
Targeting Mitochondrial Dysfunction and Oxidative Stress to Prevent the Neurodegeneration of Retinal Ganglion Cells

Abstract
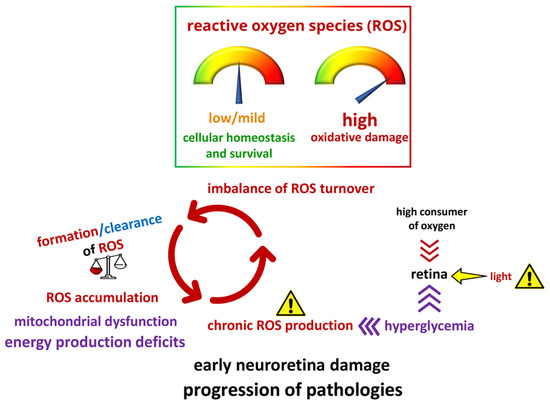
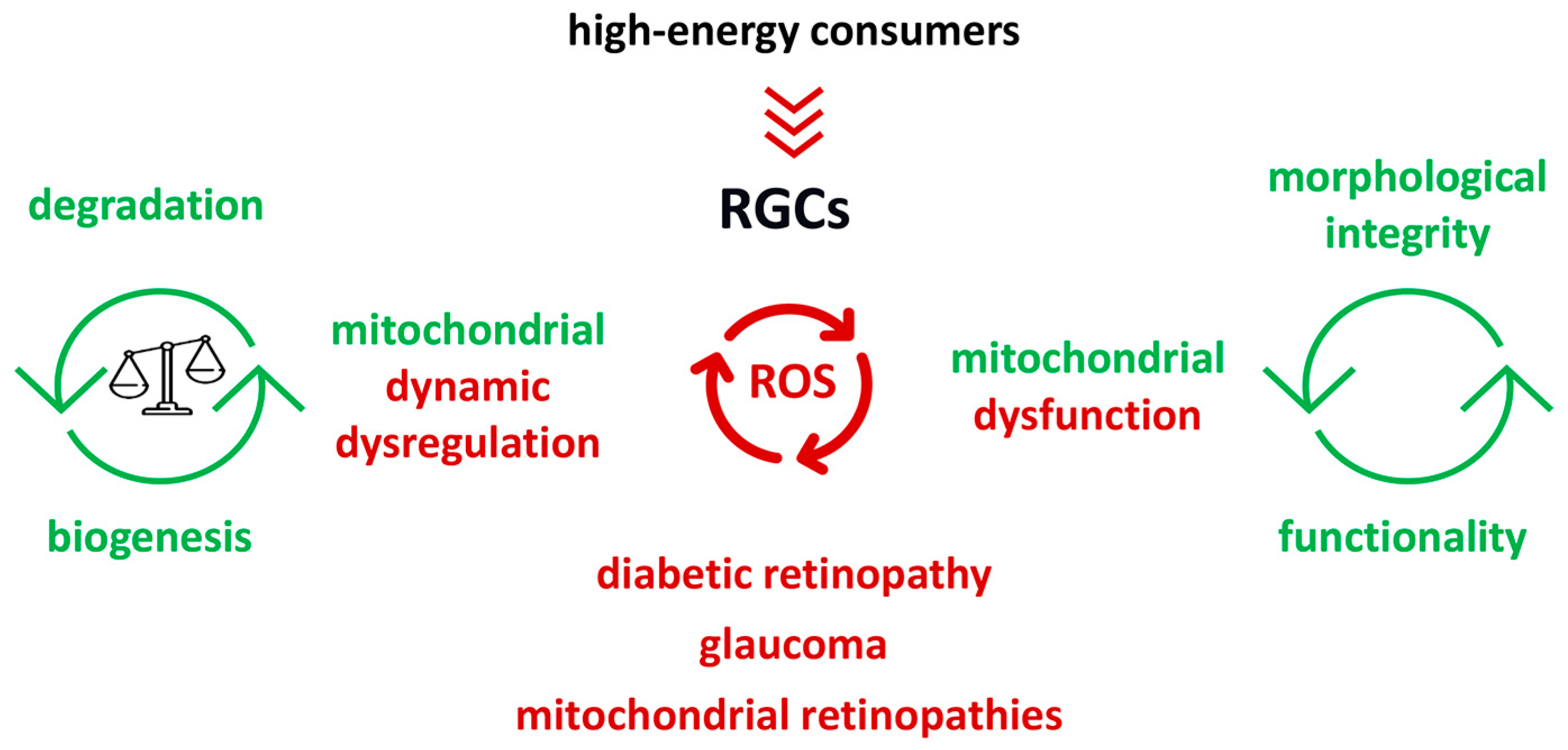
:
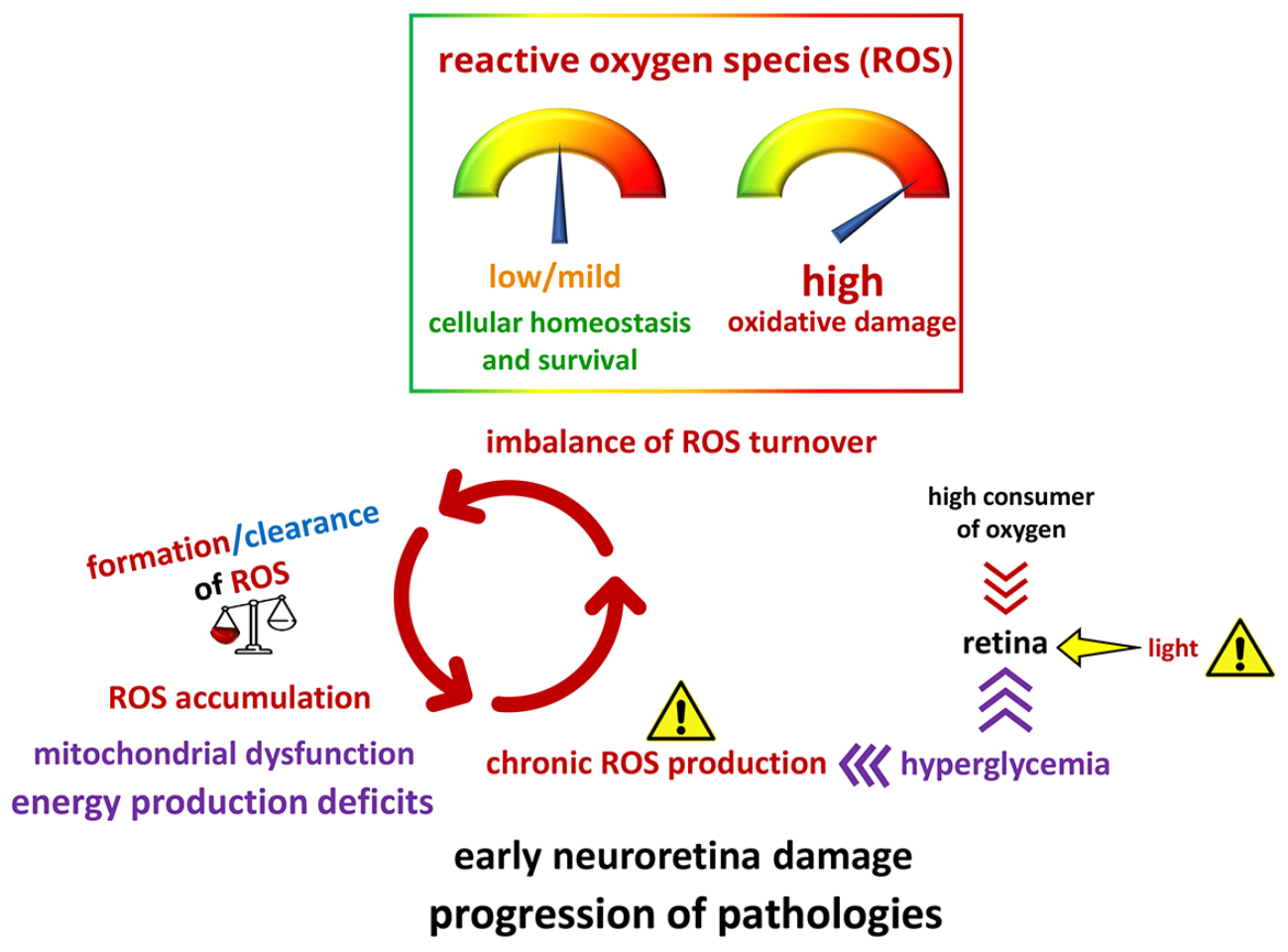{kind=link}
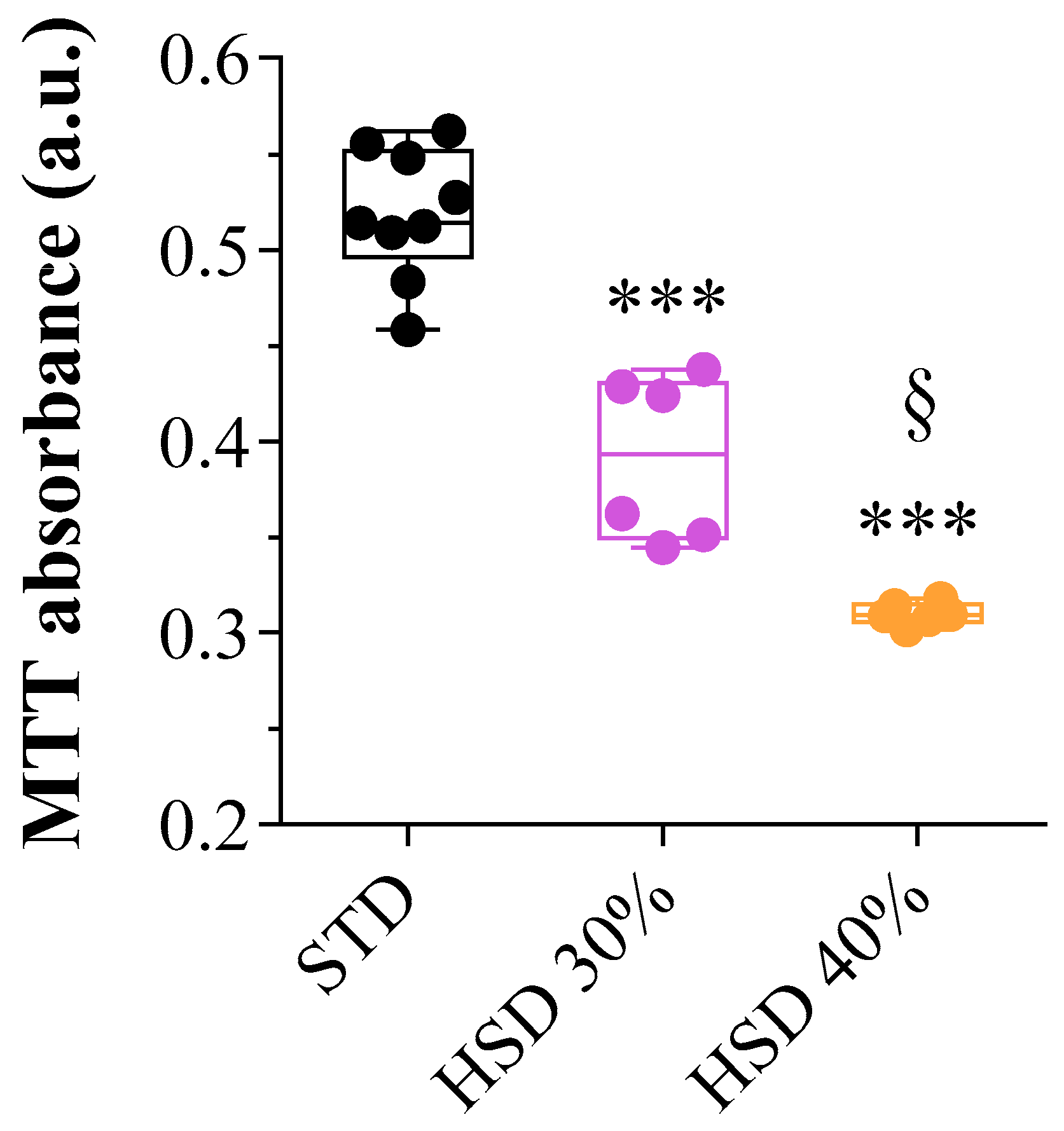{kind=link}
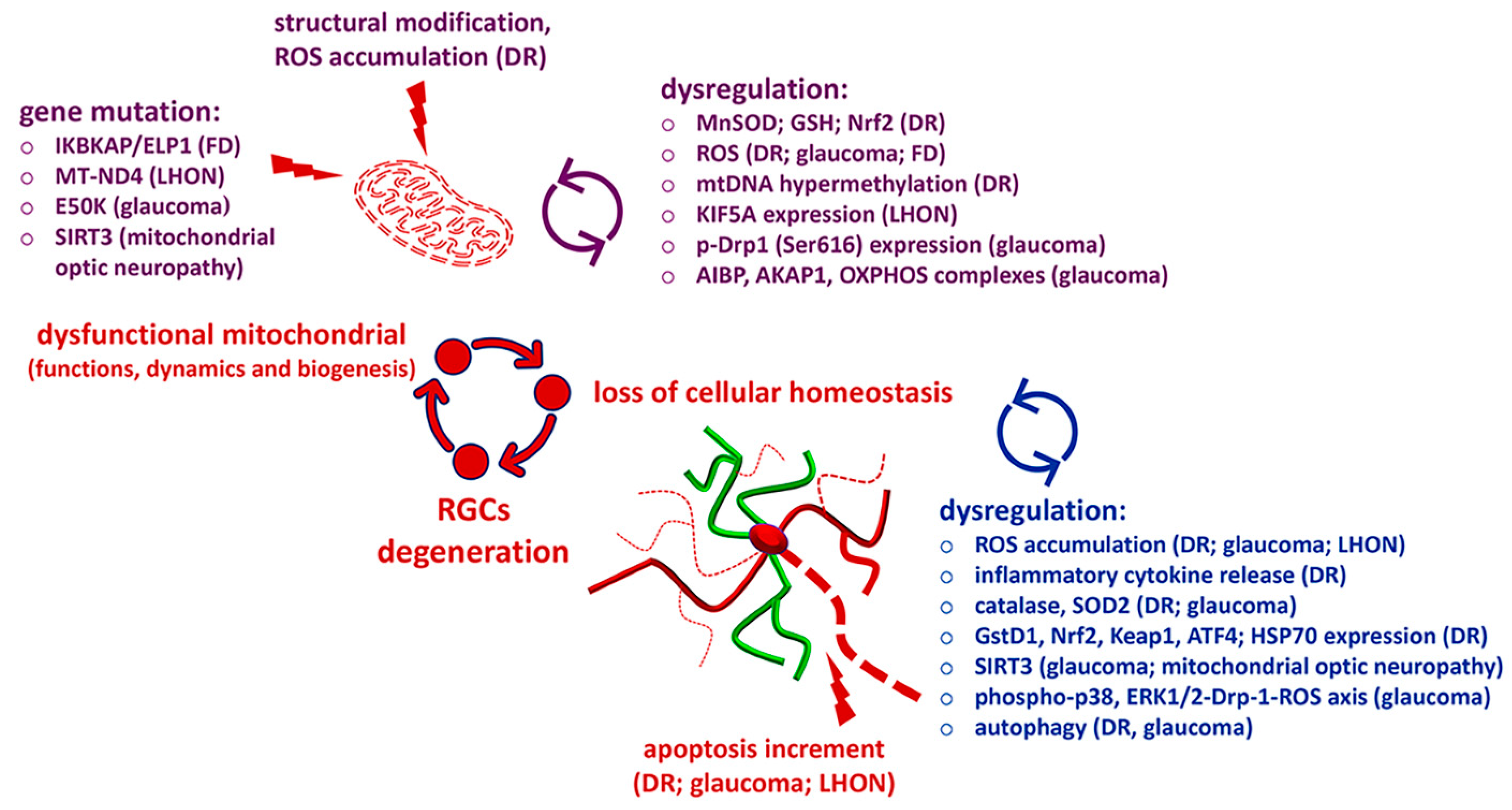{kind=link}
{kind=link}
1. Introduction
2. Mitochondrial Impairment Is Involved in RGC Degeneration
3. Mitochondria Homeostasis as a Target against RGC Degeneration
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Shu, D.Y.; Chaudhary, S.; Cho, K.S.; Lennikov, A.; Miller, W.P.; Thorn, D.C.; Yang, M.; McKay, T.B. Role of Oxidative Stress in Ocular Diseases: A Balancing Act. Metabolites 2023, 13, 187. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Fisher, C.R.; Kowluru, R.A. Mitochondrial Defects Drive Degenerative Retinal Diseases. Trends Mol. Med. 2020, 26, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Domenech, E.B.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Viegas, F.O.; Neuhauss, S.C.F. A Metabolic Landscape for Maintaining Retina Integrity and Function. Front. Mol. Neurosci. 2021, 14, 656000. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Roy, S.; Kim, D.; Sankaramoorthy, A. Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy. J. Clin. Med. 2019, 8, 1363. [Google Scholar] [CrossRef]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Tezel, G. Multifactorial Pathogenic Processes of Retinal Ganglion Cell Degeneration in Glaucoma towards Multi-Target Strategies for Broader Treatment Effects. Cells 2021, 10, 1372. [Google Scholar] [CrossRef]
- Cojocaru, K.A.; Luchian, I.; Goriuc, A.; Antoci, L.M.; Ciobanu, C.G.; Popescu, R.; Vlad, C.E.; Blaj, M.; Foia, L.G. Mitochondrial Dysfunction, Oxidative Stress, and Therapeutic Strategies in Diabetes, Obesity, and Cardiovascular Disease. Antioxidants 2023, 12, 658. [Google Scholar] [CrossRef]
