar] [CrossRef]
- Klettner, A. Oxidative stress induced cellular signaling in RPE cells. Front. Biosci. 2012, 2, 392–411. [Google Scholar] [CrossRef]
- Palestine, A.G.; Wagner, B.D.; Patnaik, J.L.; Baldermann, R.; Mathias, M.T.; Mandava, N.; Lynch, A.M. Plasma c-c chemokine concentrations in intermediate age-related macular degeneration. Front. Med. 2021, 8, 710595. [Google Scholar] [CrossRef]
- Liu, D.; Xu, Y. p53, oxidative stress, and aging. Antioxid. Redox Signal. 2011, 15, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Kim, H.E.; Cho, H.; Shi, S.; Kim, B.; Kim, O. Red light-emitting diode irradiation regulates oxidative stress and inflammation through SPHK1/NF-κB activation in human keratinocytes. J. Photochem. Photobiol. B Biol. 2018, 186, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Fietz, A.; Hurst, J.; Schnichels, S. Out of the Shadow: Blue Light Exposure Induces Apoptosis in Müller Cells. Int. J. Mol. Sci. 2022, 23, 14540. [Google Scholar] [CrossRef] [PubMed]
- Fietz, A.; Hurst, J.; Joachim, S.C.; Schnichels, S. Establishment of a primary RPE monolayer to complement retinal ex vivo cultures. STAR Protoc. 2023, 4, 102443. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, S.; Hurst, J.; Jashari, A.; Ahrens, K.; Tsai, T.; Wunderlich, I.M.; Dick, H.B.; Joachim, S.C.; Schnichels, S. A novel NMDA triggered porcine organ culture induces retinal ganglion cell apoptosis–chances for replacement of animal experiments. Altern. Lab. Anim. 2016, 44, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Schnichels, S.; Dorfi, T.; Schultheiss, M.; Arango-Gonzalez, B.; Bartz-Schmidt, K.U.; Januschowski, K.; Spitzer, M.S.; Ziemssen, F. Ex-vivo-examination of ultrastructural changes in organotypic retina culture using near-infrared imaging and optical coherence tomography. Exp. Eye Res. 2016, 147, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Schnichels, S.; Blak, M.; Hurst, J.; Dorfi, T.; Bartz-Schmidt, K.U.; Ziemssen, F.; Spitzer, M.S. Establishment of a retinal hypoxia organ culture model. Biol. Open. 2017, 6, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.; Kuehn, S.; Jashari, A.; Tsai, T.; Bartz-Schmidt, K.U.; Schnichels, S.; Joachim, S.C. A novel ex-vivo retina culture model for oxidative stress induced by H2O2. Altern. Lab. Anim. 2017, 45, 11–25. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, S.; Wu, L.; Chai, G.; Wang, H.; Chen, Y.; Sun, J.; Yu, Y.; Zhou, W.; Zheng, Q.; et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol. Cell. Biol. 2006, 7, 2782–2790. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Woods, N.; Piluso, L.G.; Lee, H.H.; Chen, J.; Bhalla, K.N.; Monteiro, A.; Liu, X.; Hung, M.C.; Wang, H.G. p53 acetylation is crucial for its transcription-independent proapoptotic functions. J. Biol. Chem. 2009, 17, 11171–11183. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Absalon, M.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Singh, M.; Selivanova, G.; Peuget, S. Pifithrin-α alters p53 post-translational modifications pattern and differentially inhibits p53 target genes. Sci. Rep. 2020, 10, 1049. [Google Scholar] [CrossRef] [PubMed]
