
Elamipretide(SS-31) Attenuates Idiopathic Pulmonary Fibrosis by Inhibiting the Nrf2-Dependent NLRP3 Inflammasome in Macrophages
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Reagents
2.3. BLM-Induced Pulmonary Fibrosis
2.4. Bronchoalveolar Lavage Fluid (BALF) Collection and Analysis
2.5. Lung Histological Assay
2.6. Hydroxyproline Assay
2.7. Primary Macrophage Culture and Treatment
2.8. Isolation of Total RNA and Quantitative PCR
| Target gene | Primer | Primer sequences (5′ -3′): |
| α-SMA | Forward | GACGCTGAAGTATCCGATAGAACACG; |
| Reverse | CACCATCTCCAGAGTCCAGCACAAT. | |
| Collagen I | Forward | TGCCGTGACCTCAAGATGTG; |
| Reverse | CACAAGCGTGCTGTAGGTGA. | |
| Fibronectin | Forward | TCTGGGAAATGGAAAAGGGGAATGG; |
| Reverse | . | CACTGAAGCAGGTTTCCTCGGTTGT |
| TNF-α | Forward | TTCTCAT TCCTGCTTGTGG; |
| Reverse | ACTTGGTGGTTTG CTACG. | |
| IL-1β | Forward | CCAGCTTCAAATCTCACAGCAG; |
| Reverse | CTTCTTTGGGTATTGCTTGGGATC. | |
| IL-6 | Forward | CCACCAAGAACGATAGTCAA; |
| Reverse | TTTCCACGATTTCCCAGA. | |
| Nrf2 | Forward | CCCGAAGCACGCTGAAGGCA; |
| Reverse | CCAGGCGGTGGGTCTCCGTA. | |
| mtDNA | Forward | GGTTCTTACTTCAGGGCCATCA; |
| Reverse | GATTAGACCCGTTACCATCGAGAT. | |
| Nrf1 | Forward | CCATCTATCCGAAAGAGACAGC; |
| Reverse | GGGTGAGATGCAGAGTACAATC. | |
| IL-18 | Forward | ACTTTGGCCGACTTCACTGT; |
| Reverse | GGGTTCACTGGCACTTTGAT. | |
| NLRP3 | Forward | GCTGTGTGAGGCACTCCAG; |
| Reverse | GGAGATGTCGAAGCAGCATT. | |
| ASC | Forward | GGAGTCGTATGGCTTGGAGC; |
| Reverse | CGTCCACTTCTGTGACCCTG. | |
| Caspase-1 | Forward | CCCACTGCTGATAGGGTGAC; |
| Reverse | GCATAGGTACATAAGAATGAACTGGA. | |
| GAPDH | Forward | TGCGACTTCAACAGCAACTC; |
| Reverse | CTTGCTCAGTGTCCTTGCTG. |
2.9. Western Blotting
2.10. Immunofluorescence Assay
2.11. Determination of Cellular Reactive Oxygen Species (ROS)
2.12. Statistical Analysis
3. Results