- Rajlic, S.; Treede, H.; Munzel, T.; Daiber, A.; Duerr, G.D. Early Detection Is the Best Prevention-Characterization of Oxidative Stress in Diabetes Mellitus and Its Consequences on the Cardiovascular System. Cells 2023, 12, 583. [Google Scholar] [CrossRef]
- Kowluru, R.A. Cross Talks between Oxidative Stress, Inflammation and Epigenetics in Diabetic Retinopathy. Cells 2023, 12, 300. [Google Scholar] [CrossRef] [PubMed]
- Mirra, S.; Marfany, G. Mitochondrial Gymnastics in Retinal Cells: A Resilience Mechanism Against Oxidative Stress and Neurodegeneration. Adv. Exp. Med. Biol. 2019, 1185, 513–517. [Google Scholar] [PubMed]
- Sanz-Morello, B.; Ahmadi, H.; Vohra, R.; Saruhanian, S.; Freude, K.K.; Hamann, S.; Kolko, M. Oxidative Stress in Optic Neuropathies. Antioxidants 2021, 10, 1538. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kim, J.; Shin, C.; Lee, S. Intersection between Redox Homeostasis and Autophagy: Valuable Insights into Neurodegeneration. Antioxidants 2021, 10, 694. [Google Scholar] [CrossRef]
- Surma, M.; Anbarasu, K.; Dutta, S.; Olivera Perez, L.J.; Huang, K.C.; Meyer, J.S.; Das, A. Enhanced mitochondrial biogenesis promotes neuroprotection in human pluripotent stem cell derived retinal ganglion cells. Commun. Biol. 2023, 6, 218. [Google Scholar] [CrossRef]
- Kang, E.Y.; Liu, P.K.; Wen, Y.T.; Quinn, P.M.J.; Levi, S.R.; Wang, N.K.; Tsai, R.K. Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration. Antioxidants 2021, 10, 1948. [Google Scholar] [CrossRef]
- Rezaie, T.; Child, A.; Hitchings, R.; Brice, G.; Miller, L.; Coca-Prados, M.; Heon, E.; Krupin, T.; Ritch, R.; Kreutzer, D.; et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef]
- Wu, Y.R.; Wang, A.G.; Chen, Y.T.; Yarmishyn, A.A.; Buddhakosai, W.; Yang, T.C.; Hwang, D.K.; Yang, Y.P.; Shen, C.N.; Lee, H.C.; et al. Bioactivity and gene expression profiles of hiPSC-generated retinal ganglion cells in MT-ND4 mutated Leber’s hereditary optic neuropathy. Exp. Cell Res. 2018, 363, 299–309. [Google Scholar] [CrossRef]