- Westlund, B.S.; Cai, B.; Zhou, J.; Sparrow, J.R. Involvement of c-Abl, p53 and the MAP kinase JNK in the cell death program initiated in A2E-laden ARPE-19 cells by exposure to blue light. Apoptosis 2009, 14, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Kuse, Y.; Tsuruma, K.; Kanno, Y.; Shimazawa, M.; Hara, H. CCR3 Is Associated with the Death of a Photoreceptor Cell-line Induced by Light Exposure. Front. Pharmacol. 2017, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Yun, J.; Yoon, Y.D.; Park, S.I.; Seo, Y.J.; Park, W.S.; Chu, H.Y.; Park, K.H.; Lee, M.Y.; Lee, C.W.; et al. Blue light effect on retinal pigment epithelial cells by display devices. Integr. Biol. 2017, 9, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Kuse, Y.; Ogawa, K.; Tsuruma, K.; Shimazawa, M.; Hara, H. Damage of photoreceptor-derived cells in culture induced by light emitting diode-derived blue light. Sci. Rep. 2014, 4, 5223. [Google Scholar] [CrossRef]
- Jaadane, I.; Villalpando Rodriguez, G.; Boulenguez, P. Retinal phototoxicity and the evaluation of the blue light hazard of a new solid-state lighting technology. Sci. Rep. 2020, 10, 6733. [Google Scholar] [CrossRef]
- Roehlecke, C.; Schumann, U.; Ader, M.; Knels, L.; Funk, R.H. Influence of blue light on photoreceptors in a live retinal explant system. Mol. Vis. 2011, 17, 876–884. [Google Scholar]
- Hu, Z.; Zhang, Y.; Wang, J.; Mao, P.; Lv, X.; Yuan, S.; Huang, Z.; Ding, Y.; Xie, P.; Liu, Q. Knockout of Ccr2 alleviates photoreceptor cell death in rodent retina exposed to chronic blue light. Cell Death Dis. 2016, 7, e2468. [Google Scholar] [CrossRef]
- Hendrickson, A.; Hicks, D. Distribution and density of medium- and short-wavelength selective cones in the domestic pig retina. Exp. Eye Res. 2002, 74, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Chandler, M.J.; Samuelson, D.A.; MacKay, E.O. Photoreceptor density of the domestic pig retina. Vet. Ophthalmol. 1999, 2, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Salminen, A.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Age-Related Macular Degeneration (AMD): Alzheimer’s Disease in the Eye? J. Alzheimers Dis. 2011, 24, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Borges, H.; Chao, C.; Xu, Y.; Linden, R.; Wang, J. Radiation-induced apoptosis in developing mouse retina exhibits dose-dependent requirement for ATM phosphorylation of p53. Cell Death Differ. 2004, 11, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.J.; Schneider, R.; Miller, J.A.; Martin, B.P. Al-Ubaidi MR, Photoreceptor cell apoptosis induced by the 2-nitroimidazole radiosensitizer, CI-1010, is mediated by p53-linked activation of caspase-3. Neurotoxicology 2006, 27, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Vuong, L.; Conley, S.M.; Al-Ubaidi, M.R. Expression and role of p53 in the retina. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Wu, J.; Spee, C.; Ryan, S.J.; Hinton, D.R. BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration. J. Biol. Chem. 2009, 284, 9529–9539. [Google Scholar] [CrossRef]
- Joo, C.K.; Choi, J.S.; Ko, H.W.; Park, K.Y.; Sohn, S.; Chun, M.H.; Oh, Y.J.; Gwag, B.J. Necrosis and apoptosis after retinal ischemia: Involvement of NMDA-mediated excitotoxicity and p53. Investig. Ophthalmol. Vis. Sci. 1999, 40, 713–720. [Google Scholar]
- Bhattacharya, S.; Chaum, E.; Johnson, D.A.; Johnson, L.R. Age-related susceptibility to apoptosis in human retinal pigment epithelial cells is triggered by disruption of p53-Mdm2 association. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8350–8366. [Google Scholar] [CrossRef]