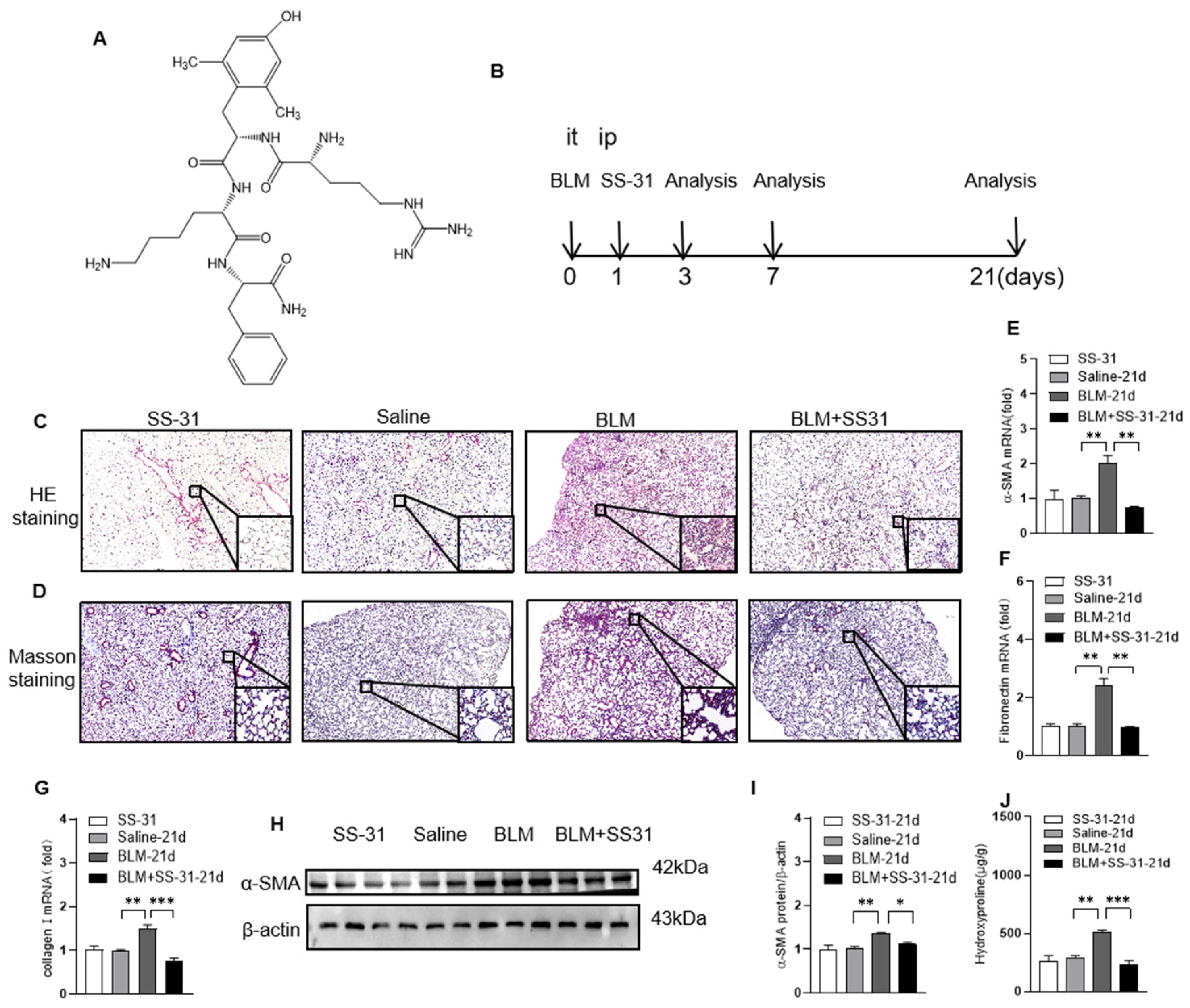
3.1. SS-31 Attenuated BLM-Induced Pulmonary Fibrosis
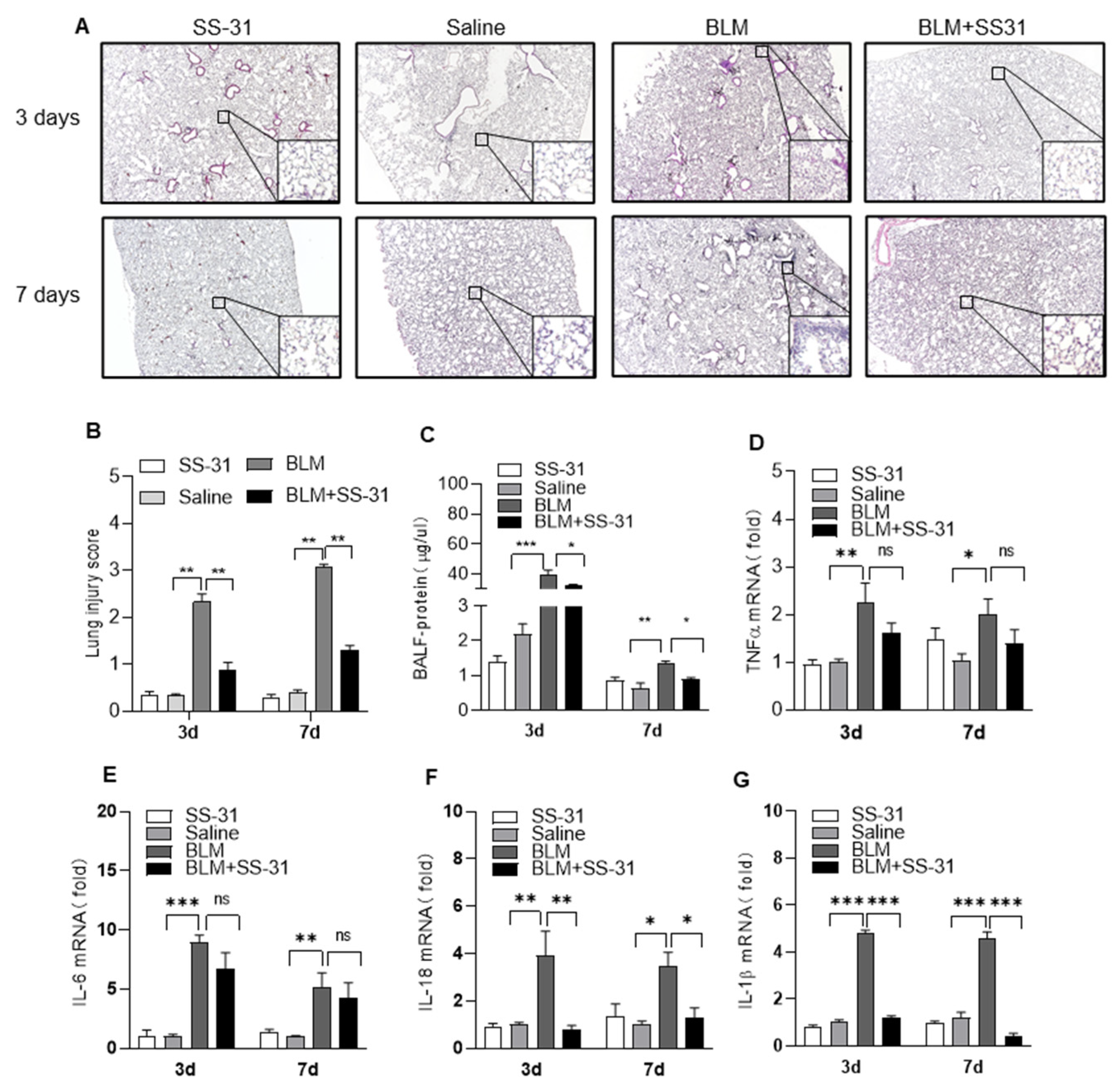
3.2. SS-31 Attenuated BLM-Induced Pulmonary Inflammation and Reduced the Expression of IL-1β and IL-18 In Vivo
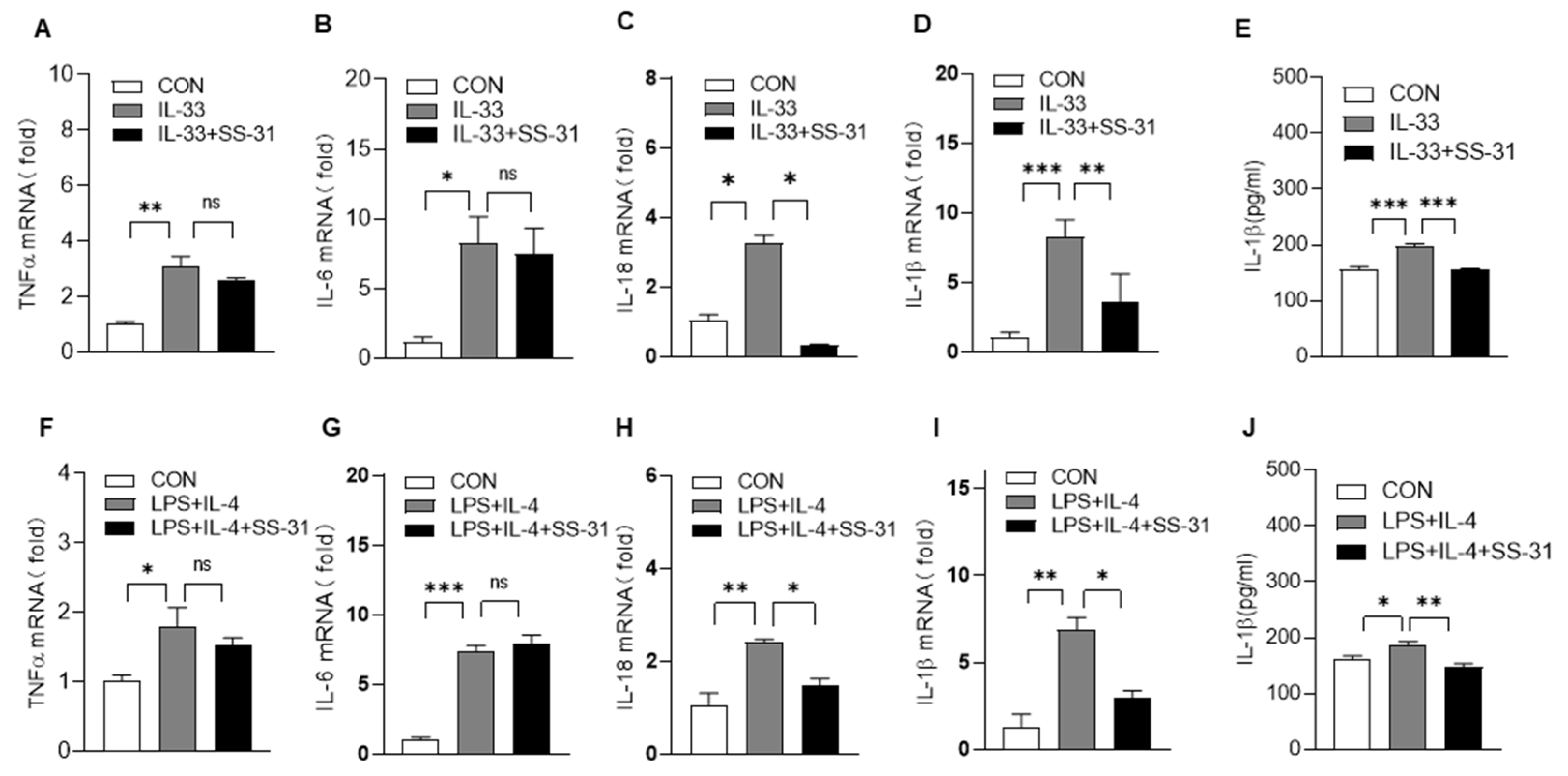
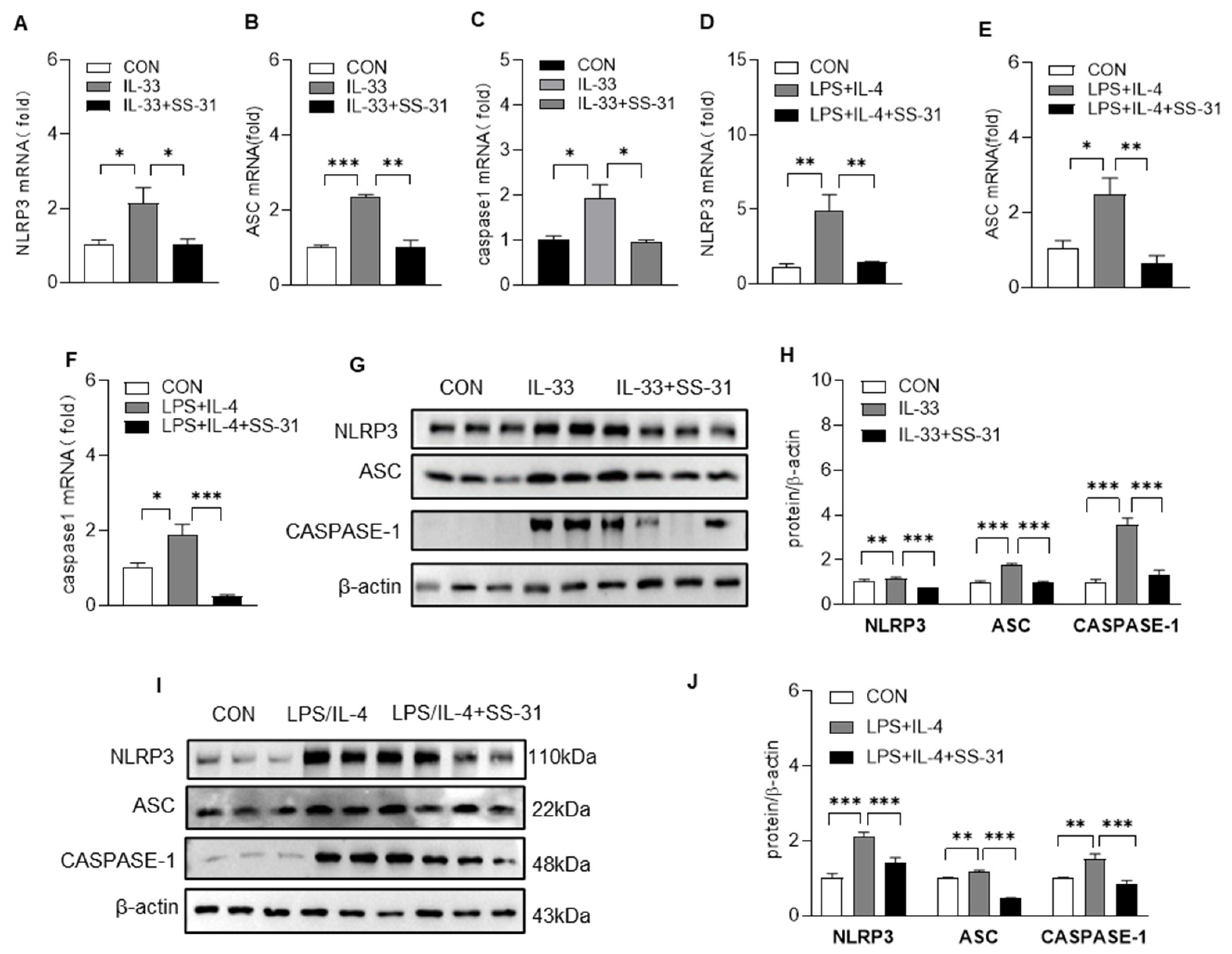
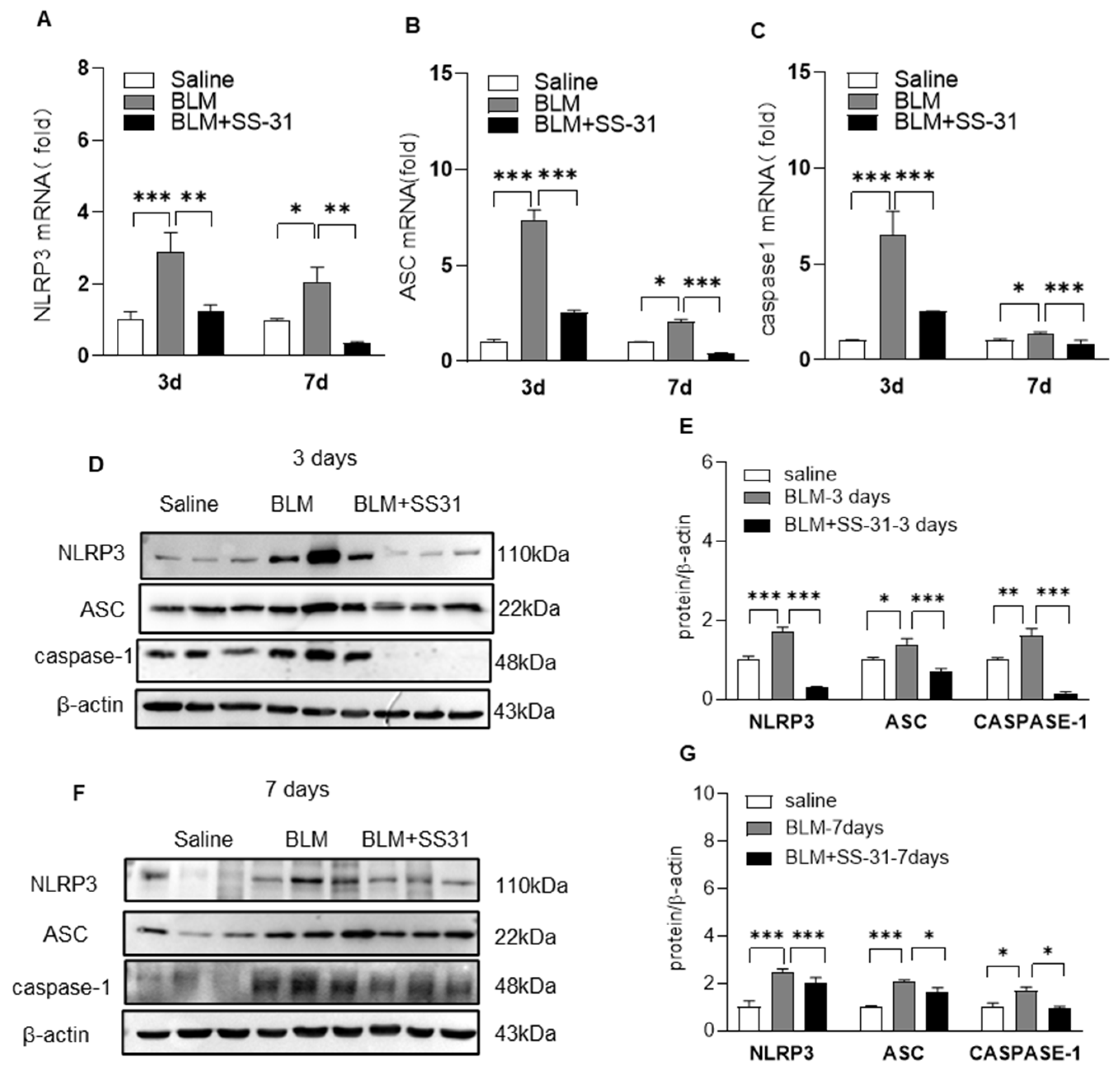
3.3. SS-31 Inhibited NLRP3 Inflammasome Activation in Macrophages and BLM-Induced Mice
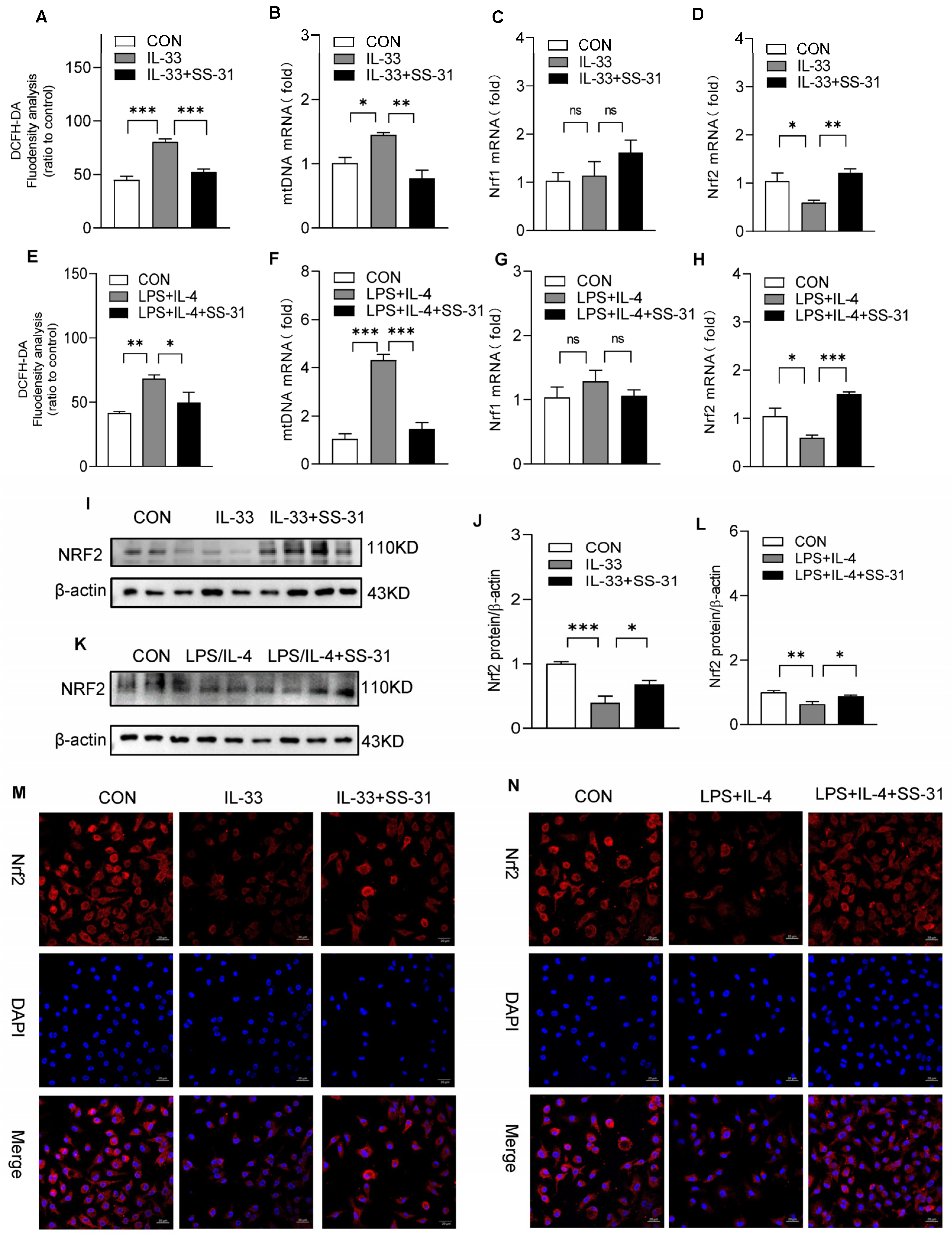
3.4. SS-31 Restored NRF2 Expression and Mitochondrial Function in Macrophages
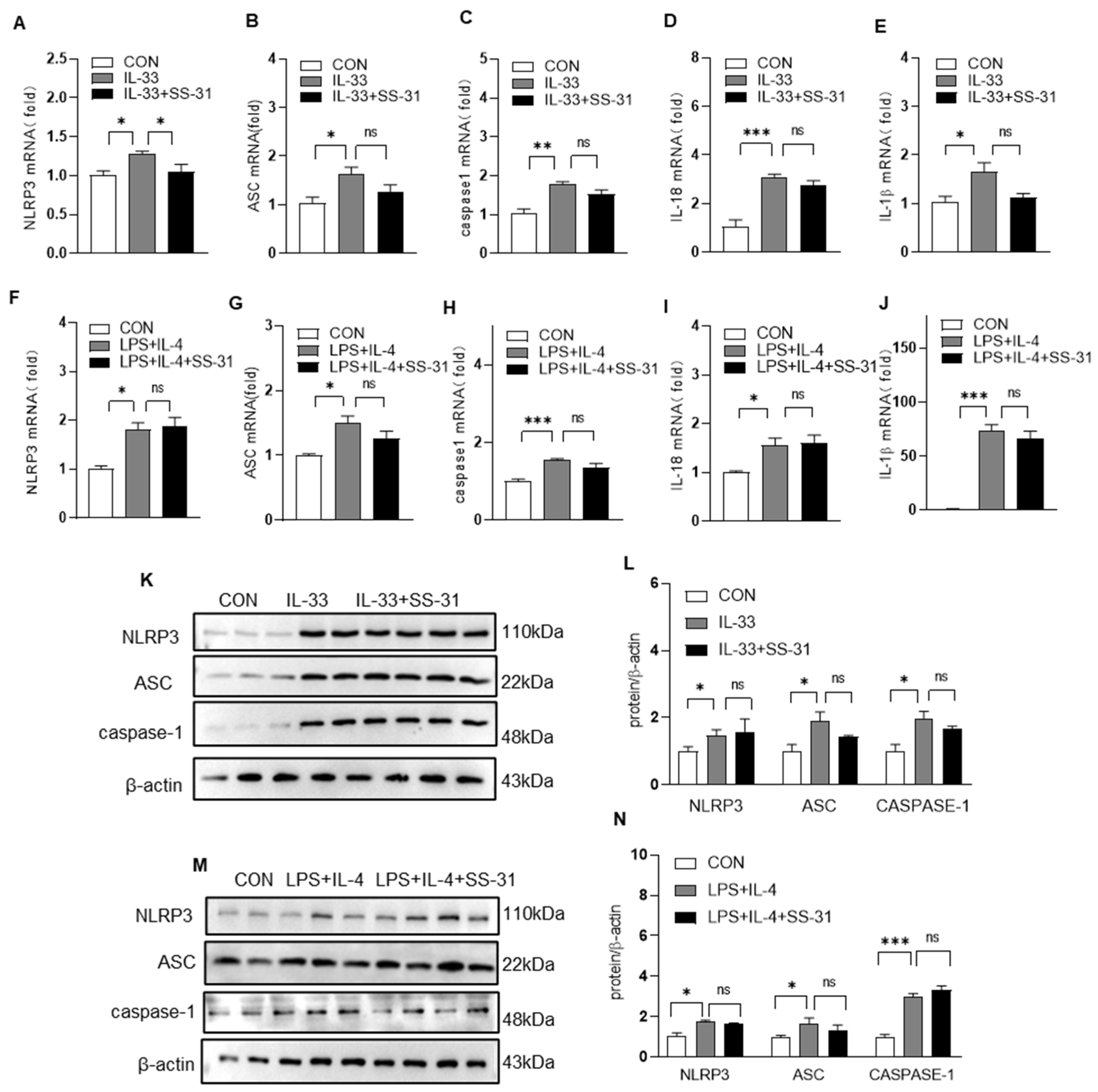
3.5. Nrf2 Knockout Reversed the Effect of SS-31 on NLRP Inflammasome Activation in Macrophages
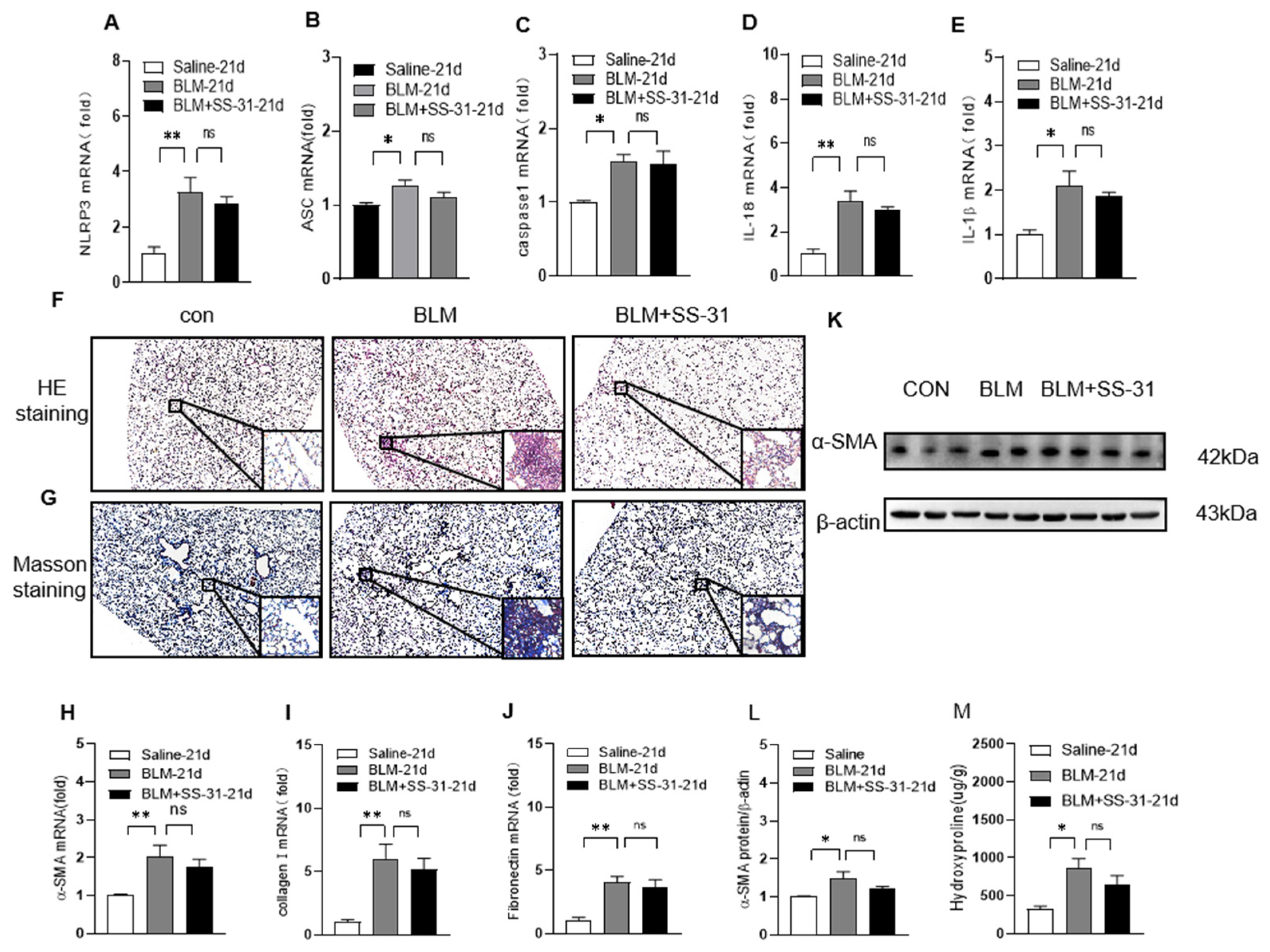
3.6. Nrf2 Knockout Reversed the Effect of SS-31 on NLRP3 Inflammasome Activation and Fibrosis in BLM-Induced Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsubouchi, K.; Araya, J.; Kuwano, K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm. Regen. 2018, 38, 18. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Chen, S.Y.; Yeh, W.S.; Li, Q.; Lee, Y.C.; Wang, A.; Raghu, G. Health care utilization and costs of idiopathic pulmonary fibrosis in U.S. Medicare beneficiaries aged 65 years and older. Ann. Am. Thorac. Soc. 2015, 12, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Morse, C.; Tabib, T.; Sembrat, J.; Buschur, K.L.; Bittar, H.T.; Valenzi, E.; Jiang, Y.; Kass, D.J.; Gibson, K.; Chen, W.; et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 54, 1802441. [Google Scholar] [CrossRef]
- Lee, J.W.; Chun, W.; Lee, H.J.; Min, J.H.; Kim, S.M.; Seo, J.Y.; Ahn, K.S.; Oh, S.R. The Role of Macrophages in the Development of Acute and Chronic Inflammatory Lung Diseases. Cells 2021, 10, 897. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, G.; Qiu, L.; Liu, X.; Zhou, S.; Wu, J. Contribution of Adiponectin/Carnitine Palmityl Transferase 1A-Mediated Fatty Acid Metabolism during the Development of Idiopathic Pulmonary Fibrosis. Oxidative Med. Cell. Longev. 2022, 2022, 5265616. [Google Scholar] [CrossRef]