- Yang, T.C.; Yarmishyn, A.A.; Yang, Y.P.; Lu, P.C.; Chou, S.J.; Wang, M.L.; Lin, T.C.; Hwang, D.K.; Chou, Y.B.; Chen, S.J.; et al. Mitochondrial transport mediates survival of retinal ganglion cells in affected LHON patients. Hum. Mol. Genet. 2020, 29, 1454–1464. [Google Scholar] [CrossRef]
- Zeng, Z.; You, M.; Fan, C.; Rong, R.; Li, H.; Xia, X. Pathologically high intraocular pressure induces mitochondrial dysfunction through Drp1 and leads to retinal ganglion cell PANoptosis in glaucoma. Redox Biol. 2023, 62, 102687. [Google Scholar] [CrossRef]
- Giovarelli, M.; Zecchini, S.; Martini, E.; Garre, M.; Barozzi, S.; Ripolone, M.; Napoli, L.; Coazzoli, M.; Vantaggiato, C.; Roux-Biejat, P.; et al. Drp1 overexpression induces desmin disassembling and drives kinesin-1 activation promoting mitochondrial trafficking in skeletal muscle. Cell Death Differ. 2020, 27, 2383–2401. [Google Scholar] [CrossRef] [PubMed]
- Bastola, T.; Perkins, G.A.; Kim, K.Y.; Choi, S.; Kwon, J.W.; Shen, Z.; Strack, S.; Ju, W.K. Role of A-Kinase Anchoring Protein 1 in Retinal Ganglion Cells: Neurodegeneration and Neuroprotection. Cells 2023, 12, 1539. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Perkins, G.A.; Kim, K.Y.; Kong, Y.; Lee, Y.; Choi, S.H.; Liu, Y.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Zangwill, L.; et al. Loss of AKAP1 triggers Drp1 dephosphorylation-mediated mitochondrial fission and loss in retinal ganglion cells. Cell Death Dis. 2020, 11, 254. [Google Scholar] [CrossRef]
- Ueki, Y.; Shchepetkina, V.; Lefcort, F. Retina-specific loss of Ikbkap/Elp1 causes mitochondrial dysfunction that leads to selective retinal ganglion cell degeneration in a mouse model of familial dysautonomia. Dis. Model. Mech. 2018, 11, dmm033746. [Google Scholar] [CrossRef] [PubMed]