- Grimm, C.; Wenzel, A.; Williams, T.P.; Rol, P.O.; Hafezi, F.; Remé, C.E. Rhodopsin-mediated blue-light damage to the rat retina: Effect of photoreversal of bleaching. Investig. Ophthalmol. Vis. Sci. 2001, 42, 497–505. [Google Scholar]
- Hunter, J.J.; Morgan, J.; Merigan, W.H.; Sliney, D.H.; Sparrow, J.R.; Williams, D.R. The susceptibility of the retina to photochemical damage from visible light. Prog. Retin. Eye Res. 2012, 31, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Ham, W.T., Jr.; Mueller, H.; Ruffolo, J.J., Jr.; Millen, J.E.; Cleary, S.F.; Guerry, R.K.; Guerry, D., 3rd. Basic mechanisms underlying the production of photochemical lesions in the mammalian retina. Curr. Eye Res. 1984, 3, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Franze, K.; Grosche, J.; Skatchkov, S.N.; Schinkinger, S.; Foja, C.; Schild, D.; Uckermann, O.; Travis, K.; Reichenbach, A.; Guck, J. Müller cells are living optical fibers in the vertebrate retina. Proc. Natl. Acad. Sci. USA 2007, 104, 8287–8292. [Google Scholar] [CrossRef] [PubMed]
- Labin, A.M.; Safuri, S.K.; Ribak, E.N.; Perlman, I. Müller cells separate between wavelengths to improve day vision with minimal effect upon night vision. Nat. Commun. 2014, 5, 4319. [Google Scholar] [CrossRef] [PubMed]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sánchez, M.C. A Journey Into the Retina: Müller Glia Commanding Survival and Death. Eur. J. Neurosci. 2018, 47, 1429–1443. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Kefalov, V.J. The cone-specific visual cycle. Prog. Retin. Eye Res. 2011, 30, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Medeiros, N.E.; Millican, C.L. Photoreceptor loss in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1236–1249. [Google Scholar]
- Cao, S.; Ko, A.; Partanen, M.; Pakzad-Vaezi, K.; Merkur, A.B.; Albiani, D.A.; Kirker, A.W.; Wang, A.; Cui, J.Z.; Forooghian, F.; et al. Relationship between systemic cytokines and complement factor H Y402H polymorphism in patients with dry age-related macular degeneration. Am. J. Ophthalmol. 2013, 156, 1176–1183. [Google Scholar] [CrossRef]
- Rath, P.C.; Aggarwal, B.B. TNF-induced signaling in apoptosis. Clin. Immunol. 1999, 19, 350–364. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Luo, J.; Brooks, C.L.; Gu, W. Acetylation of p53 inhibits its ubiquitination by Mdm2. Biol. Chem. 2002, 277, 50607–50611. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.I.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Marti, A.; Hafezi, F.; Lansel, N.; Hegi, M.E.; Wenzel, A.; Grimm, C.; Niemeyer, G.; Reme, C.E. Light-induced cell death of retinal photoreceptors in the absence of p53. Investig. Ophthalmol. Vis. Sci. 1998, 5, 846–849. [Google Scholar]
- The Vision Council. ANSI Z80.36-2016; Ophthalmics—Light Hazard Protection for Ophthalmic Instruments. American National Standards Institute (ANSI): New York, NY, USA, 2016.
- Núñez-Álvarez, C.; Osborne, N.N. Blue light exacerbates and red light counteracts negative insults to retinal ganglion cells in situ and R28 cells in vitro. Neurochem. Int. 2019, 125, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Moyano, D.B.; Sola, Y.; González-Lezcano, R.A. Blue-Light Levels Emitted from Portable Electronic Devices Compared to Sunlight. Energies 2020, 13, 4276. [Google Scholar] [CrossRef]
- The Vision Council. Eyes Overexposed: The Digital Device Dilemma: Digital Eye Strain Report; The Vision Councile: Alexandria, VA, USA, 2016. [Google Scholar]