- Rao, L.Z.; Wang, Y.; Zhang, L.; Wu, G.; Zhang, L.; Wang, F.X.; Chen, L.M.; Sun, F.; Jia, S.; Zhang, S.; et al. IL-24 deficiency protects mice against bleomycin-induced pulmonary fibrosis by repressing IL-4-induced M2 program in macrophages. Cell Death Differ. 2021, 28, 1270–1283. [Google Scholar] [CrossRef]
- Wang, H.; Wen, Y.; Wang, L.; Wang, J.; Chen, H.; Chen, J.; Guan, J.; Xie, S.; Chen, Q.; Wang, Y.; et al. DDR1 activation in macrophage promotes IPF by regulating NLRP3 inflammasome and macrophage reaction. Int. Immunopharmacol. 2022, 113, 109294. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, C.; Xia, Z.; Song, B.; Hu, W.; Cui, Y.; Xue, Y.; Xia, M.; Xu, D.; Zhang, S.; et al. Nano-designed CO donor ameliorates bleomycin-induced pulmonary fibrosis via macrophage manipulation. J. Control. Release Off. J. Control. Release Soc. 2022, 341, 566–577. [Google Scholar] [CrossRef]
- Sari, E.; He, C.; Margaroli, C. Plasticity towards Rigidity: A Macrophage Conundrum in Pulmonary Fibrosis. Int. J. Mol. Sci. 2022, 23, 11443. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Sun, L.; Wu, Y.; Yang, Y.; Wang, J.; He, H.; Hu, Y.; Chang, Y.; Liang, Q.; Zhu, J.; et al. AKT2 Regulates Pulmonary Inflammation and Fibrosis via Modulating Macrophage Activation. J. Immunol. 2017, 198, 4470–4480. [Google Scholar] [CrossRef] [PubMed]