- Chun, B.Y.; Choi, J.M.; Hwang, S.K.; Rhiu, S. Sirtuin 3 mutation-induced mitochondrial dysfunction and optic neuropathy: A case report. BMC Ophthalmol. 2023, 23, 118. [Google Scholar] [CrossRef]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef]
- Jiang, D.; Xiong, G.; Feng, H.; Zhang, Z.; Chen, P.; Yan, B.; Chen, L.; Gandhervin, K.; Ma, C.; Li, C.; et al. Donation of mitochondria by iPSC-derived mesenchymal stem cells protects retinal ganglion cells against mitochondrial complex I defect-induced degeneration. Theranostics 2019, 9, 2395–2410. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, K.Y.; Perkins, G.A.; Phan, S.; Edwards, G.; Xia, Y.; Kim, J.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Ellisman, M.H.; et al. AIBP protects retinal ganglion cells against neuroinflammation and mitochondrial dysfunction in glaucomatous neurodegeneration. Redox Biol. 2020, 37, 101703. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Kiyama, T.; Chen, C.K.; Wang, S.W.; Pan, P.; Ju, Z.; Wang, J.; Takada, S.; Klein, W.H.; Mao, C.A. Essential roles of mitochondrial biogenesis regulator Nrf1 in retinal development and homeostasis. Mol. Neurodegener. 2018, 13, 56. [Google Scholar] [CrossRef]
- Nascimento-Dos-Santos, G.; de-Souza-Ferreira, E.; Lani, R.; Faria, C.C.; Araujo, V.G.; Teixeira-Pinheiro, L.C.; Vasconcelos, T.; Goncalo, T.; Santiago, M.F.; Linden, R.; et al. Neuroprotection from optic nerve injury and modulation of oxidative metabolism by transplantation of active mitochondria to the retina. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165686. [Google Scholar] [CrossRef] [PubMed]
- Chadderton, N.; Palfi, A.; Maloney, D.M.; Carrigan, M.; Finnegan, L.K.; Hanlon, K.S.; Shortall, C.; O’Reilly, M.; Humphries, P.; Cassidy, L.; et al. Optimisation of AAV-NDI1 Significantly Enhances Its Therapeutic Value for Correcting Retinal Mitochondrial Dysfunction. Pharmaceutics 2023, 15, 322. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.; Catalani, E.; Dal Monte, M.; Cammalleri, M.; Cervia, D.; Casini, G. Morpho-functional analysis of the early changes induced in retinal ganglion cells by the onset of diabetic retinopathy: The effects of a neuroprotective strategy. Pharmacol. Res. 2022, 185, 106516. [Google Scholar] [CrossRef] [PubMed]
- Catalani, E.; Cervia, D. Diabetic retinopathy: A matter of retinal ganglion cell homeostasis. Neural Regen. Res. 2020, 15, 1253–1254. [Google Scholar] [CrossRef] [PubMed]
- Cervia, D.; Fehlmann, D.; Hoyer, D. Native somatostatin sst2 and sst5 receptors functionally coupled to Gi/o-protein, but not to the serum response element in AtT-20 mouse tumour corticotrophs. Naunyn Schmiedebergs Arch. Pharmacol. 2003, 367, 578–587. [Google Scholar] [CrossRef]
- Pavan, B.; Fiorini, S.; Dal Monte, M.; Lunghi, L.; Biondi, C.; Bagnoli, P.; Cervia, D. Somatostatin coupling to adenylyl cyclase activity in the mouse retina. Naunyn Schmiedebergs Arch. Pharmacol. 2004, 370, 91–98. [Google Scholar] [CrossRef]
- Cammalleri, M.; Bagnoli, P.; Bigiani, A. Molecular and Cellular Mechanisms Underlying Somatostatin-Based Signaling in Two Model Neural Networks, the Retina and the Hippocampus. Int. J. Mol. Sci. 2019, 20, 2506. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, N.; Li, Q.; Hu, X.; Wang, L.; Sun, J.G.; Wang, Z.; Sun, X.H. Neuroprotective effect of the somatostatin receptor 5 agonist L-817,818 on retinal ganglion cells in experimental glaucoma. Exp. Eye Res. 2021, 204, 108449. [Google Scholar] [CrossRef]
- Gomes-Porras, M.; Cardenas-Salas, J.; Alvarez-Escola, C. Somatostatin Analogs in Clinical Practice: A Review. Int. J. Mol. Sci. 2020, 21, 1682. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Wu, N.; Yin, N.; Sun, X.H.; Wang, Z. Activation of somatostatin receptor 5 suppresses T-type Ca2+ channels through NO/cGMP/PKG signaling pathway in rat retinal ganglion cells. Neurosci. Lett. 2019, 708, 134337. [Google Scholar] [CrossRef]