- Cougnard-Gregoire, A.; Merle, B.M.; Aslam, T.; Seddon, J.M.; Aknin, I.; Klaver, C.C.; Garhöfer, G.; Layana, A.G.; Minnella, A.M.; Silva, R.; et al. Blue Light Exposure: Ocular Hazards and Prevention—A Narrative Review. Ophthalmol. Ther. 2023, 12, 755–788. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company Primary AB | Dilution Primary AB | Company Secondary AB | Dilution Secondary AB |
|---|---|---|---|---|
| acetylated (Lys382) p53 | Cell Signaling (Danvers, MA, USA) | 1:100 | Thermo Fisher Scientific (Invitrogen; Karlsruhe, Germany) | 1:1000 Alexa Fluor 488 |
| GFAP | BD Bioscience (BP Pharmigen, Heidelberg, Germany) | 1:200 | Thermo Fisher Scientific (Karlsruhe, Germany) | 1:1000 Alexa Fluor 555 |
| Rhodopsin | Abcam (Cambridge, UK) | 1:200 | Thermo Fisher Scientific (Invitrogen; Karlsruhe, Germany) | 1:1000 Alexa Fluor 594 |
| Primary Antibody | ||
|---|---|---|
| Target | Company | Dilution |
| β-Actin | Cell Signaling, Danvers, MA, USA | 1:1000 |
| HSP70 | Santa Cruz, Dallas, TX, USA | 1:500 |
| NF-kB | Elabscience, Hamburg, Germany | 1:500 |
| Rhodopsin | Abcam, Cambridge, UK | 1:200 |
| BAX | Cell Signaling, Danvers, MA, USA | 1:100 |
| BCL2 | Cell Signaling, Danvers, MA, USA | 1:100 |
| Secondary Antibody | ||
| Target | Company | Dilution |
| mouse | IRDye® 680RD, LI-COR, Homburg, Germany | 1:10,000 |
| rabbit | IRDye® 800CW, LI-COR, Homburg, Germany | 1:10,000 |
| Gene | Sequence 5′-3′ | |
|---|---|---|
| Actin-β for | CACGCCATCCTGCGTCTGGA | XM_003357928.4 |
| Actin-β rev | AGCACCGTGTTGGCGTAGAG | |
| RPL4 for | CAAGAGTAACTACAACCTTC | XM_005659862.3 |
| RPL4 rev | GAACTCTACGATGAATCTTC | |
| GFAP for | GGAGAAGCCTTTGCTACACG | NM_001244397.1 |
| GFAP rev | TCTTCACTCTGCCTGGGTCT | |
| Opsin (mws) for | GGGGAGCATCTTCACCTACA | NM_001011506.1 |
| Opsin (mws) rev | GATGATGGTCTCTGCCAGGT | |
| Rhodopsin for | TCCAGGTACATCCCAGAAGG | NM_214221.1 |
| Rhodopsin rev | GCTGCCCATAGCAGAAGAAG | |
| β-III-tubulin for | CAGATGTTCGATGCCAAGAA | AK391872.1 |
| β-III-tubulin rev | GGGATCCACTCCACGAAGTA | |
| TNF-α for | CCACCAACGTTTTCCTCACT | JF831365.1 |
| TNF-α rev | CCAAAATAGACCTGCCCAGA | |
| PKCa for | ACCGAACAACAAGGAACGAC | XM_021066740.1 |
| PKCa rev | CTGAGCTCCACGTTTCCTTC | |
| HSP70 for | ATGTCCGCTGCAAGAGAAGT | NM_001123127.1 |
| HSP70 rev | GGCGTCAAACACGGTATTCT | |
| NF-kB for | AGGATGGGATCTGCACTGTC | NM_001048232.1 |
| NF-kB rev | ATCAGGGTGCACCAAAAGTC | |
| BAX for | AAGCGCATTGGAGATGAACT | XM_003127290.5 |
| BAX rev | AAAGTAGAAAAGCGCGACCA | |
| Bcl-2 for | AATTACCATCGGCGTAGTGC | XM_021099593.1 |
| Bcl-2 rev | CGTTTCAGCCACCGTAAAAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fietz, A.; Corsi, F.; Hurst, J.; Schnichels, S. Blue Light Damage and p53: Unravelling the Role of p53 in Oxidative-Stress-Induced Retinal Apoptosis. Antioxidants 2023, 12, 2072. https://doi.org/10.3390/antiox12122072
Fietz A, Corsi F, Hurst J, Schnichels S. Blue Light Damage and p53: Unravelling the Role of p53 in Oxidative-Stress-Induced Retinal Apoptosis. Antioxidants. 2023; 12(12):2072. https://doi.org/10.3390/antiox12122072
Chicago/Turabian StyleFietz, Agnes, Francesca Corsi, José Hurst, and Sven Schnichels. 2023. "Blue Light Damage and p53: Unravelling the Role of p53 in Oxidative-Stress-Induced Retinal Apoptosis" Antioxidants 12, no. 12: 2072. https://doi.org/10.3390/antiox12122072