- Faas, M.; Ipseiz, N.; Ackermann, J.; Culemann, S.; Gruneboom, A.; Schroder, F.; Rothe, T.; Scholtysek, C.; Eberhardt, M.; Bottcher, M.; et al. IL-33-induced metabolic reprogramming controls the differentiation of alternatively activated macrophages and the resolution of inflammation. Immunity 2021, 54, 2531–2546.e2535. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Zhai, X.; Li, J.; Sun, A.; Che, H.; Christman, J.W.; Chai, G.; Zhao, P.; Karpurapu, M. NFATc3 Promotes Pulmonary Inflammation and Fibrosis by Regulating Production of CCL2 and CXCL2 in Macrophages. Aging Dis. 2023, 14, 1441–1457. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Chen, X.; Xu, H.; Song, Y.; Wang, H.; Gao, Y.; Wang, J.; Du, R.; Lou, H.; Dong, T. Mitochondrial metabolism mediated macrophage polarization in chronic lung diseases. Pharmacol. Ther. 2022, 239, 0163–7258. [Google Scholar] [CrossRef] [PubMed]
- Willenborg, S.; Sanin, D.E.; Jais, A.; Ding, X.; Ulas, T.; Nuchel, J.; Popovic, M.; MacVicar, T.; Langer, T.; Schultze, J.L.; et al. Mitochondrial metabolism coordinates stage-specific repair processes in macrophages during wound healing. Cell Metab. 2021, 33, 2398–2414.e2399. [Google Scholar] [CrossRef]
- Eapen, M.S.; Sharma, P.; Sohal, S.S. Mitochondrial dysfunction in macrophages: A key to defective bacterial phagocytosis in COPD. Eur. Respir. J. 2019, 54, 1901641. [Google Scholar] [CrossRef]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2020, 19, 57–75. [Google Scholar] [CrossRef]
- Sweetwyne, M.T.; Pippin, J.W.; Eng, D.G.; Hudkins, K.L.; Chiao, Y.A.; Campbell, M.D.; Marcinek, D.J.; Alpers, C.E.; Szeto, H.H.; Rabinovitch, P.S.; et al. The mitochondrial-targeted peptide; SS-31, improves glomerular architecture in mice of advanced age. Kidney Int. 2017, 91, 1126–1145. [Google Scholar] [CrossRef]
- Chavez, J.D.; Tang, X.; Campbell, M.D.; Reyes, G.; Kramer, P.A.; Stuppard, R.; Keller, A.; Zhang, H.; Rabinovitch, P.S.; Marcinek, D.J.; et al. Mitochondrial protein interaction landscape of SS-31. Proc. Natl. Acad. Sci. USA 2020, 117, 15363–15373. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, C.P.; Pirisinu, M.; Vlachos, E.N.; Langel, U. Novel cell-penetrating peptide targeting mitochondria. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 4589–4599. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; She, P.; Zhao, Z.; Ma, B.; Li, G.; Wang, Y. Duplex Responsive Nanoplatform with Cascade Targeting for Atherosclerosis Photoacoustic Diagnosis and Multichannel Combination Therapy. Adv. Mater. 2023, 35, e2300439. [Google Scholar] [CrossRef]
- Nickel, A.G.; von Hardenberg, A.; Hohl, M.; Loffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, Y.J.; Liu, Z.R.; Tang, D.D.; Chen, X.W.; Chen, Y.H.; Zhou, R.N.; Chen, S.Q.; Niu, H.X. Role of mitochondrial dysfunction in renal fibrosis promoted by hypochlorite-modified albumin in a remnant kidney model and protective effects of antioxidant peptide SS-31. Eur. J. Pharmacol. 2017, 804, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1beta and IL-18 and Arrests CKD. J. Am. Soc. Nephrol. JASN 2017, 28, 1437–1449. [Google Scholar] [CrossRef]
- Shang, L.; Ren, H.; Wang, S.; Liu, H.; Hu, A.; Gou, P.; Lin, Y.; Zhou, J.; Zhu, W.; Shi, X. SS-31 Protects Liver from Ischemia-Reperfusion Injury via Modulating Macrophage Polarization. Oxidative Med. Cell. Longev. 2021, 2021, 6662156. [Google Scholar] [CrossRef]
- Gong, Q.; Li, Y.; Ma, H.; Guo, W.; Kan, X.; Xu, D.; Liu, J.; Fu, S. Peiminine Protects against Lipopolysaccharide-Induced Mastitis by Inhibiting the AKT/NF-kappaB, ERK1/2 and p38 Signaling Pathways. Int. J. Mol. Sci. 2018, 19, 2637. [Google Scholar] [CrossRef]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: An open-label extension study. The Lancet. Respir. Med. 2019, 7, 657–664. [Google Scholar] [CrossRef]