- Amato, R.; Catalani, E.; Dal Monte, M.; Cammalleri, M.; Di Renzo, I.; Perrotta, C.; Cervia, D.; Casini, G. Autophagy-mediated neuroprotection induced by octreotide in an ex vivo model of early diabetic retinopathy. Pharmacol. Res. 2018, 128, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Catalani, E.; Del Quondam, S.; Brunetti, K.; Cherubini, A.; Bongiorni, S.; Taddei, A.R.; Zecchini, S.; Giovarelli, M.; De Palma, C.; Perrotta, C.; et al. Neuroprotective role of plumbagin on eye damage induced by high-sucrose diet in adult fruit fly Drosophila melanogaster. Biomed. Pharmacother. 2023, 166, 115298. [Google Scholar] [CrossRef]
- Catalani, E.; Fanelli, G.; Silvestri, F.; Cherubini, A.; Del Quondam, S.; Bongiorni, S.; Taddei, A.R.; Ceci, M.; De Palma, C.; Perrotta, C.; et al. Nutraceutical Strategy to Counteract Eye Neurodegeneration and Oxidative Stress in Drosophila melanogaster Fed with High-Sugar Diet. Antioxidants 2021, 10, 1197. [Google Scholar] [CrossRef] [PubMed]
- Catalani, E.; Silvestri, F.; Bongiorni, S.; Taddei, A.R.; Fanelli, G.; Rinalducci, S.; De Palma, C.; Perrotta, C.; Prantera, G.; Cervia, D. Retinal damage in a new model of hyperglycemia induced by high-sucrose diets. Pharmacol. Res. 2021, 166, 105488. [Google Scholar] [CrossRef]
- Navneet, S.; Zhao, J.; Wang, J.; Mysona, B.; Barwick, S.; Ammal Kaidery, N.; Saul, A.; Kaddour-Djebbar, I.; Bollag, W.B.; Thomas, B.; et al. Hyperhomocysteinemia-induced death of retinal ganglion cells: The role of Muller glial cells and NRF2. Redox Biol. 2019, 24, 101199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, T.; Zeng, S.; Zhang, X.; Zhou, F.; Gillies, M.C.; Zhu, L. The Role of Nrf2/sMAF Signalling in Retina Ageing and Retinal Diseases. Biomedicines 2023, 11, 1512. [Google Scholar] [CrossRef] [PubMed]
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 2008, 205, 1227–1242. [Google Scholar] [CrossRef]