- Tanner, L.; Single, A.B.; Bhongir, R.K.V.; Heusel, M.; Mohanty, T.; Karlsson, C.A.Q.; Pan, L.; Clausson, C.M.; Bergwik, J.; Wang, K.; et al. Small-molecule-mediated OGG1 inhibition attenuates pulmonary inflammation and lung fibrosis in a murine lung fibrosis model. Nat. Commun. 2023, 14, 643. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, G.M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef]
- Zhu, Y.; Luo, M.; Bai, X.; Li, J.; Nie, P.; Li, B.; Luo, P. SS-31, a Mitochondria-Targeting Peptide, Ameliorates Kidney Disease. Oxidative Med. Cell. Longev. 2022, 2022, 1295509. [Google Scholar]
- Nashine, S. Potential Therapeutic Candidates for Age-Related Macular Degeneration (AMD). Cells 2021, 10, 2483. [Google Scholar] [CrossRef]
- Ding, X.W.; Robinson, M.; Li, R.; Aldhowayan, H.; Geetha, T.; Babu, J.R. Mitochondrial dysfunction and beneficial effects of mitochondria-targeted small peptide SS-31 in Diabetes Mellitus and Alzheimer’s disease. Pharmacol. Res. 2021, 171, 105783. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.; Herrmann, S.M.S.; Eirin, A.; Ferguson, C.M.; Glockner, J.F.; Bjarnason, H.; McKusick, M.A.; Misra, S.; Lerman, L.O.; Textor, S.C. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) during Stent Revascularization in Patients With Atherosclerotic Renal Artery Stenosis. Circ. Cardiovasc. Interv. 2017, 10, e005487. [Google Scholar] [CrossRef] [PubMed]
- Vasse, G.F.; Nizamoglu, M.; Heijink, I.H.; Schleputz, M.; van Rijn, P.; Thomas, M.J.; Burgess, J.K.; Melgert, B.N. Macrophage-stroma interactions in fibrosis: Biochemical, biophysical, and cellular perspectives. J. Pathol. 2021, 254, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Pakshir, P.; Alizadehgiashi, M.; Wong, B.; Coelho, N.M.; Chen, X.; Gong, Z.; Shenoy, V.B.; McCulloch, C.A.; Hinz, B. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat. Commun. 2019, 10, 1850. [Google Scholar] [CrossRef]
- Byrne, A.J.; Mathie, S.A.; Gregory, L.G.; Lloyd, C.M. Pulmonary macrophages: Key players in the innate defence of the airways. Thorax 2015, 70, 1189–1196. [Google Scholar] [CrossRef]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. 2018, 16, 89. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Zhong, Z.; Stubelius, A.; Sweeney, S.R.; Booshehri, L.M.; Antonucci, L.; Liu-Bryan, R.; Lodi, A.; Terkeltaub, R.; Lacal, J.C.; et al. Choline Uptake and Metabolism Modulate Macrophage IL-1beta and IL-18 Production. Cell Metab. 2019, 29, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Nunez, G. The NLRP3 inflammasome: Activation and regulation. Trends Biochem. Sci. 2023, 48, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Lasithiotaki, I.; Giannarakis, I.; Tsitoura, E.; Samara, K.D.; Margaritopoulos, G.A.; Choulaki, C.; Vasarmidi, E.; Tzanakis, N.; Voloudaki, A.; Sidiropoulos, P.; et al. NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur. Respir. J. 2016, 47, 910–918. [Google Scholar] [CrossRef]
- Traba, J.; Sack, M.N. The role of caloric load and mitochondrial homeostasis in the regulation of the NLRP3 inflammasome. Cell. Mol. Life Sci. CMLS 2017, 74, 1777–1791. [Google Scholar] [CrossRef]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Nuclear control of respiratory gene expression in mammalian cells. J. Cell. Biochem. 2006, 97, 673–683. [Google Scholar] [CrossRef]
- Sekine, H.; Motohashi, H. Roles of CNC Transcription Factors NRF1 and NRF2 in Cancer. Cancers 2021, 13, 541. [Google Scholar] [CrossRef]
- Chan, J.Y.; Kwong, M.; Lu, R.; Chang, J.; Wang, B.; Yen, T.S.; Kan, Y.W. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 1998, 17, 1779–1787. [Google Scholar] [CrossRef]
- Sant, K.E.; Hansen, J.M.; Williams, L.M.; Tran, N.L.; Goldstone, J.V.; Stegeman, J.J.; Hahn, M.E.; Timme-Laragy, A. The role of Nrf1 and Nrf2 in the regulation of glutathione and redox dynamics in the developing zebrafish embryo. Redox Biol. 2017, 13, 207–218. [Google Scholar] [CrossRef]