- Pitale, P.M.; Gorbatyuk, O.; Gorbatyuk, M. Neurodegeneration: Keeping ATF4 on a Tight Leash. Front. Cell. Neurosci. 2017, 11, 410. [Google Scholar] [CrossRef]
- Wang, N.; Luo, Z.; Jin, M.; Sheng, W.; Wang, H.T.; Long, X.; Wu, Y.; Hu, P.; Xu, H.; Zhang, X. Exploration of age-related mitochondrial dysfunction and the anti-aging effects of resveratrol in zebrafish retina. Aging 2019, 11, 3117–3137. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Luo, L.J.; Yang, C.J.; Lai, J.Y. Highly Retina-Permeating and Long-Acting Resveratrol/Metformin Nanotherapeutics for Enhanced Treatment of Macular Degeneration. ACS Nano 2023, 17, 168–183. [Google Scholar] [CrossRef]
- Zeviani, M.; Carelli, V. Mitochondrial Retinopathies. Int. J. Mol. Sci. 2021, 23, 210. [Google Scholar] [CrossRef] [PubMed]
- Birtel, J.; von Landenberg, C.; Gliem, M.; Gliem, C.; Reimann, J.; Kunz, W.S.; Herrmann, P.; Betz, C.; Caswell, R.; Nesbitt, V.; et al. Mitochondrial Retinopathy. Ophthalmol. Retina 2022, 6, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jiang, K.; McIlmoyle, B.; To, E.; Xu, Q.A.; Hirsch-Reinshagen, V.; Mackenzie, I.R.; Hsiung, G.R.; Eadie, B.D.; Sarunic, M.V.; et al. Amyloid Beta Immunoreactivity in the Retinal Ganglion Cell Layer of the Alzheimer’s Eye. Front. Neurosci. 2020, 14, 758. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.O.; Acosta, M.L.; Di Angelantonio, S.; Salt, T.E. Editorial: Seeing Beyond the Eye: The Brain Connection. Front. Neurosci. 2021, 15, 719717. [Google Scholar] [CrossRef] [PubMed]
- Catalani, E.; Bongiorni, S.; Taddei, A.R.; Mezzetti, M.; Silvestri, F.; Coazzoli, M.; Zecchini, S.; Giovarelli, M.; Perrotta, C.; De Palma, C.; et al. Defects of full-length dystrophin trigger retinal neuron damage and synapse alterations by disrupting functional autophagy. Cell Mol. Life Sci. 2021, 78, 1615–1636. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.; Schnichels, S. Editorial: Brain vs. retina—Differences and commonalities: The role of oxidative stress in neurodegenerative diseases. Front. Neurosci. 2023, 17, 1171235. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, T.; Deore, S.L.; Kide, A.A.; Shende, B.A.; Sharma, R.; Dadarao Chakole, R.; Nemade, L.S.; Kishor Kale, N.; Borah, S.; Shrikant Deokar, S.; et al. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and Amyotrophic Lateral Sclerosis—An updated review. Mitochondrion 2023, 71, 83–92. [Google Scholar] [CrossRef]
- Tarozzi, A. Oxidative Stress in Neurodegenerative Diseases: From Preclinical Studies to Clinical Applications. J. Clin. Med. 2020, 9, 1223. [Google Scholar] [CrossRef]
- Domanskyi, A.; Parlato, R. Oxidative Stress in Neurodegenerative Diseases. Antioxidants 2022, 11, 504. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Aslkhodapasandhokmabad, H.; Henninger, N.; Demillard, L.J.; Nikanfar, M.; Nourazarian, A.; Ren, J. Targeting autophagy in neurodegenerative diseases: From molecular mechanisms to clinical therapeutics. Clin. Exp. Pharmacol. Physiol. 2021, 48, 943–953. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catalani, E.; Brunetti, K.; Del Quondam, S.; Cervia, D. Targeting Mitochondrial Dysfunction and Oxidative Stress to Prevent the Neurodegeneration of Retinal Ganglion Cells. Antioxidants 2023, 12, 2011. https://doi.org/10.3390/antiox12112011
Catalani E, Brunetti K, Del Quondam S, Cervia D. Targeting Mitochondrial Dysfunction and Oxidative Stress to Prevent the Neurodegeneration of Retinal Ganglion Cells. Antioxidants. 2023; 12(11):2011. https://doi.org/10.3390/antiox12112011
Chicago/Turabian StyleCatalani, Elisabetta, Kashi Brunetti, Simona Del Quondam, and Davide Cervia. 2023. "Targeting Mitochondrial Dysfunction and Oxidative Stress to Prevent the Neurodegeneration of Retinal Ganglion Cells" Antioxidants 12, no. 11: 2011. https://doi.org/10.3390/antiox12112011