- Kang, L.; Wang, X.; Wang, J.; Guo, J.; Zhang, W.; Lei, R. NRF1 knockdown alleviates lipopolysaccharide-induced pulmonary inflammatory injury by upregulating DKK3 and inhibiting the GSK-3beta/beta-catenin pathway. Clin. Exp. Immunol. 2023. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Healy, Z.; Zobi, F.; Kraft, B.D.; Welty-Wolf, K.; Smith, J.; Barkauskas, C.; Piantadosi, C.A. Nuclear respiratory factor-1 negatively regulates TGF-beta1 and attenuates pulmonary fibrosis. iScience 2022, 25, 103535. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Deng, J.; Zhou, X.; Cai, B.; Zhang, B.; Chen, X.; Chen, Z.; Wang, W. Sitagliptin activates the p62-Keap1-Nrf2 signalling pathway to alleviate oxidative stress and excessive autophagy in severe acute pancreatitis-related acute lung injury. Cell Death Dis. 2021, 12, 928. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nie, Y.; Li, J.; Zhai, X.; Wang, Z.; Wang, J.; Wu, Y.; Zhao, P.; Yan, G. Elamipretide(SS-31) Attenuates Idiopathic Pulmonary Fibrosis by Inhibiting the Nrf2-Dependent NLRP3 Inflammasome in Macrophages. Antioxidants 2023, 12, 2022. https://doi.org/10.3390/antiox12122022
Nie Y, Li J, Zhai X, Wang Z, Wang J, Wu Y, Zhao P, Yan G. Elamipretide(SS-31) Attenuates Idiopathic Pulmonary Fibrosis by Inhibiting the Nrf2-Dependent NLRP3 Inflammasome in Macrophages. Antioxidants. 2023; 12(12):2022. https://doi.org/10.3390/antiox12122022
Chicago/Turabian StyleNie, Yunjuan, Jiao Li, Xiaorun Zhai, Zhixu Wang, Junpeng Wang, Yaxian Wu, Peng Zhao, and Gen Yan. 2023. "Elamipretide(SS-31) Attenuates Idiopathic Pulmonary Fibrosis by Inhibiting the Nrf2-Dependent NLRP3 Inflammasome in Macrophages" Antioxidants 12, no. 12: 2022. https://doi.org/10.3390/antiox12122022